

1 Introduction
Oak heartwood is rich in lignin, 25–30% of the weight of dry wood [1–4]. Oak wood lignins, like other angiosperms, are polymers formed principally by the copolymerization of two phenylpropenoic alcohols: hydroxy-4-methoxy-3-cinnamic alcohol (guaiacyl structure; coniferyl alcohol), hydroxy-4-dimethoxy-3.5-cinnamic alcohol (syringyl structure; synapylic alcohol). Structure and composition of industrial woods are now well known, in particular the natural lignins from Betula sp. [5,6], Populus sp. [7–10], Ginkgo biloba [11], Pinus sp. [12], for major contribution and synthetic lignins [13]. There is less information concerning oak wood lignins and its NMR studies are very limited. Oak wood lignins have a very important impact on the wood properties for wine and spirit ageing, especially the hydrophobicity of lignins which contribute to the impermeability and decrease the frequency of leakage [14,15]. Lignins, regarding their composition represent a wide source of aromatic compounds produced during the toasting treatment. In the particular area of spirit production, lignans derived from lignin degradation, contribute to the colour of the final commercial product [16].
NMR techniques permit qualitative characterization of native or acetylated lignins, the principal intermonomeric units and the side chain structures [17–19]. The 1H NMR technique gives limited information, only focusing on the different types of protons present in the phenolic groups and side chains. Despite its lower sensitivity, the 13C NMR technique is much more powerful for the structural investigation of macromolecules. In addition, under specific experimental conditions for spectral recording, it is possible to perform quantitative structural analyses [20]. Recent development of 2D homo- and heteronuclear techniques increases the performance of these techniques.
Our work has focused on the fractionation and structural analysis of three lignin fractions from oak wood using 1H and 13C NMR homo- and heteronuclear 2D techniques.
2 Materials and methods
2.1 Materials
Samples were taken from heartwood of Q. petraea from the Allier region of France and Q. robur from the Limousin region of France. Their botanical characteristics were checked [21]. Two sets of samples were made from wood from the same regions but from different suppliers. The trees from which the two sets of Q. petraea samples were taken were around 150–175 years old and those for the Q. robur samples were 80–110 years old. The samples representing only the heartwood part were planed to obtain woodchips, and then ground into sawdust using liquid nitrogen. No distinction was made between the different heartwood areas: near the sapwood, at the centre of growth or near the middle of the tree. They were all mixed and constituted an average representative sample of the whole heartwood.
2.2 Lignin extraction and purification
Three lignin fractions were selected for their different characteristics: lignan fraction (Ln), native Brauns lignin or ethanol lignin (LE), and dioxan lignin (LD).
2.2.1 Extraction and purification of Ln
Sawdust (100 g) was extracted after 2 months at 20 °C (± 2 °C) with a hydroalcoholic solution (1 l, 12% vol. EtOH, 5 g/l tartaric acid, NaOH to adjust pH to 3.5). The solution was then filtered and evaporated in a rotary evaporator (40 °C). The dry extract was placed in a minimum quantity of water and deposited onto a LH20 column (Pharmacia, 80 × 3 cm column), eluted successively with water (1 l), MeOH with 20% water (1 l) and finally MeOH (1 l). The last fraction was collected, and the methanol was eliminated in the rotary evaporator; the dry residue was then placed in water (100 ml) and extracted with dichloromethane (2 × 100 ml, 1 × 50 ml). The dichloromethane was then evaporated and the resin extract was washed with chloroform (3 × 50 ml). The fraction was then purified with water (3 × 50 ml). After each stage the precipitate was centrifuged (3500 rpm, 15 min). The Ln fraction thus obtained, corresponding to the water precipitate, was freeze-dried and stored at –20 °C. Then a second step of purification was performed in order to separate the two major lignans for NMR studies. The Ln fraction thus obtained was analysed by cellulose HPTLC with chloroform/ethyl acetate/formic acid (50:40:10, v/v/v). After migration, spectrometric reading at 340 nm revealed the existence of 6 distinct bands of Rf: 1 (0.17), 2 (0.31), 3 (0.35), 4 (0.51), 5 (0.62), 6 (0.80). The colours of these bands varied from scarlet to orange. Each band was recovered from the stationary phase by scraping and eluted with MeOH. After centrifugation and concentration using a rotary evaporator, the products were applied onto a C18 column. Elution was successively with water, MeOH/water (1:1, v/v) then MeOH. The methanol fraction was collected, dried in a rotary evaporator, then precipitated once with water then lyophilised. The UV–visible spectra for fractions 1–4 gave a double shoulder at 330 nm and 380 nm. The last two fractions, 5 and 6, gave a single shoulder at 340 nm. All fractions collected absorbed at 280 nm. The chromatographic purity of the samples was verified by silica HPLC analysis (solvent: dioxan/water, 1:1, v/v). Only fraction 5, the most abundant, was chromatographically pure. The other fractions required further purification. Fraction 5 was, therefore, studied using LSIMS and NMR. The purified band consisted of a 60/40 mixture of two lignans. The molecular weights were determined as 463 (2 or 3 units, Ln1) and 610 (3 or 4 units, Ln2) by LSIMS in positive mode. It was not possible to separate these two molecules under our conditions, probably due to their similar structures.
2.2.2 Extraction and purification of LE
The extractable part of sawdust (100 g) was removed by maceration in water (3 × 1 l, 40 °C, 12H), followed by chloroform (1 × 1 l, 20 °C, 12H). The sawdust was then oven-dried (30 °C, 24H) and macerated in 95% vol. EtOH (5 l) adjusted to pH = 5 with acetic acid (24 months at room temperature in the dark). The extract was filtered, concentrated to 100 ml in a rotary evaporator (40 °C) and purified by five successive precipitations in water (precipitation: 5 × 1 l of water at 5 °C, centrifugation: (3500 rpm, 15 min). The powder thus obtained was washed with diethyl ether (2 × 50 ml), freeze-dried and stored in a desiccator. The powder obtained is of a light beige colour.
2.2.3 Extraction and purification of LD
The extractable part of sawdust (100 g) was removed by maceration in water (3 × 1 l, 40 °C, 12H), followed by chloroform (1 × 1 l, 20 °C, 12H) and finally by EtOH/H2O (2 × 1 l, 9:1, 20 °C, 12H). The sawdust was then oven-dried (30 °C, 24H) prior to 6H extraction with dioxan (1 l) containing 12 N HCl (100 ml). The mixture was kept under reflux. The extracted liquid was filtered and evaporated in the rotary evaporator (40 °C) before being processed in the same way as LE. The powder obtained is a light buff-colour.
2.3 NMR experiments
All the 1D and 2D NMR spectra were performed using a Bruker DPX 400 apparatus equipped with an inverse broad band 5 mm diameter probe.
2.3.1 Sample preparation
The different fractions of acetylated lignins of LD and LE (pyridine/glacial acetic acid, 1:1, v/v at room temperature, isolation performed by precipitation twice in water, twice in methanol and finally dissolved twice in chloroform with rotary evaporation and were freeze-dried before analysis) were dissolved in CDCl3, or in CH3OD for native lignan (Ln) in a final volume of 0.6 ml. Approximately 20 mg of the different lignin fractions were used for the experiments.
2.3.2 NMR spectra
Standard proton spectra were usually obtained using the following parameters: a spectral width of 4800 Hz (12 ppm), a pulse width of 7 μs (corresponding to a 90° flip angle), a repetition time of 7.4 s (3.4 s for the acquisition time and 4s for the interpulse delay), and 64 scans. Standard 13C spectra were obtained using a 90° pulse angle (8 μs) using garp decoupling throughout the experiment in order to generate the ‘NOE’ effect. The repetition time was close to 2 s (0.5 s for the acquisition time and 1.5 s interpulse delay).
2.3.3 1H–1H 2D experiments
Phase-sensitive TOCSY spectra were acquired over a 4800 Hz window using a LKX512 matrix. 256 increments were obtained with 16 scans per tl increment. The MLEV-17 spin-lock period of 40 and 100 ms was preceded and followed by 2.5 ms trim-pulses. Prior to the Fourier transform, a sine-bell squared function shifted by π/3 was applied upon the two dimensions.
2.3.4 1H–13C 2D experiments
The phase-sensitive 1H detected HMQC experiments were performed using a B0 gradient field with the following parameters: the F2 windows were close to 4800 Hz (12 ppm) and a F1 window of 22 000 Hz (210 ppm) with a LKX256 matrix (zero-filled to lk in the F1 dimension) and 64 scans per tl increment. The delay for polarization transfer was set for an assumed lJCH of 130 Hz (0.0038 s). Prior to the Fourier transform, a sine-bell function of 3π was applied upon the two dimensions.
3 Results and discussion
3.1 LE and LD lignins
One-dimensional 1H NMR gave little structural information (Fig. 1). This spectrum has the form of large envelopes grouping a family of protons. Comparison of signals for syringyl (Hs) and guaiacyl (Hg) aromatic protons indicates that LD's signal is mainly attributable to Hs, whereas LE has both Hs and Hg signals at approximately equal intensity. These results are in agreement with previous thioacydolysis studies [22]. Taking into account work by other authors [9,17–19,23–25], the 13C NMR signals obtained could be attributed by comparison with the numerous standard synthetic molecule 13C NMR spectra (Fig. 2) gives more information. The two 13C NMR spectra can be divided into three distinct zones: 200–165 ppm for carbonyl C groups, 165–100 ppm for aromatic C groups and 100–50 ppm for lateral chain C. It is evident that for equivalent lignin amounts, the resolution of the LE spectrum is better than that of LD. This difference is probably due to the higher molecular weight of LD [26]. The aromatic carbon signals for syringyl units are greater than those for guaiacyl for LD compared with LE. For LE, the proportion of both aromatic C groups is the same. As was seen with the proton spectra, contaminant polysaccharide signals are present, which appear to be more abundant for LE than LD. This is probably due to extraction conditions.

1H NMR (CDCl3) spectra of paracetylated lignin fractions. S: solvent; P: polysaccharide contaminant; a.ar: aromatic acetates; a.al: aliphatic acetates; Hs, Hg: aromatic protons of syringyl and guaiacyl motifs, respectively.
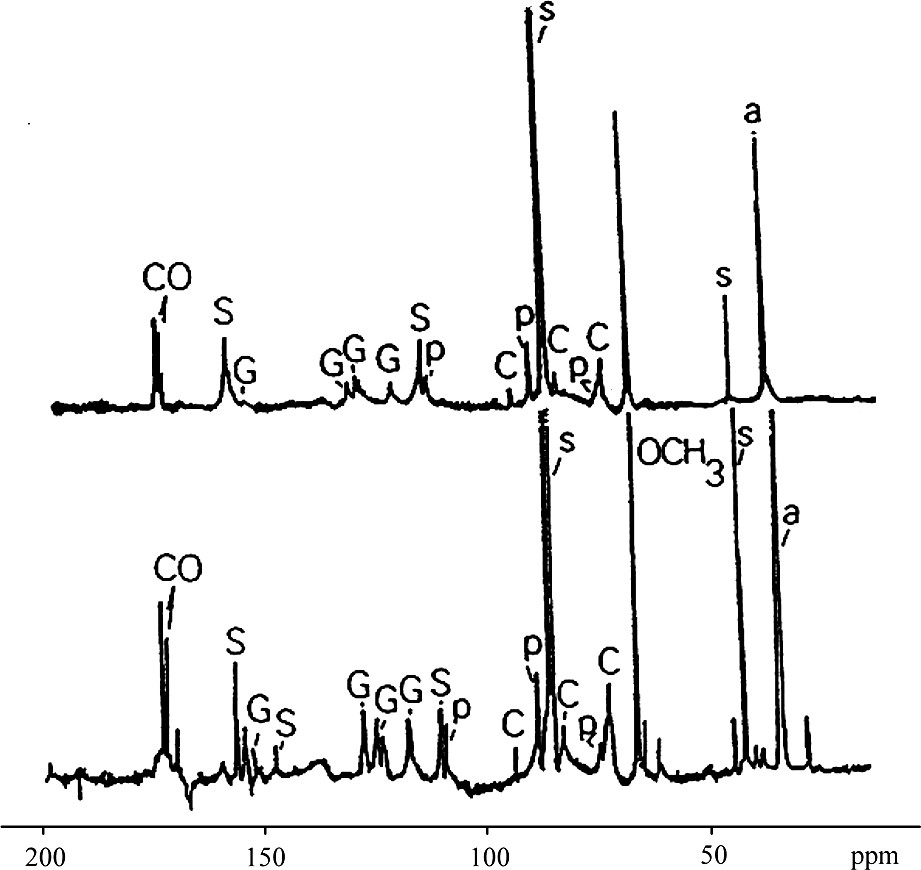
13C spectra (CDCl3) of LE and LD paracetylated lignin fractions.
S: solvent; P: polysaccharide contaminant; a: acetates; CO: carbonyls
Signals are attributed from right to left:
● C, carbons of side chains of aryl-glycerol and aryl ether structures.
● S, aromatic carbons of syringyl groups (2,6 with αCHOO), (4 etherified), and (3,5 etherified).
● G, aromatic carbons of guaiacyl groups 2, 5, 6 and 3.
All 1H and 13C resonances were assigned by the aid of 2D 1H{13C}HMQC, and TOCSY spectra (Table 1).The choice of TOCSY rather than COSY experiments was due to the extremely short relaxation time of the signals generally observed in large molecules. By a judicious choice of a pair of spin-lock mixing times (a short one around 40 ms and a longer one close to 100 ms) we could assign, on one hand, 3J 1H–1H indirect spin–spin coupling and, on the other hand, a total spin system. By the combination of data obtained by the different NMR techniques, LD and LE lignin structures of oak wood could be analysed qualitatively and quantitatively.
1H and 13C NMR attribution of lignin fractions LE (chemical shifts are given in ppm relative to TMS)
| No. | δ | Attributions | |
| 1H | 13C | ||
| 1 | – | 171 | Acetyl COOH |
| 2 | – | 153 | S3,5 |
| 3 | 7.0 | 123 | G6 |
| 4 | 6.9–7.0 | 119 | G5 |
| 5 | 7.05 | 112 | G2 |
| 6 | 6.6 | 104 | S2,6 |
| 7 | 6.0 | 74 | α(β-O-4)a |
| 8 | 4.75 | 86 | α(β–β) |
| 9 | 4.6 | 81 | β(β-O-4)a |
| 10 | 3.9–4.3 | 72 | γ(β–β)b |
| 11 | 4.1–4.4 | 63 | γ, γ(β-O-4)a |
| 12 | 3.7 | 56 | OCH3 |
| 13 | 3.1 | 54 | β(β–β) |
| 14 | 2.3 | 21 | Acetyl phenolic CH3 |
| 15 | 2.0 | 21 | Acetyl CH3 of aliphatic chains |
a With both erythro–threo forms.
b Axial and equatorial.
3.1.1 Side-chain structure
1D 1H spectra of high molecular weight lignin always give the same pattern (Fig. 1) making analysis of such a spectrum difficult. Nevertheless, the TOCSY experiment is a powerful tool to analyse the side chain structure of lignin, since the coupling network can give access to the type of C3 units. Two major types of C3 units were revealed in the two high molecular weight lignin LE and LD; Fig. 3 shows an example of the spectra obtained with the LE fraction:
- ● β-O-4 aryl ether (Fig. 4), which exists in the erythro and the threo forms. These two forms are revealed in the proton resonance of the proton linked to the Cα carbon of the unit; the α resonance of the erythro isomer has a resonance close to 6.0 ppm and the α proton of the threo isomer has a resonance close to 6.1 ppm. The ratio of these two isomers, determined from the 1D 1H spectrum, is close to 1 for LE and LD. This type of unit represents the major C3 unit component for both species (Table 2).
- ● Another observable C3 type unit is the pinoresinol residue (Fig. 4). Its presence was revealed by the TOCSY experiment, since we were able to observe four resonances on the 2D TOCSY contour plot, (Fig. 3). These resonances exibit characteristic chemical shifts of proton α, β and the two γ protons, reflecting the axial and equatorial protons of this type of residue.
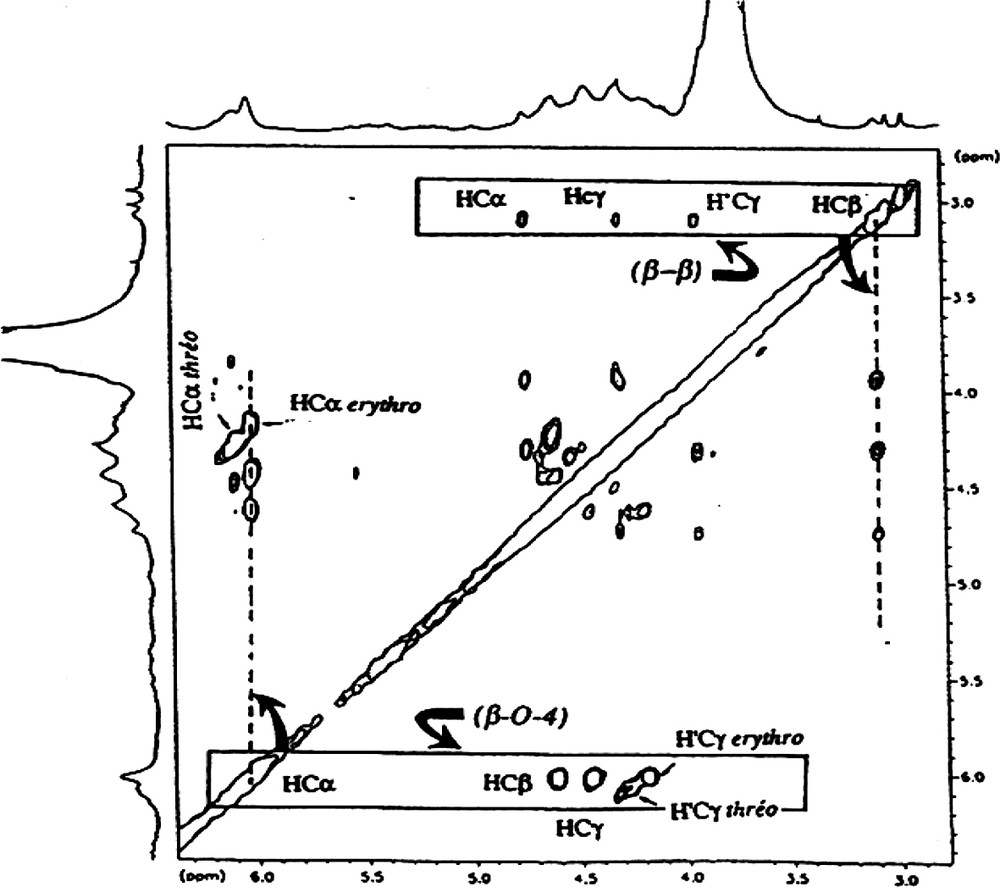
Part of TOCSY spectrum of LE fraction. Attribution of symbols to the protons: α, β, γ correspond to Hα, Hβ and hγ, respectively. Attribution of symbols to the side chains; (β−β), pinoresinol linkage; (β-O-4), β-O-4 linkage.
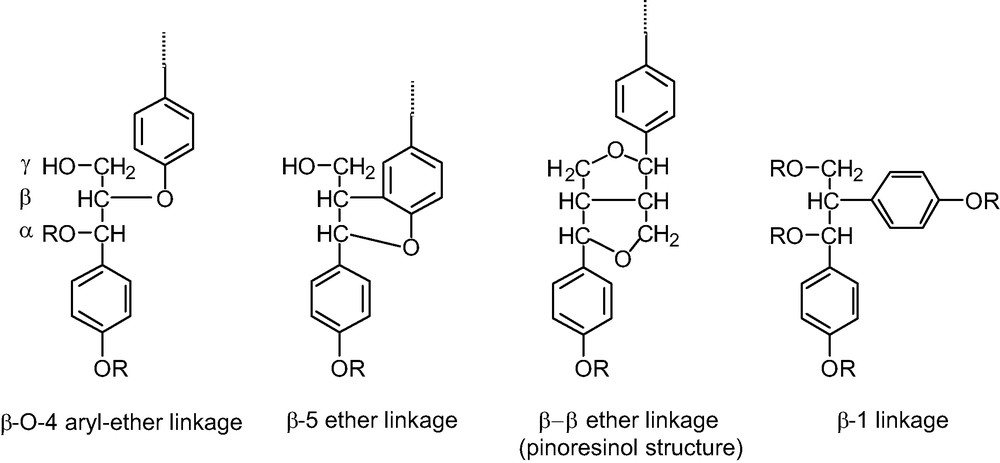
Main typical side-chain structure of lignins.
Determination of the proportion of the different elements constituting lignins LE and LD
| LE | LD | ||
| Side chains | nβ-O-4a | 10 | 9 |
| β-O-4 erythro–threo | 1.0 | 1.0 | |
| nβ–βa | 0.3 | 1 | |
| Aromatic ring | nSa | 8 | 16 |
| nGa | 7 | 6 | |
| S/Gb | 1.1 | 2.7 | |
| β-O-4/G | 1.3 | 1.5 | |
| β–β/G | 0.05 | 0.2 |
a The number of β-O-4, β–β linkages and the G and S aromatic units is calculated from the MW determined by LSIMS (MW: LE = 2900, LD = 3900).
b Ratio of syringyl/guaiacyl units.
No unattached side chain was observed in these two lignin fractions, such as coniferyl alcohol or aldehyde structures. Therefore LE and LD have very characteristic structures of the ‘end-wise lignin’.
3.1.2 Phenolic groups
Two methods were used to determine the structure of phenolic groups in LE and LD. The characteristic 1H chemical shift of the two major types of phenolic groups, syringyl and guaiacyl groups are slightly different (close to 6.6 ppm for syringyl units and close to 7.1 ppm for guaiacyl units). It is therefore possible to observe and to quantify the presence of these two groups (Fig. 1). The 13C chemical shifts of these two phenolic groups are also characteristic, particularly the carbon linked to a proton which are easily observable in 13C spectrum (close to 108 ppm for C2 and C6 of the syringyl type unit, and close to 116 ppm for C2 and C5 and. 122 ppm for C6 of guaiacyl units). 1H{13C} HMQC experiments revealed these two types of phenolic group (Appendix A). These spectra could not demonstrate the presence of phenolic groups characterised by the presence of three resonances of carboxyl linked to acetyl groups [19].
3.1.3 Quantitative aspect
The assignment done, it was then possible to quantify the ratio between syringyl and guaiacyl units and the ratio of the two side chains observed, β-O-4 aryl ether and pinoresinol in the two types of lignins LE and LD (Table 2). The quantification was determined from the proton spectra and shows that the LD fraction exhibits a higher syringyl to guaiacyl ratio than the LE fraction (1.1 and 2.7, for LE and LD, respectively). Such a result is in accordance with the ratio measured from thioacidolysis [22] and probably reflects the origin of the lignin fraction (fibre secondary wall for LD and middle lamella for LE). On the other hand, the ratio β-O-4 aryl ether and pinoresinol for the guaiacyl unit was also determined and is close to 1.3 and 0.05 for LE, and 1.5 and 0.2 for LD, for β-O-4 aryl ether and pinoresinol, respectively. The ratio (guaiacyl + 2 × syringyl)/OMe is close to 0.8 for both LE and LD and not 1 as expected. Such a value most likely reflects experimental errors since the resonances used to determine ratios are broad enough to generate measurement errors. Knowing the molecular weight average of each fraction determined by the LSIMS technique (2900 Da for LE and 3900 Da for LD) [26], and the different ratios obtained from NMR spectra, after the elucidation of the structure (Table 2), it is possible to calculate the approximate number of each structure present (β-O-4 aryl ether and pinoresinol side chains, syringyl and guaiacyl aromatic groups) for an average lignin molecule. These values are reported in Table 3 and demonstrate the higher degree of polymerisation of LD compared with LE.
Structural characteristics of LE and LD. (results given as % from Table 2)
| LE | LD | ||
| Aromatic ring substitution | S | 53 | 73 |
| G | 47 | 27 | |
| Nature of side chain linkage | β-O-4 | 97 | 90 |
| β–β | 3 | 10 | |
| Lignin structure | SG (β-O-4) | S(β-0-4) |
3.2 Lignan Ln
The total lignan fraction was too complex to allow the structural elucidation of the compounds present. Therefore, after a second purification step, we focused analysis uniquely on one major fraction of the two lignans Ln1 and Ln2 (60:40, w/w).
3.2.1 Side chains
Three different types of side chains are present:
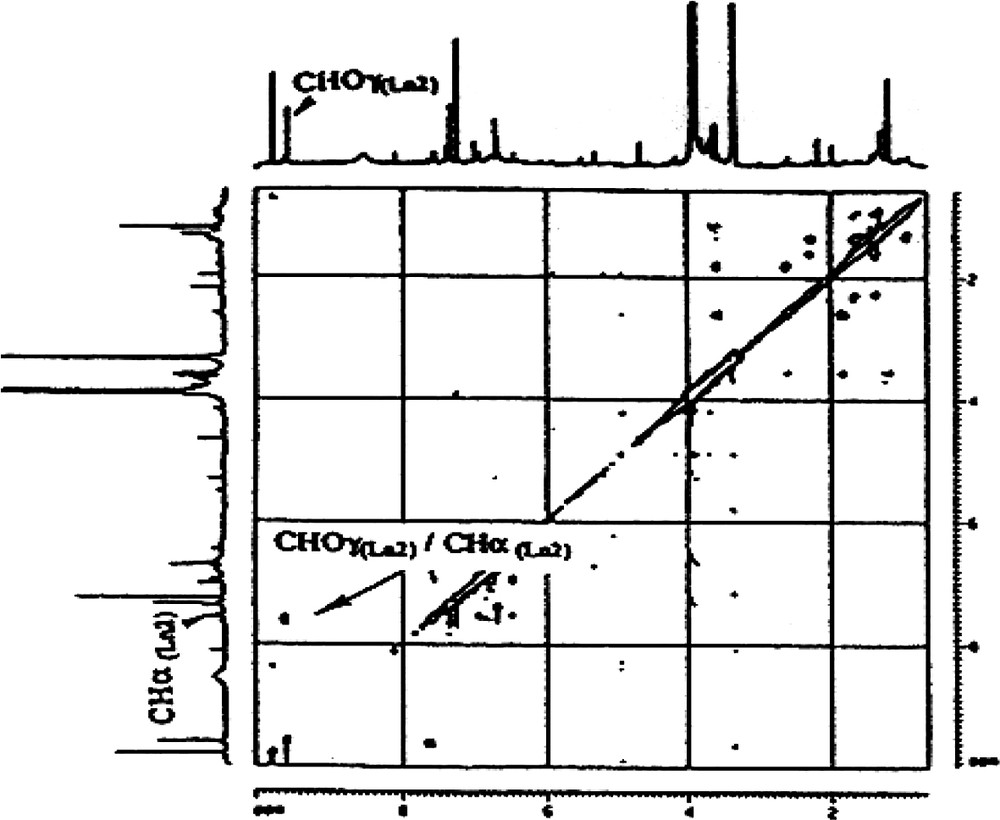
- ● cinnamaldehyde side chains (CHO–CH=CH–). These side chains are present in both lignans isolated, Ln1 and Ln2. The presence of two CHO groups was revealed by both 1H NMR (9.8 and 9.6 ppm, for Ln1 and Ln2, respectively) and by 13CNMR (192.0 and 178.6). The DEPT spectrum (Appendix B) and the 1H–13C correlation are in agreement with these attributions (Fig. 5). The presence of two highly exposed ethyl-CHs is characteristic of the two α carbons of a cinnamaldehyde type of side chain (δ 13C: 155.5 and 144.0; δ 1H: 7.6 and 7.5 for Ln1 and Ln2, respectively, Appendix B). There is a correlation between CHα and CHOγ on the TOCSY map with a mixing time of tm = 100 ms (Appendix C), indicating a linkage between the two protons in Ln2. Finally these protons belong to the CHβ resonating around 7 ppm. On the COSY spectrum (Appendix D), a correlation between CHOγ and the CHβ of Ln2 is evident. This type of coupling is not revealed for Ln1, which could suggest the absence of such a structure in Ln1. Therefore, the presence of a CHO−φ type of unit is possible, but the CH resonating at 155.5 ppm remains to be explained. In general, this latter type of structure can be attributed to the presence of artefacts in the lignin preparation, either due to oxidation of a coniferyl-alcohol group, to sample preparation method or to the solvent used during the NMR experiments [13,27];
- ● φ−OCH2−φ side chains. These side chains were revealed by 1H and 13C chemical shifts: δ 1H, 4.7 ppm measured from the spectrum and 5.3 ppm calculated using Shoolery’s additivity rules; δ 13C, 64.3 ppm. This attribution was then confirmed by the absence of coupling of CH2 with other protons (Appendix E);
- ● other side chains. The DEPT spectrum (Fig. 5) revealed 3 types of CH2, one of which is attributable to φ−OCH2−φ (δ 1H, 4.7 ppm; δ 13C, 64.3 ppm). The other two are linked to less exposed protons (δ 1H, 3.7 and 3.6 ppm; δ 13C, 61.2 and 72.5 ppm for each CH2, respectively). These CH2s are probably involved in β-O-4-type side chains (Fig. 6). In this particular case, the Cα proton resonates around 7 ppm and is evident on the 2D 1H–13C. This proton correlates with a carbon at 106.5 ppm. The CH nature of the whole is confirmed by the DEPT spectrum.
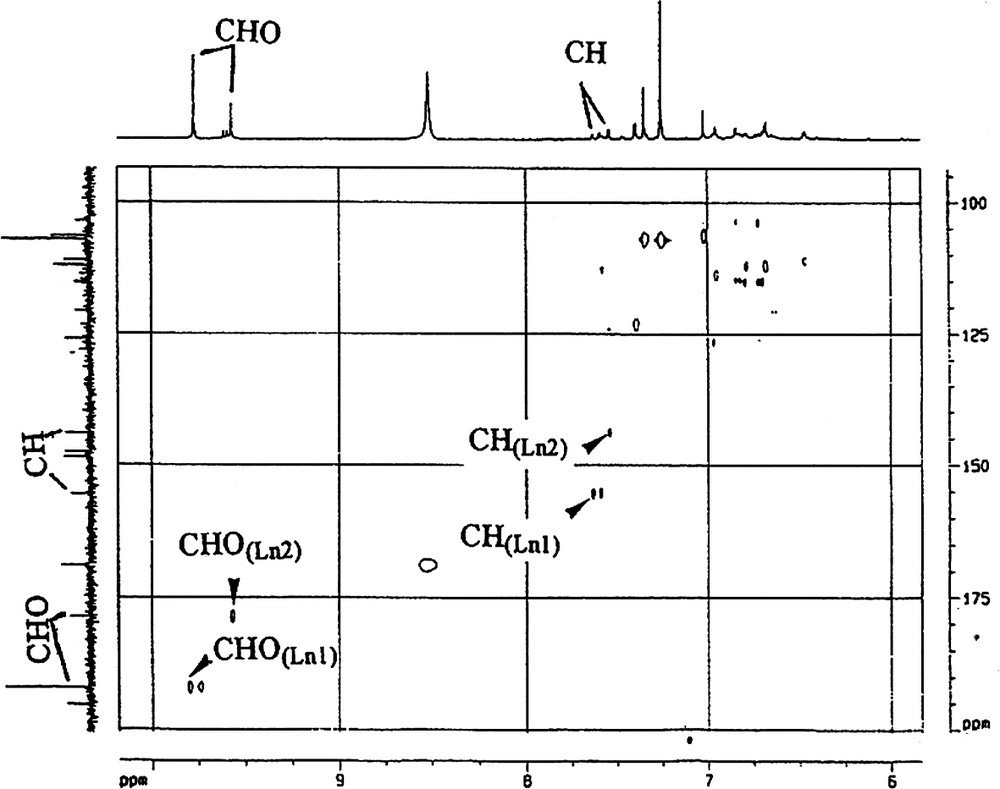
Attribution by inverse correlation 1H–13C of lateral Ln1 and Ln2 chains CHO–CH=CH–.
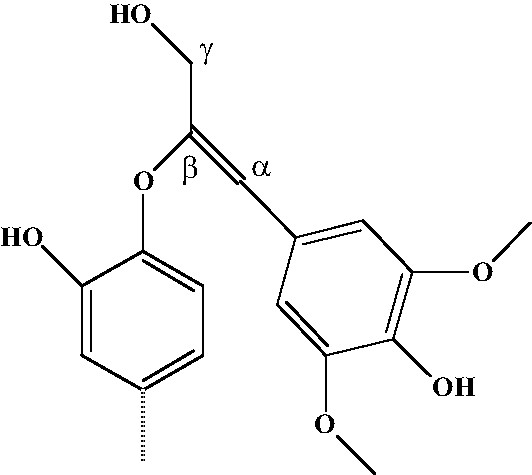
Hypothetical structure of lignan’s linkage.
3.2.2 Aromatic groups
Two types of aromatic groups are present: syringyl and guaiacyl groups. No hydroxyphenyl group was detected.
- ● Syringyl units (Appendices B and F) were easily identified by proton NMR. Protons 2 and 6 resonate as a singlet at the same frequency. Two major syringyl groups were detected, one at 7.2 ppm and the other at 7.3 ppm. Taking into account their area, the first belongs to Ln1 and the second to Ln2. From the 1H–13C inverse correlation, carbons 2 and 6 were also identified: 107.1 and 107.2 ppm for Ln2 and Ln1, respectively. These chemical shift values are in general agreement with those observed for such units. Quaternary carbons 3 and 5, which are also magnetically equivalent, resonate at 148.6 ppm and 147.6 ppm. However, with direct correlation experiments, they could not be attributed. Experiments such as HMBC are planned in order to attribute these carbons. Finally, it had been calculated that carbons 1 and 4 should resonate between, 120 and 140 ppm. Quaternary carbons are present in this area, but could not be attributed for the same reasons.
- ● Guaiacyl units (Appendices B and F) were assigned based on chemical characteristics for these types of units. From the 13C we find G6 at 126.1 and 123.5 ppm for Ln1 and Ln2, respectively; G2 at 112.1 and 111.1 ppm for Ln1 and Ln2, respectively. The 1H–13C inverse correlation confirms attributions of G2; in the case of G6 the signal is weaker, especially for Ln2 of G6 which did not allow the correlation spots to be visualised.
4 Conclusion
Because, the lignin represents one of the major polymers of oak heartwood, this study presents a scientific and technological interest. For example the problem of lignification can influence the water impregnation and finally some technological accident as the leak of barrels [15]. The lignins represent a major aroma precursor for wine or spirit ageing in barrels. Revelation of a large variety of flavouring components is provided by pyrolysis reactions of lignin during a crucial step of barrel making: the toasting [28]. So the lignin, have a high impact in wine aroma. The composition and molecular mass influence largely the quantity and the diversity of aroma [29].
Heterogeneity of the lignins is an important parameter for the variability of the composition and structure between species [30,31], in a same species [32–34] and even regarding to the localisation in the tissue [35]. This is the reason why the different works devoted on the lignins structures were no extended because they depend on the nature of the sample, to the aims of the study and to the specificity of the extraction technique. The specific structure of oak wood extractable lignins present a S/G structure (S/G ≥ 1), with a large amount of β-O-4 and β–β linkages. For other angiosperm trees such as Populus sp., heartwood lignin present a S/G structures with a majority of β-O-4 linkages and secondary β–β [36]. For angiosperm trees such as Pinus thunbergii, the lignins of stems from newly formed xylem [12] found a typical G/H structure with mainly, β-O-4 linkages. In comparison Ginkgo biloba shoots lignins were S/H lignins with large part of β-O-4 linkages [11]. This tree is one of the oldest tree species, very similar to modern conifers, but with gymnosperms-type leafs.
Concerning the lignan fraction, in relation to the complexity of the composition, additional isolation and purification procedures need to be applied to give definitive structural information on the variability and characteristics of oak wood extractive lignans. Their participation to the wood colour can be envisaged for future investigation.
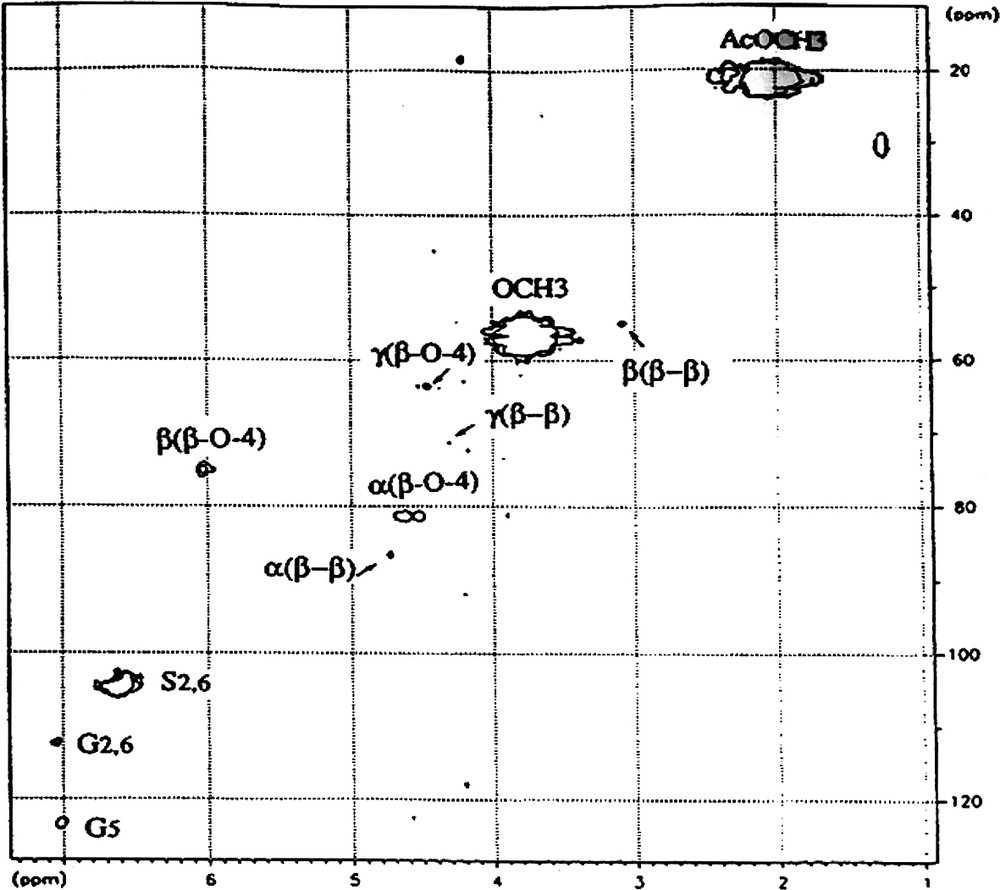
Appendix A
Part of 2D HMQC spectrum of LD fraction.
Attribution of symbols to the carbons: α, β, γ correspond to Cα, Cβ and Cγ, respectively.
Attribution of symbols to the side chains; (β–β), pinoresinol linkage; (β-O-4), β-O-4 linkage.
Attribution of symbols to the phenolic group: S2,6, carbons 2 and 6 of syringyl motifs; G2,6, carbons 2 and 6 of guaiacyl motifs; G5 carbon 5 of guaiacyl motifs.
OMe, methoxyl of phenolic groups; AcMe, methyl carbon of acetylated groups.
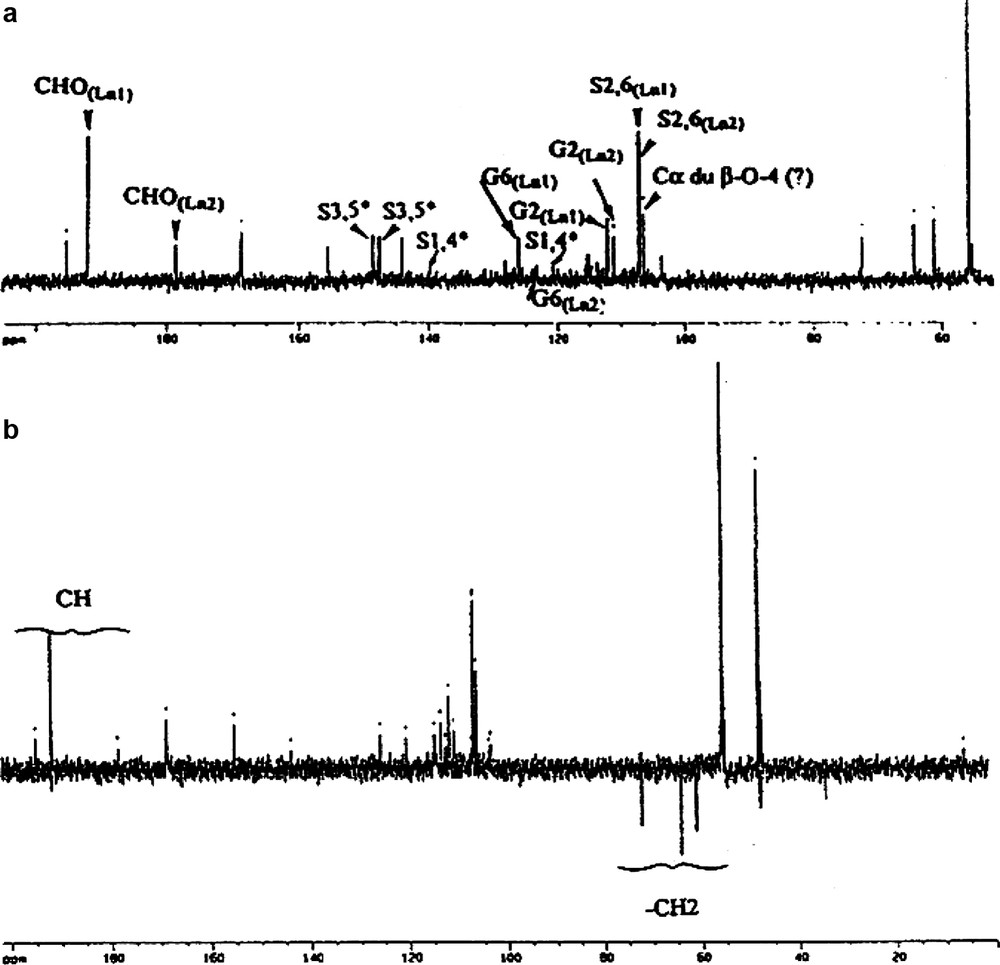
Appendix B
13C (a) and DEPT (b) spectra (* reverse attribution of Ln1/Ln2).
Appendix C
Part of TOCSY spectrum (mixing time of 100 ms) of Ln2 fraction.
Appendix D
Part of COSY 1H–1H spectrum of Ln2 fraction.

Appendix E
Inverse correlation 1H–13C spectrum allowing the attribution of the principal Ln1 and Ln2 lateral chains.
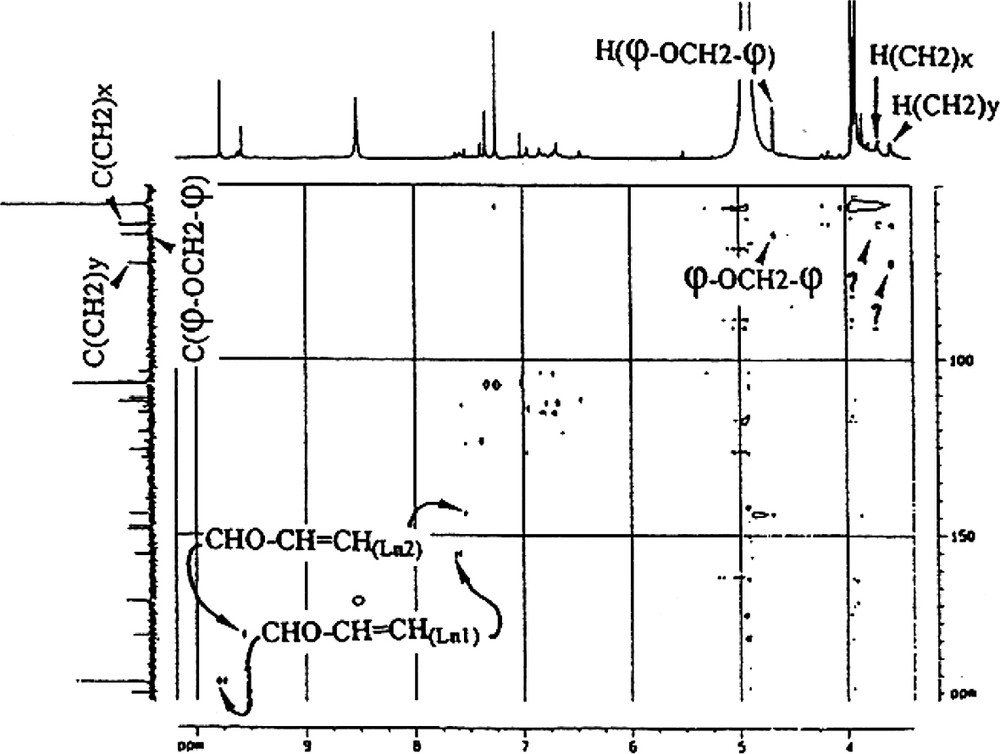
Appendix F
Inverse correlation 1H–13C showing Ln1, Ln2 syringyl and guaiacyl units.
