

1 Introduction
Different routes may be used in order to obtain – by reactive extrusion – polymer blends with monitored and stabilized morphology. This includes crosslinking of the dispersed phase, crosslinking of the major elastomeric phase with phase inversion for thermoplastic vulcanizates (TPV) elaboration, polymerization of monomer in molten polymeric matrix, copolymer synthesis in situ at the interface through reactions between polymer chains.
The chemical reactions involved should exhibit kinetics compatible with the time scale of reactive processing. As a consequence, the choice of the catalyst plays a key role for obtaining short reaction times in the appropriate temperature domain. A good candidate is expected to be well dispersed in the polymer matrix, not destroyed during the process, and finally “at the right place at the right time”.
In the last decade, we investigated a large range of interchange reactions involving ester groups, such as ester/alcohol, ester/ester, ester/carbonate, ester/amide ester/alkoxysilanes etc., in the presence of tin catalysts.
In this context, our interest was focused on four directions:
- ● the assessment of the required blend stabilization after the reaction;
- ● the characterization of the reactions involved using model compounds of the polymer reactive groups (very low amount of tin compound added);
- ● the comparison between different catalytic tin compounds;
- ● the characterization of the reactions involved between the ‘catalyst’ and the models of the polymer reactive groups (esters and catalyst in equal molar ratio).
For a long time, tin compounds proved to be excellent catalysts in interchange reactions involving esters: the catalytic efficiency of alkoxy trialkyl tin compounds in ester/alcohol interchange reaction was discovered in the 1970 by Pereyre et al. [1], that of dibutyltin derivatives such as diacetate, dichloride, dioxide, sulfide in ester/alcohol reactions by Poller and Retout [2] and more recently Otera et al. [3–6] showed the quite higher efficiency of R4Sn2X2O dimers. These distannoxanes (DS) are crystalline compounds and are obtained by partial hydrolysis-condensation of Bu2SnX2. They display a two-dimensional centrosymmetric dimeric structure with an essentially planar central ring and a global ‘staircase’ structure in some cases or ‘ladder-like’ (entirely planar) in others (Fig. 1). The four tin atoms are pentacoordinated in two different ways (endo and exo Fig. 1). X-ray, 119Sn NMR and Mössbauer discriminate these two different types of tin sites.

Typical dimeric distannoxane (DS)2 structure.
2 Results
Some mechanistic questions already arose in 1990 when we started our work in this field: what was at the origin of the catalytic efficiency of the dibutyltinoxide (DBTO) in reactions involving esters, why was the efficiency of R2SnX2 lower than that of the corresponding distannoxane structure [R4Sn2X2O]2? Which mechanism may be plausibly proposed? To explain the catalytic efficiency of the dimeric distannoxane in alcoholysis of esters, Otera and Nozaki [6] assumed an exchange at the endo tin site (with Sn-X giving Sn-OR) and then coordination of the ester on the neighboring tin site.
2.1 Reaction of esters with DBTO evidenced with model compounds [7]
In this context, our laboratory first showed that DBTO reacts with esters [octylacetate as an ethylene-co-vinylacetate (EVA) model has been chosen and methyl nonanoate as an ethylene-co-methyl acrylate (EMA) model] [7]. The reaction products were fully characterized by NMR (1H, 13C and 119Sn) and Mössbauer spectroscopy. These compounds are not crystallizable and X-ray cannot be used for their characterization. But in that case, Bonetti et al. [7] have clearly shown that this reaction –2 mol of DBTO and 1 mol of ester—gives a very simple and unique asymmetric dimeric distannoxane compound in which the acyl group (donor group in the dimer structure) is found at the endocyclic tin sites. The four tin atoms—two endo and two exo sites equally populated—are pentacoordinated in two different ways (endo and exo) as shown by 119Sn NMR 1D and 2D spectra (Fig. 2).
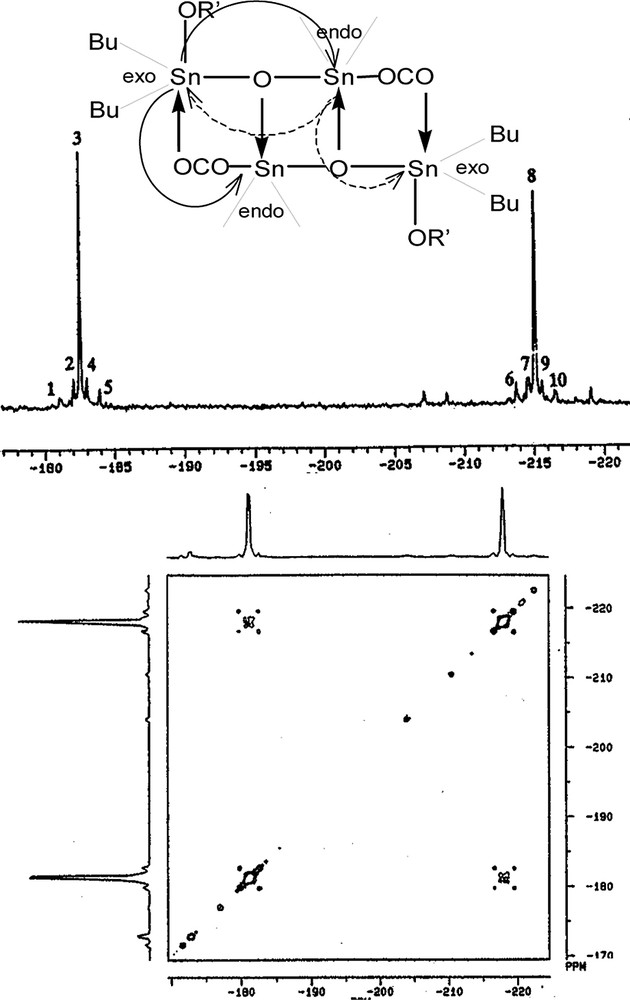
119Sn NMR (74.6 MHz) spectra (top: 1D, bottom: 2D COSY Sn–Sn) of dimeric (alkoxy, acyloxy distannoxane) structure obtained by reaction of an ester RCOOR’ with Bu2SnO at 200 °C (ester model compound: methyl nonanoate) [7].
It was also shown by addition of an ester R2COOR′2 to a solution of the distannoxane dimer of ester R1COOR′1, that continuous ligand exchange occurs above 110 °C between the free ester and the ester involved in the dimeric distannoxanne at both alkoxy and acyloxy tin sites. These ligand exchanges evidenced by Espinasse et al. [8] by NMR spectroscopy (Fig. 3) accounted for the random distribution of the four esters obtained and the random distribution of the alkoxy and acyloxy ligands at the exo and endo tin sites, respectively.
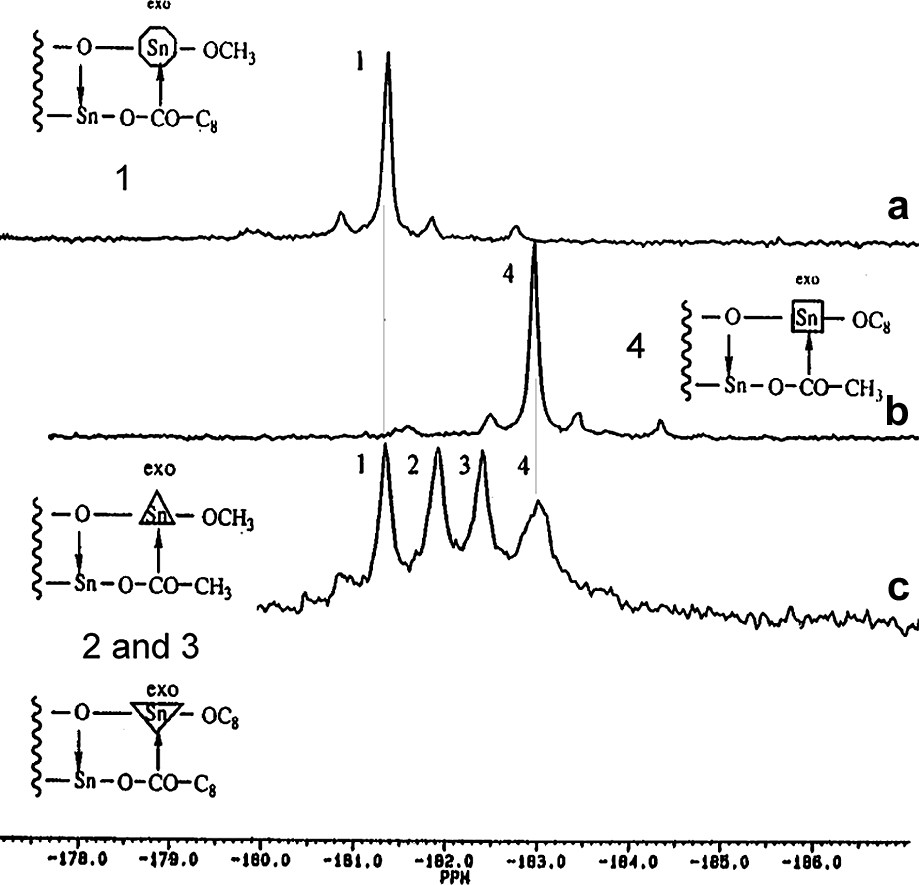
119Sn NMR (74.6 MHz) spectra: enlargement of exocyclic tin sites region for slightly different 1,3-alkoxy,acyloxy distannoxanes; (a) 1,3-methoxy, nonanyloxy distannoxane, (b) 1,3-octyloxy, acetoxy distannoxane; (c) mixture of equimolar amounts of the previous distannoxanes [8].
At ambient temperature, in solution, after mixing the two dimeric distannoxanes (DS1)2 and (DS2)2 in presence of corresponding residual model esters 1 and 2, namely octylacetate and methylnonanoate, it was shown that only intermolecular dimeric rearrangement occurred giving a third mixed dimer (DS1) (DS2) without any ester/ester exchange reaction.
The reaction of DBTO with model esters explained very well the crosslinking by a dimeric DS of EVA and EMA at 230°, attested by the increase of the storage modulus as shown by Fig. 4.

Storage modulus versus reaction time at 230 °C: □, EVA + Bu2SnO; Δ, EMA + Bu2SnO; EVA + EMA + Bu2SnO [7].
Reactions observed in presence of the two model esters (EVA and EMA models) allowed to conclude that with a blend of EVA and EMA in presence of DBTO at 230 °C, a third mixed distannoxane dimer (DSEVA)(DSEMA) is expected to be present at the interface giving ‘a mobile’ co-crosslinked interphase. But – as expected from the investigation on models – the most important phenomenon was covalent co-crosslinking EVA/EMA by ester/ester exchange reaction. This co-crosslinking was attested by both the methylacetate formation and a very important increase of the storage modulus (Fig. 4).
This EVA/EMA covalent crosslinking by ester/ester interchange reaction also occurred with direct addition of the alkoxy, acyloxy DS, but higher storage modulus was obtained when dibutyltin oxide was used, i.e. when the alkoxy, acyloxy DS was formed in situ in the polymer matrix.
Then, we focused our interest on the mechanism of distannoxane catalyst in ester–ester interchange reactions. But it is worth highlighting that some authors investigated other aspects like alcohol–ester exchange reactions [10] or biphasic esterification and transesterification catalyzed by fluoroalkyldistannoxane [11].
The catalytic efficiency of different DS towards ester–ester interchange reactions has been investigated with model esters by Espinasse [9] at different temperatures with catalytic amount of tin compounds. As shown on Fig. 5, at 140 °C for example, diacetoxy and dichloro DS have nearly the same efficiency (Fig. 5: curves 4 and 5, respectively), much higher than dibutyltin diacetate (curve 2), but lower than that observed with the acetoxy, octyloxy DS (curve 7).

Transesterification between methyl myristate and stearyl acetate catalyzed by different tin compounds (5% weight): conversion rate versus time [9]. 1: 140 °C, without catalyst; 2: 120 °C, dibutyl Sn diacetate; 3: 140 °C, dibutyl Sn diacetate; 4: 120 °C, diacetoxy DS; 5: 140 °C, dichloro DS; 6: diacetoxy DS; 7: 140 °C, acetoxy, octyloxy DS.
Moreover, to clarify whether ligand exchanges at the tin sites are essential to the observed DS catalytic effect, or whether coordination of the reactants to neighboring tin sites is sufficient, ligand exchanges at the tin sites have been monitored at different temperatures with a model ester and stoichiometric amounts of tin compounds, without any solvent. It has been shown with a dimethoxy DS (2Sn-OCH3/1ester) that rapid ligand exchange alkoxy/alkoxy occurred (90% conversion rate in 20 min at 140 °C, whereas acyloxy/acyloxy exchange with a diacetoxy DS (2Sn-OCOCH3/1ester) was much slower even at higher temperature (20% in 20 min at 160 °C and almost null at 140 °C). With a dichloro DS, in presence of an ester, no ligand exchange at all was observed after 15 min at 200 °C.
From these two sets of observations, the following conclusions were possible: the dichloro and diacetoxy DS catalytic efficiency – without any ligand exchange observed at 140 °C – indicates that ligand exchange is not essential and coordination of the reactants on the neighboring tin sites (exo and endo) of the dimeric DS entity is probably the driving force of the mechanism. But the higher efficiency of the acetoxy, octyloxy DS is probably due to additional alkoxy ligand exchange which appeared to be quite rapid at this temperature.
This direct, rapid and simple synthesis of bi-functional DS – obtained directly in the polymer matrix – has opened a new route for many applications in reactive processing involving ester interchange reactions. Some of the applications investigated by our laboratory with model compounds – and later on with parent polymers – are given here.
2.2 Compatibilization of PETG/EVA blends by formation of a grafted polymer (EVA-g-PETG) at the interface [12–14]
In such blends, EVA is the dispersed phase in a continuous phase of polyethylene terephthalate (PETG). In presence of DBTO at 210 °C compatibilization of the blend occurs and has been assessed via change in mechanical properties [12,13].
The mechanistic path is derived from the conclusions of the previous study with the model compounds and supported by complex viscosity measurements. It has been shown first that DS formation occurred with PETG models, namely dimethylterephthalate [13]. With PETG the distannoxane formation corresponds to [Sn(Bu)2–O–Sn(Bu)2–O–] insertion in the polymer chain, between alkoxy [R] and acyl groups [–C(=O)–R] of the PET ester.
On the one hand alkoxy, acyloxy DS dimers are sensitive to moisture, and complete elimination of water traces in polyesters being impossible, chain scissions may occur by DS hydrolysis. On the other hand the ester/ester interchange reaction at the interface catalyzed by the dimeric DS formed in each phase (EVA and PETG) gives a grafted copolymer EVA-g-PETG with PETG chain scission and formation of shortened PETG chains with acetate end groups. In order to reduce the extent of the first scission, samples were pre-dried during 24 h. Whereas an initial decrease of the complex viscosity was observed in the presence of residual water, on the contrary, with previous drying, Lacroix et al. [13] observed the almost complete suppression of the initial decrease. The complex viscosity of the blend is much larger than that of the matrix either due to some crosslinking of the dispersed EVA phase or to formation of the grafted copolymer combined with the co-crosslinking of both phases.
The compatibilization of the blend EVA/PETG was observed in the presence of DBTO at 210 °C. The graft polymer formed at the interface acting as a core-shell structure for the dispersed EVA phase.
2.3 Morphology control of PE/PBT blends by formation of EVA-g-PBT copolymer during processing [15]
In that case poly butylene terephthalate (PBT) was the dispersed phase in a continuous phase of polyethylene (PE). In order to compatibilize these polymers, EVA was added as a third polymer. It was shown previously by Ray and Khastgir [16] that EVA (11.2% mole of vinylacetate units) is partially miscible with low density PE. Pesneau et al. [15] have shown that the miscibility increased with decreasing vinyl acetate ratio in the EVA copolymer, which was well explained by the increase of the average sequence length of ethylene units (LE) between two acetate units. By adding both EVA (3.12% mole of vinyl acetate units) and catalytic amounts of Bu2SnO, compatibilization of PE/PBT blends was obtained during processing.
The ester/ester exchange reaction at the interface between EVA and PBT was expected to produce a graft polymer acting as a compatibilizer between PE and PBT. In each phase, DS formation was expected to induce crosslinking. At the interface, the ligand exchange (alkoxy and acyloxy exchanges at this temperature) at the DS tin sites should accelerate ester–ester interchange reaction giving a graft copolymer EVA-g-PBT and shortened PBT chains with acetate end groups coming from transesterification. PE does not take any part in the DS formation and transesterification reaction.
These reactions, expected from previous NMR studies on model compounds, have been directly evidenced by Pesneau et al. [15] who separated the EVA-g-PBT grafted polymer (12% by weight) of the two parent polymers by a series of extractions. This copolymer exhibited all the NMR resonances of PBT and EVA sequences and additional resonances that have been specifically assigned to grafting as shown on Fig. 6.
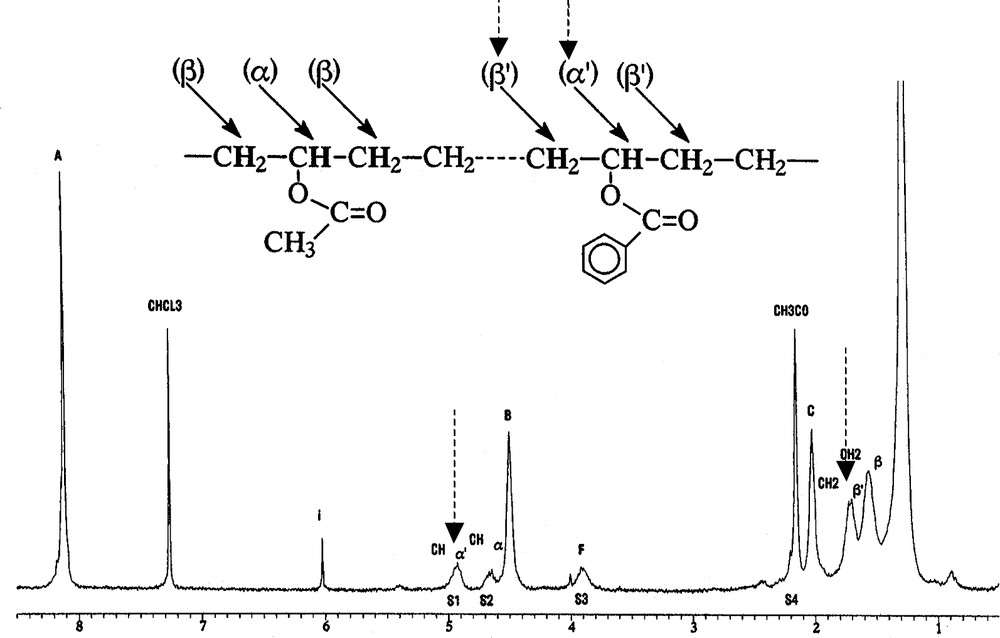
1H NMR (250 MHz) spectra in CF3COOD + CDCl3 (1/1 by volume) of the EVA-g-PBT polymer soluble fraction in tetrachloroethane, obtained after selective extractions of PBT in dichloroacetic acid and EVA in toluene. Specific β′ and α′ resonances are in good agreement with those observed after reaction of methyl benzoate with EVA ester groups (δβ′ = 1.640 and δα′ = 5.075 ppm) [15].
Moreover, part of the PBT grafts were found to bear acetyl end-groups and an insoluble crosslinked residue (6% by weight) is also obtained. These two observations being well explained by a second transesterification occurring on the PBT graft either in position (1) or (2) (Scheme 1) giving an acetyl end group or a crosslink, respectively.
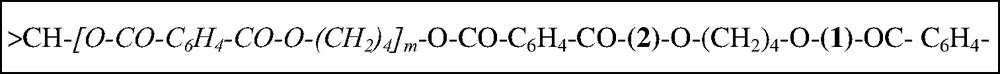
Chemical structure of EVA-g-PBT after transesterification on EVA acetate groups and positions of a second possible transesterification giving either an acetyl ended PBT graft (1) or a crosslink (2).
The morphology of these blends has been examined by scanning electron microscopy (SEM) for different PBT contents (from 10% to 30% weight) by measurement of the size of the dispersed phase. The results have shown an increased adhesion. With EVA and DBTO added, if the contrast was too bad then PBT was extracted and the measure of the holes corresponding to the PBT nodules was carried out afterwards (Fig. 7-middle). It is worth noticing that when EVA was added, without DBTO, as well as with DBTO alone, the particle size of the PBT dispersed phase was unchanged.

SEM micrographs of blends containing 20% weight PBT: left: PBT/PE 20/80, middle: PBT/PE/EVA/DBTO 20/74/5/1, right: PBT/PE/DBTO 20/79/1 (× 2000). Average sizes of the PBT nodules are from left to right: 1.8, 0.5 and 1.6 nm, respectively [15].
2.4 New copolymer PVC(VA)-g-PC not directly accessible by a direct polymerization route [17]
Poly[vinylchloride-co-vinylacetate] (3.12% VA units) and bisphenol A polycarbonate (PC) are incompatible polymers and their Tg values are 72 and 145 °C, respectively. A 50/50 polymer blend processed in presence of Bu2SnO gave a polymer with a unique transition (at 83 °C). This compatibilization resulted from the formation in situ of a grafted copolymer by transesterification ester/carbonate as shown by a preliminary investigation of the reactions on model compounds.
The reactions on model compounds, namely octyl acetate for VA units in PVC(VA) and diphenyl carbonate for carbonate groups in poly[bisphenol A carbonate], have been characterized by NMR. The conclusions were the following:
- ● Diphenyl carbonate with DBTO gave – after 10 min at 160 °C – a crystallizable symmetric diphenoxy tetrabutyldistannoxane (PF = 146 °C), together with carbon dioxide formation. We fully characterized this compound, which was already mentioned in the literature as a catalyst in transesterification reactions and was obtained by reaction of DBTO and phenol. The rapid reaction of DBTO with carbonate is only a new route and above all it shows that during processing of PVC(VA) and PC blends in the presence of DBTO, two dimeric DS are expected to be rapidly formed in situ the first one with acetate units and the second one with carbonate units by [Sn(Bu)2–O–Sn(Bu)2–O–] insertion in the PC chain.
- ● Diphenyl carbonate and octyl acetate in the presence of DBTO provided by transesterification the phenyl acetate and two new carbonates: the phenoxy, alkoxy(C8–O) and the dialkoxy(C8–O)2. An important decarboxylation (35% in weight) occurred and the presence of some diphenyl ether and phenol was also detected.
- ● In an attempt to limit decarboxylation in this ester/carbonate transesterification, the influence of four added tin compounds was compared: dichloro DS, diphenoxy DS, alcoxy, acyloxy DS and DBTO. The corresponding decarboxylation amounts were 10%, 10%, 20% and 35%, respectively. It was found that the ability to produce ligand exchange at tin sites of the three above DS was related with the extent of decarboxylation: in fact – as shown previously by Espinasse et al. [8] – no ligand exchange occurred between an ester and the dichloro DS, the same is observed with the diphenoxy DS, whereas both alkoxy and acyloxy exchanges occurred with the third one.
- ● Concerning the catalytic efficiency of these three DS [17] towards ester/carbonate transesterification, in all cases 56% of the ‘mixed’ carbonate was obtained for 100 mol of initial octyl acetate. This mixed carbonate octyl–O–CO–O–phenyl is the model of the aimed structure [>CH–O–CO–O–PC] in the grafted polymer PVC(VA)-g-PC. This showed that ligand exchange at the tin site was not essential for catalyst efficiency, coordination of the reactants to neighboring tin sites being probably sufficient. Moreover, in this specific case, formation of a [Sn–O–CO–O–phenyl] sequence is probably the driving force for the decarboxylation.
This conclusion concerning the catalytic mechanism is in good agreement with Espinasse's [9] previous work about ester/ester interchange, in which efficient catalysis of diacetoxy and dichloro DS was observed at 140 °C – without any ligand exchange observed at this temperature – and a higher efficiency of the alkoxy, acyloxy DS at the same temperature when rapid alkoxy ligand exchange is observed.
3 Conclusion
The bis[alkoxyacyloxytetrabutyldistannoxane], [RCOO–(C4H9)2Sn–O–(C4H9)2Sn–OR′]2, which is rapidly formed in situ by reaction of DBTO with polymer ester groups (pendant or in the backbone) was shown to act as a very efficient catalyst in numerous reactions between these ester groups and other reactive groups, esters and carbonates for example. Such reactions were successfully used to control the morphology of polymer blends in which efficient compatibilization was produced by formation of a graft copolymer at the interface.
Recently, Bounor-Legare et al. [18] extended this type of catalysis to ester/alkoxysilane exchange reactions between polymer–ester pendant groups and a tetraalkoxysilane giving access to hybrid crosslinked materials easily obtained during processing. In this case, the reactions with model compounds have shown that the catalytic efficiency was due to very efficient exchanges between alkoxy groups of catalytic exo tin sites and the alkoxy silane crosslinking agent. These exchanges occur without any alteration of the ladder structure of the DS structure. NMR proved to be a powerful tool in a view to investigating the mechanisms of both the reaction and the catalytic mode.