

1 Introduction
Reactive extrusion (REx) is an attractive route for polymer processing in order to carry out melt-blending, and various reactions including polymerization, grafting, branching and functionalization as well [1–3]. The economics of using the extruder for conducting chemical modifications or polymerization reactions has shown to be an efficient way for low cost production and processing methods, which enhances the commercial viability and cost-competitiveness of these materials. For instance, the obvious advantages of an extrusion polymerization are as follows:
- ● solvent free melt process;
- ● continuous processing, starting from monomer and resulting in polymer or the finished product;
- ● control over residence time and residence time distribution;
- ● integration of other extrusion streams along with the polymerization process.
In essence, an extruder is a versatile equipment that can be used in a variety of applications. However, regardless of the application, the primary factor that determines whether the desired objective of the extrusion is met is the screw geometry used in the extruder. The screw geometry controls the residence time, the power input, and the extent of mixing imparted to the melt in the extruder. In this contribution, we will show how REx, more specifically performed in a well-designed twin-screw co-rotating extruder, can find application in continuous production of new biodegradable polymeric materials.
Growing environmental concerns worldwide demand the reduction of environmentally unfavorable waste as well as the addition of biodegradability or recyclability of the product as the design criteria [4]. Therefore and in order to produce environmental friendly materials, various types of biodegradable plastics have been developed including poly(ε-caprolactone) (PCL), polylactides (PLAs) and other aliphatic–aromatic polycondensates such as poly(butylene adipate-co-terephtalate). Although the mechanism and rate of degradation of each polymer vary, they were all found to be converted to carbon dioxide and water under adequate conditions of temperature, humidity and microbial activity [5].
High molecular weight aliphatic polyesters such as PCL and PLAs are usually prepared by ring-opening polymerization (ROP) of their respective cyclic ester, i.e. ε-caprolactone and lactide (the cyclic dimer of lactic acid) (see Fig. 1) [6]. This method provides sufficient polymerization control, which results in polymers of the required molecular weights and the desired end-groups. Both polyesters are actually produced at the industrial scale: PCL by Solvay Interox in UK, Dow Union Carbide in US and Daicel in Japan, whereas PLA is supplied by, e.g., Cargill Dow in US, Mitsui Toatsu and Shimadzu in Japan. While ε-caprolactone is a petrochemical monomer, lactic acid and thus lactide are interestingly produced from non-fossil renewable natural resources by fermentation of polysaccharides or sugar, e.g. from corn and beet, therefore allowing the biological cycle to be closed owing to the PLA biodegradation together with the photosynthesis process [5].

Chemical structure of some commercial biodegradable polyesters and related synthetic pathways. PCL coined for poly(ε-caprolactone), PLA for polylactide and PTMAT for poly(tetramethylene adipate-co-therephalate), i.e. Eastar-Bio® and Ecoflex®.
Beside PCL and PLAs, a series of biodegradable aliphatic polyesters have been developed on the basis of the traditional polycondensation reactions. Most notable ones are the poly(alkylene succinate)s manufactured by Showa Denko, trademarked Bionolle®. In addition, the mechanical strength of biodegradable polyesters obtained by polycondensation may be increased by substituting more rigid aromatic diacids for some aliphatic diacid building blocks (Fig. 1). Eastman Company and BASF have developed such aliphatic–aromatic copolyesters, so-called Eastar-Bio® and Ecoflex®, respectively, which retain their biodegradability with a maximum amount of aromatic compounds (terephthalic acid) at about 40 wt.% [7].
However, the use of these biodegradable (co)polyesters as bulk materials is still restricted by their relatively high cost, and poor mechanical properties compared to commodity plastics such as polyethylene and polypropylene. Combination of these biodegradable polymers with cheap inorganic or organic fillers such as silicate-type particles or starch granules, respectively, represents a useful way for reducing the cost and for optimizing the properties of aliphatic polyesters or aliphatic–aromatic copolyesters. Unfortunately, simple polyester composites/blends have poor properties because of lack of interfacial adhesion [8]. Indeed it has been reported that the mechanical properties of the aliphatic polyester/filler blends generally become poorer at higher particle filling level. This can be attributed to the incompatibility between the hydrophobic polyester and the usually hydrophilic fillers. Thus, it is expected that the properties of the polyester-based blends/composites could be improved by proper interface compatibilization [9].
This paper aims at reviewing various strategic pathways to produce biodegradable polyesters and related compatibilized melt blends by REx (all steps conducted in a twin-screw extruder):
- ● polyester synthesis by continuous catalyzed ROP;
- ● polyester chemical modification (self-branching) and chain-functionalization (maleation);
- ● compatibilization of polyester melt blends with different naturally occurring fillers.
2 Production of biodegradable aliphatic polyesters by REx: REx-polyesters
The requirements needed to develop an extrusion process to continuously polymerize cyclic esters like ε-caprolactone or lactide monomers and form high molecular weight REx-polyesters are as follows:
- ● accurate metering of the monomer and initiator/catalyst streams under an inert atmosphere;
- ● adequate length of the extrusion zone to meet residence time requirements needed to convert more than 95% of the monomer to Rex-polymer;
- ● suitable screw configuration in order to meet residence time requirements, melt pumping needs and mixing requirements;
- ● good melt temperature control;
- ● reasonable product throughputs of high molecular weight polyesters.
First, bulk polymerization of either δ-caprolactone (CL) or another γ-functionalized ε-lactone, i.e. 1,4-dioxan-2-one (PDX), has been carried out by initiation with Al(OsecBu)3 in a co-rotating twin-screw extruder through a fast single-step process. Then attention has been paid to lactide polymerization.
2.1 Homopolymerization of CL: Rex-PCL
In a first series of experiments, the bulk polymerization of CL initiated by Al(OsecBu)3, an aluminum alkoxide known to initiate fast and controlled ROP of lactones [6], was carried out in a co-rotating closely intermeshing twin-screw extruder with a screw diameter of 30 mm and a length-to-diameter (L/d) ratio of 15 (Fig. 2). Three heating/cooling sections constituted the extruder barrel. The temperature of the barrel sections and two-strand hole die was kept at 150 °C, while the screw rotation speed was fixed at 130 rpm. CL and Al(OsecBu)3 (in anhydrous toluene solution) were simultaneously and continuously introduced under a nitrogen flow into the first feed throat of the twin-screw extruder. The relative feeding rates in monomer and initiator allowed reaching various initial [CL]0/[Al(OsecBu)3] molar ratios of 1170 and 1710. The feeding systems were continuously purged with nitrogen, as well as the enrance section of the extruder in order to avoid unnecessary contact of lactone, initiator and growing polyester chains with moisture.
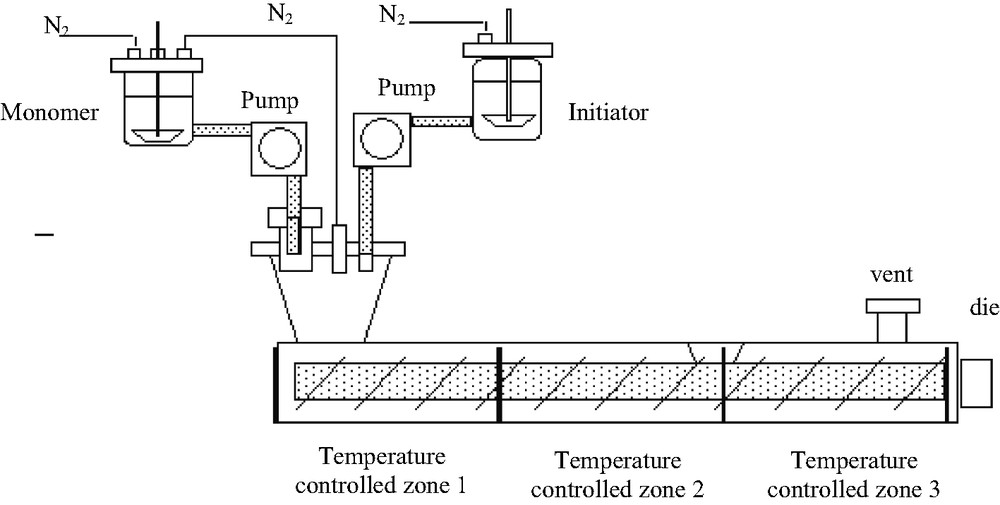
Overall scheme of the investigated co-rotating closely intermeshing twin-screw extruder and feeding ports.
Table 1 shows the polymerization yield and molecular parameters of the as-recovered PCL samples. It comes out that CL is completely converted into polyester chains (polym. yield ~ 99%) within a maximum residence time of ca. 2 min. More interestingly, it must be emphasized that there is a good agreement between theoretical and experimental molecular weights. Finally, it is worth pointing out that polydispersity indices remain quite narrow (Mw/Mn ≤ 1.7) [10,11].
Polymerization yield and molecular parameters of PCL as obtained by ROP of CL initiated by Al(OsecBu)3 in a co-rotating twin-screw extruder (T = 150 °C, screw rotation speed = 130 rpm and residence time of ca. 2 min)
| Entry | [CL]0/[Al(OsecBu)3] | Polymerization yielda (%) | MnPCL (g/mol) | Mw/Mn | |
| Theoreticalb | Experimentalc | ||||
| 1 | 1170 | 99 | 44 400 | 39 500 | 1.7 |
| 2 | 1710 | 99 | 65 300 | 63 000 | 1.6 |
a As determined by gravimetry after solubilization of crude polymerizing mixture in toluene, precipitation in heptane, and drying until a constant weight.
b Mntheor = [CL]0/[Al(OsecBu)3] × yield × (MwCL/3).
c Number average molecular weight and molecular weight distribution of PCL as determined by SEC in THF according to universal calibration.
The ROP of CL initiated by aluminum alkoxide active species is thus well controlled and proceeds via the so-called ‘coordination–insertion’ mechanism, which yields polyester chains end-capped by an active aluminum alkoxide bond [6]. As a result of the tri-functionality of Al(OsecBu)3 which allows for initiation and propagation of three growing polyester chains per aluminum atom (Fig. 3), 3-arm star shaped REx-PCL with a number average molecular weight of each arm as high as 200 000 g/mol have been also successfully produced in the extruder within a mean residence time of less than 5 min (monomer conversions in excess of 95%) [10]. Such REX-PCL blown films were found to display significantly better dart and tear properties as compared to commercially available linear PCL.
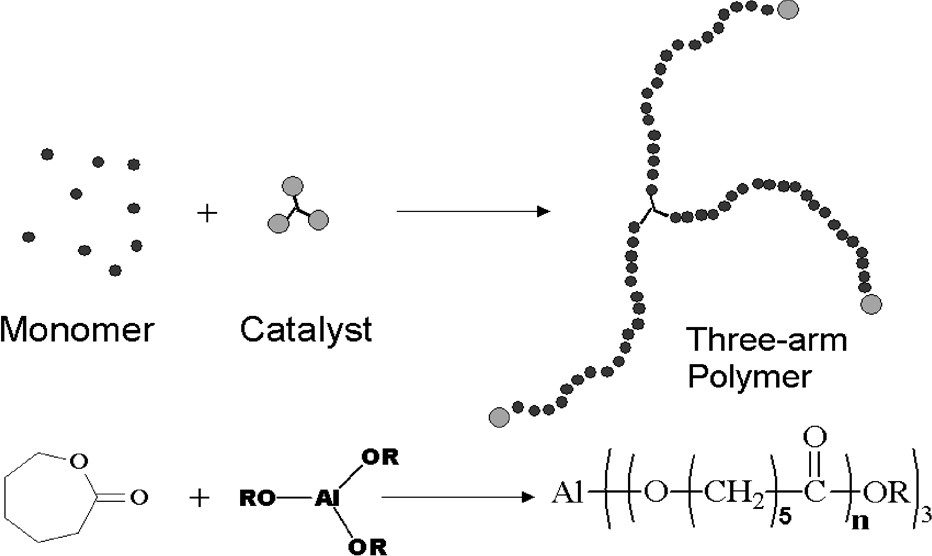
3-Arm star-shaped REx-PCL as synthesized by ROP of CL initiated with Al(OsecBu)3 via REx.
2.2 (Co)polymerization of PDX: REx-PPDX
Poly(1,4-dioxan-2-one) (PPDX) is a biodegradable and biocompatible semi-crystalline poly(ester-alt-ether) copolymer with attractive thermal properties (glass transition temperature (Tg) at –10 °C and melting temperature (Tm) at 110 °C) with regard to processing conditions and available service temperatures [12]. Furthermore, PPDX has proven to be tougher than polylactides and even HDPE with a tensile strength close to 50 MPa for an ultimate elongation ranging from 500% to 600%. In this respect, PPDX is a potential candidate as biodegradable substitute for non-degradable petrochemical-based thermoplastics in, e.g., single-use applications. However, the use of PPDX as bulk material is hampered by its low ceiling temperature [13] favoring, in the melt state, the unzipping depolymerization reactions from the hydroxyl end-group (Fig. 4).

Unzipping depolymerization mechanism of ω-hydroxyl PPDX chains.
Aluminum trialkoxides represent also very efficient initiators for the synthesis of high molecular weight PPDX through a fast and continuous process in a twin-screw extruder [11]. However and as aforementioned, a key parameter in the production and development of PPDX as biodegradable thermoplastics is the reduction of its thermal degradation that occurs essentially by unzipping, without compromising the semi-crystalline properties of PPDX (high melting temperature slightly above 100 °C). In this respect, various experiments have been carried out to initiate the bulk copolymerization of mixtures of PDX and CL (in limited relative content) by Al(OsecBu)3 in a co-rotating twin-screw extruder through a fast single-step process. Indeed, it comes out that the best method to prevent or at least limit the thermal degradation, i.e. unzipping reaction, of PPDX chains is to randomly distribute limited amount of CL all along the PPDX backbone [13].
(Co)polymerization of PDX (with CL) initiated by Al(OsecBu)3 was carried out by REx for various molar fractions in CL ranging from 0 to 16 mol%. The extruder and screw configurations were kept unchanged but the three available feeding ports were used for adding PDX, CL and the initiator solution, respectively. The copolymerization temperature was fixed at 130 °C, while the screw rotation speed was maintained at 130 rpm. PDX was introduced as a liquid at 50 °C into the first feed throat, while CL was fed by a second feed throat. At the same time, the initiator solution (5 wt.% of Al(OsecBu)3 previously prepared in anhydrous toluene) was introduced via a third feed throat. The feeding rates in both monomers and initiator allowed reaching initial molar fractions in CL of 0, 8, 11 and 16 mol-% for an initial (([PDX]0+[CL]0)/[Al(OsecBu)3]) molar ratio of ca. 2500. Table 2 shows the polymerization yields, final contents in CL within the extruded copolymers and their inherent viscosity [11].
Polymerization yield, final molar fraction in CL (FCL) and intrinsic viscosity of P(PDX-co-CL) copolymers as obtained by (co)polymerization of PDX and CL (added in various initial molar fractions: fCL) initiated by Al(OsecBu)3 in a co-rotating twin-screw extruder (T = 130 °C, screw rotation speed = 130 rpm and residence time of ca. 2 min)
| Entry | fCL | FCL | Polymer yielda (%) | [η]b (dl/g) |
| 1 | 0 | 0 | 65 | 0.77 |
| 2 | 0.08 | 0.11 | 99 | 0.65 |
| 3 | 0.11 | 0.16 | 97 | 0.55 |
| 4 | 0.16 | 0.20 | 100 | 0.37 |
a As determined by gravimetry after solubilization of crude polymerizing mixture in hot 1,2-dichloroethane, precipitation in heptane, and drying until a constant weight.
b As determined by solution viscometry in 1,1,2,2-tetrachloroethane/phenol (3:2) at 25 °C.
As far as the homopolymerization of PDX is concerned (entry 1 in Table 2), the limited polymerization yield (yield = 65%) is explained by the monomer–polymer thermodynamic equilibrium highly shifted toward monomer at the processing temperature of 130 °C in agreement with previously published data performed in bulk and in sealed glass ampoules [12]. Very interestingly, the polymerization yield for the P(PDX-co-CL) copolymers increases up to complete conversion as the molar fraction in CL within the copolymer increases. Such a behavior is of the utmost importance as far as the economical viability of the process is concerned and gives credit to the formation of a blocky-like copolymer structure (see section hereafter). Composition of the resulting copolymers has been determined by 1H NMR spectroscopy. It comes out that the CL contents in the feed and in the recovered copolymer are very close from each other (actually slightly lower in the copolyester chains), which is in favor of the complete incorporation of CL into PPDX chains. In addition, the distribution of CL all along PPDX chains has been analyzed by 13C NMR spectroscopy, which indicates that the copolyester chains are composed by multiple short homosequences PPDX separated from each other by CL unit(s). Moreover, it is worth pointing out that the sharp decrease in intrinsic viscosity by the incorporation of CL into PPDX chains might reflect a substantial change of the copolymer conformation in solution rather than an abrupt decrease of the molecular weight. Unfortunately it has not been possible to determine the actual molecular weight of the copolymers by SEC or osmometry because of the poor solubility of the as-obtained copolymers in conventional organic solvents.
Thus, both homopolymerizations of PDX and CL as well as their copolymerization proceed very rapidly and reach almost complete (co)monomer(s) conversion as soon as 8 mol% of CL are added in the feed. Even tough PPDX is known to thermally degrade mainly through unzipping depolymerization promoted from the hydroxyl end-groups and therefore yielding PDX monomer, it comes out from TGA experiments that the thermal stability of PPDX chains is largely improved by the copolymerization of PDX with limited amounts of CL [13]. Indeed, a substantial reduction of the time-dependence weight loss recorded under isothermal conditions at 180 °C, i.e. at PPDX processing temperature, is detected in the presence of 11 mol% of CL or more. Interestingly, DSC analysis of the so-obtained P(PDX-co-CL) copolymers has demonstrated that a CL molar fraction as high as 11 mol% does not prevent the crystallization of the resulting copolymer, which retains a melting temperature as high as 95 °C. This last observation has been explained by the formation of a blocky-like copolymer structure, in which short PPDX and (P)CL sequences are randomly distributed.
2.3 Homopolymerization of LA: REX-PLA
Similarly, a new polymerization process has been developed for the continuous production of PLA using REx polymerization based on another catalytic system, i.e. tin octoate added with one equivalent of triphenylphosphine (as co-catalyst), which not only enhances the kinetics of lactide ROP but also suppresses (or at least delays) any side and degradation reactions such as transesterification reactions [14]. Actually, it has been shown that the addition of one equimolar amount of a Lewis base like triphenylphosphine on 2-ethylhexanoic tin(II) salt (Sn(Oct)2), significantly enhances the lactide polymerization rate in bulk. This kinetic effect has been accounted for the coordination of the Lewis base onto the tin atom, making easier the insertion of the monomer into the metal alkoxide bond of the initiator/propagation active species [15]. This tin alkoxide bond is formed in situ by reaction of alcohol and the tin(II) dicarboxylate. As reported in polymerization of CL and PDX initiated by aluminum trialkoxides (see above), the lactide ROP proceeds via the same ‘coordination–insertion’ mechanism involving the selective oxygen-acyl cleavage of the cyclic ester monomer. The addition of 1 equiv of P(C6H5)3 onto Sn(Oct)2 allows for reaching an acceptable balance between propagation and depolymerization rates, so that the polymerization is fast enough to be performed through a continuous one-stage process in an extruder [16]. Melt stable PLA has been produced, the molecular weight of which being tunable by the addition of alcohol.
Owing to this catalytic complex, it has been possible to design a suitable processing and screw concept using a closely intermeshing co-rotating twin-screw extruder (with a screw diameter of 25 mm and a L/d ratio of 48) equipped with a static mixer at the tip of the die [15]. In Table 3, PLAs produced in glass ampoules using bulk batch polymerization technology and in single-stage continuous REx polymerization are compared on the basis of molecular parameters. The comparison of two polyesters polymerized under such different conditions is very difficult, although the same starting polymerization conditions were used. The resulting conversions of 98.5% and 99% show that, in both cases, the polymerization reaction has been finished, and reach the monomer–polymer thermodynamic equilibrium, though for the ROP carried out in glass ampoules, the time required to reach this conversion is approximately 40 min, compared to ca. 7 min in the REx process. This difference can be explained by the fact that as soon as high molecular weight, i.e. a high melt viscosity is reached, the rate of the reaction is limited not by the reactivity of the chemicals, but by the diffusion of the monomers (and other low molecular weight compounds) inside the high viscous melt to find a reactive partner. This physical movement is limited to the Brownian movement in the glass ampoule, but is supported in the twin-screw extruder by the mixing elements and by the shearing of the polymer inside the intermeshing zone. The difference in molecular weight can be explained by the fact that lactide involved in the polymerization carried out in glass ampoule has been freshly recrystallized, while it has been used as received for the REx polymerization, and by the fact that contact of LA with humid air can not be totally avoided during the transfer of the monomer from the gravimetry feeder to the twin-screw extruder.
Comparison between PLAs as produced in bulk either in traditional batch processing or in REx polymerization in a co-rotating closely intermeshing twin-screw extruder, both promoted by an equimolar Sn(Oct)2·P(C6H5)3 complex with a [L – LA]o/[Sn] ratio of 5000, at 180 °C (extrusion throughput rate ~ 1 kg/h)
| Entry | Process | Time for conversion (min) | Polymer conversiona (%) | Mnb | Mw/Mnb |
| 1 | Batch | 40 | 98.5 | 246 000 | 1.9 |
| 2 | REx | ~7 | 99 | 91 100 | 1.8 |
a Monomer conversion as determined by FTIR spectroscopy [16].
b Number average molecular weight (Mn) and molecular weight distribution (Mw/Mn) as determined by size exclusion chromatography with regard to a polystyrene calibration.
The as-recovered REx-PLA materials are melt-stable enough so as it can be used directly for further melt processing using any processing device. Indeed it has been possible to reduce the amount of catalytic complex ([L – LA]o/[Sn] = 5000) so that residual catalyst can remain inside the polymer without displaying negative effects on the melt stability of the resulting polyester during further melt processing. An additional stabilizer, i.e. Ultranox 626, which does not influence the course of the polymerization reaction, further enhances the melt stability of the resulting PLAs. PLA-based block copolymers with hydroxyl-terminated oligo-PCL or poly(ethylene glycol) have been produced by single-stage REx as well [14].
3 Chemical modification of biodegradable polyesters by REx
PLA chains have been chemically modified either by self-branching or by maleation reaction both induced by a free-radical process performed in bulk through REx. Actually the polyesters chains have been functionalized by maleation reaction in order to compatibilize the PLA matrix with hydrophilic fillers like starch granules. It was expected to get reactive functions pending all along the polyester chains and able to strongly interact with the hydroxyl groups available at the surface of the polysaccharide dispersed phase.
3.1 Self-branched REx-PLA
The self-branching reaction has been conducted in a co-rotating intermeshing twin-screw extruder (screw diameter of 30 mm and L/d ratio of 14) allowing a mean residence time ranging from 4 to 5 min at a temperature ranging from 180 to 190 °C. 2,5-Dimethyl-2,5-di-(tert-butylperoxy)hexane (Lupersol L101) was used as the free-radical initiator [17]. Lupersol L101 is FDA approved as food additive and interestingly is characterized by a half life below 1 min in the extrusion temperature range, thus around 180 °C. In absence of L101, the simple extrusion of PLA triggers a drastic degradation of the polyester chains as evidenced by the decrease of both molecular weight (Mn from ca. 120 000 down to ca. 80 000) and melt viscosity (Table 4). This degradation takes place whatever the extrusion temperature studied and more likely results from thermo-hydrolysis and back-biting side-reactions.
Effect of the PLA extrusion temperature and L101 relative content on the PLA molecular weight (Mn), polydispersity index (Mw/Mn), and melt flow index under 2.16-kg load at 190 °C (MI2)
| Extrusion temperature (°C) | L101 (wt.%) | Mn | Mw/Mn | MI2 (g/10 min) |
| –a | – | 121 600 | 1.4 | 12.76 |
| 180 | 0 | 81 000 | 1.3 | 31.92 |
| 0.1 | 104 700 | 1.3 | 13.93 | |
| 0.25 | 129 000 | 1.5 | 6.99 | |
| 190 | 0 | 82 900 | 1.3 | 82.67 |
| 0.1 | 114 500 | 1.3 | 19.65 | |
| 0.5 | 153 600 | 520b | ND |
a Not extruded PLA sample.
b Highly branched PLA chains, incl. formation of microgel domains.
It is very interesting to point out that the simple addition of L101 to the extruded PLA allows to reduce the lost of molecular weight by a free-radical self-branching reaction. This is nicely evidenced by the extrusion of PLA conducted at 180 °C in the presence of 0.25 wt.% L101 leading to PLA chains with molecular weight and molecular weight distribution very similar to the values measured for the pristine (not extruded) PLA chains. It is however worth noting that at higher temperature and free-radical initiator content (Table 1, last entry), highly branched PLA chains (even with microgel formation) are formed as shown by SEC with triple detection which displays a much larger radius of gyration, a large fraction of chains with apparent molecular weight above 1000 000 Da and a Mark–Houwink parameter a below 0.5, attesting for the highly branched structure and even the presence of microgels of limited solubility in THF at 25 °C. Actually the free-radical chain branching of PLA more likely occurs by hydrogen radical abstraction in α position of carbonyl groups followed by radical addition onto the carbon–carbon double bond of the enolate forms in equilibrium within the polyester chains (Fig. 5). At larger extent, cross-linking and microgel formation may take place. Furthermore, free-radical chain scissions might occur and participate in the chain branching process as well.
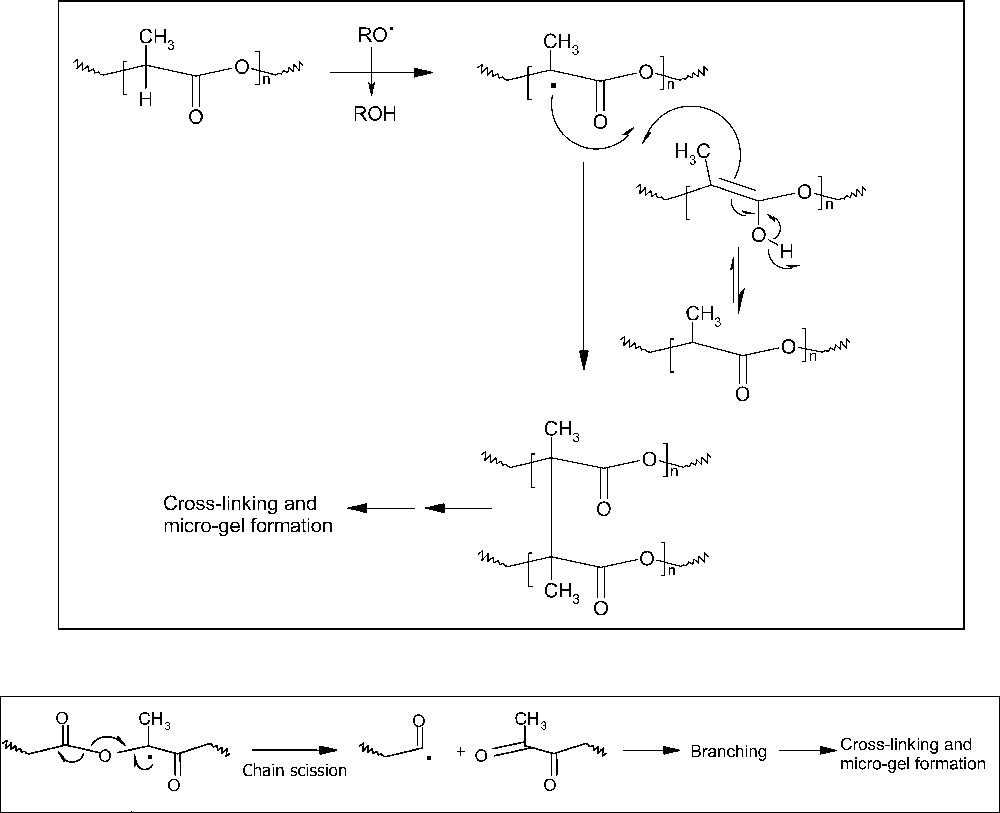
Free-radical reactions of REx-PLA including self-branching, cross-linking and chain scissions.
3.2 Maleated REx-PLA
The grafting reaction of maleic anhydride (MA) onto the PLA backbone has been performed through a free-radical process again conducted by REx and using very similar conditions as aforementioned (Fig. 6). The relative content in MA was maintained at 2 wt.% on PLA basis. Clearly, by increasing the free-radical initiator content from 0 to 0.5 wt.%, the content of MA moieties grafted onto PLA chains does increase as determined by back titration of an excess of morpholine with HCl (Table 5) [18]. The molecular weight remains quite constant at a value around 85 000, which is more likely explained by a competition between the molecular weight increase through chain branching and molecular weight decrease by β-chain scission triggered by grafted MA.
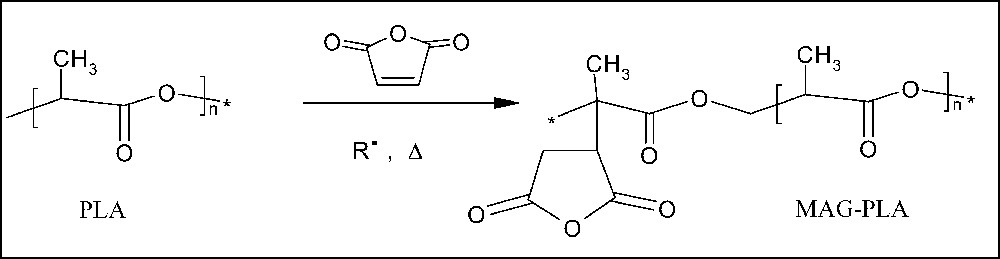
Maleation reaction or REx-PLA via a free-radical process.
Maleation reaction of PLA chains carried out by REx in the presence of 2 wt.% MA at 180 °C. Effect of L101 relative content on the MAG-PLA molecular weight (Mn), polydispersity index (Mw/Mn), and the content of grafted MA
| L101 (wt.%) | Maleation (wt.%) | Mn | Mw/Mn |
| 0 | 0.07 | 81 200 | 1.2 |
| 0.1 | 0.23 | 95 300 | 1.3 |
| 0.25 | 0.47 | 89 000 | 1.3 |
| 0.5 | 0.65 | 85 100 | 1.3 |
4 Compatibilization of polyester melt blends by REx
As a first example, MA-grafted PLA (MAG-PLA) with 0.65 wt.% MA has been melt blended with granular corn starch in an internal mixer [18]. A cryofracture surface as observed by SEM (not shown here) displays improved PLA/starch interfacial interactions by using MA-grafted polyester chains. Compared to non-compatibilized starch/PLA blends, surface dewetting is no longer observed around the starch particles. This likely results from the strong interactions between carboxylic anhydrides and the hydroxyl functions of the polysaccharide chains.
Interestingly enough, this strategy of compatibilizing the interface between aliphatic polyester and hydrophilic fillers has been very recently extended to other biodegradable matrices and naturally occurring fillers [19]. Actually, poly(butylene adipate-co-terephtalate) copolyesters (Ecoflex®) have been first treated with 3 wt.% MA in the presence of 0.05 wt.% L101 at 180 °C in a co-rotating intermeshing twin-screw extruder (screw diameter of 30 mm and L/d ratio of 42) with a screw rotation speed of 130 rpm. MA-grafted Ecoflex® (MAG-Ecoflex®) has been recovered with 0.65 wt.% MA grafted along the polyester chain. MAG-Ecoflex® has been then melt blended with talc microparticles (average diameter of 6 μm) by REx under the same extrusion conditions as for performing the maleation reaction, i.e. within a mean residence time of 4–5 min. The tensile properties of the so-obtained melt blends have been measured on films blown at 150 °C using a Killion single-screw extruder (Table 6). By comparison with the reference Ecoflex®/talc melt blend, i.e. without any compatibilization through previous maleation reaction, one can observe a twofold increase of both Young modulus (thus material stiffness) and yield/ultimate stress while maintaining high elongation at break, meaning high ductility. However such an improvement is detected only in the presence of 0.5 wt.% tin octoate actually added as an esterification catalyst for promoting the reaction between talc and the polyester-grafted cyclic anhydride moeities (see Fig. 7).
Blown film tensile properties of Ecoflex®/talc melt blends (with 30 wt.% talc) as produced by REx. Effect of maleation (MAG-Ecoflex®) and addition of an esterification catalyst (tin octoate). Tensile testing were performed following norm ASTM D-882
| Entry | Copolyester | Esterification catalyst | Young modulus (MPa) | Yield stress (MPa) | Ultimate stress (MPa) | Elongation at break (%) |
| 1a | Ecoflex | – | 117.3 | 12.7 | 14.2 | 600 |
| 2 | MAG-Ecoflex | – | 110.0 | 18.6 | 18.8 | 460 |
| 3 | MAG-Ecoflex | 0.5 wt.% Sn(Oct)2 | 227.5 | 27.2 | 27.2 | 630 |
a Simple melt blend (without any compatibilization).

Sketch of the catalyzed interfacial grafting reaction between MAG-Ecoflex® and talc microparticles.
It is worth pointing out that the unique possibility to perform simultaneously in a continuous one-stage REx process both maleation reaction of the poly(butylene adipate-co-terephtalate) copolyester chains, fine dispersion of talc particles and catalyzed interfacial grafting reaction allowing the compatibilization between talc and in situ generated MAG-Ecoflex®. Thus in this case, the free-radical functionalization of the aliphatic–aromatic copolyesters occurs along with the simultaneous filler dispersion within the molten matrix. In addition, a catalytic grafting reaction takes place between the particle surface and the in situ generated reactive functions pending onto the polyester chains. A much improved filler/matrix reactive compatibilization results and again allows for largely enhancing the thermo-mechanical properties of the pristine copolyester matrix.
5 Conclusions
This contribution has emphasized that biodegradable materials can be effectively produced by performant single-stage continuous reaction extrusion processes. When combined to both an adapted chemistry, i.e. by the right selection of the catalytic system, and finely tuned and well tailored extrusion parameters, co-rotating twin-screw extruder particularly demonstrated remarkable ability in (i) synthesis of aliphatic polyesters by catalyzed ROP; (ii) chemical modification of these polyesters such as self-branching or maleation reaction, and (iii) production of compatibilized melt blends between biodegradable polyesters and different naturally occurring fillers. There is no doubt that the above experiments represent only some illustrations of the huge potential of REx, a solvent free melt process, in the field of biodegradables and more largely in the industry of polymeric and composite materials.
Acknowledgements
Authors are very grateful to the ‘Région wallonne’ and the European Community (FEDER, FSE) for general support in the frame of ‘Objectif 1-Hainaut: Materia Nova’. This work was partly supported by the Belgian Federal Government Office of Science Policy (SSTC-PAI 5/3). J.-M. Raquez is much indebted to FRIA (Fonds pour la formation à la recherche dans l'industrie et dans l'agriculture) for his Ph.D. grant, and to the ‘Ministère de la Communauté française de Belgique’ for financial support in the frame of his research stay at MSU.