

In 1997 Hubert et al. reported the solid-state synthesis1 of a new boron subnitride B6N by the reaction of boron with hexagonal graphite-like boron nitride (hBN) at 7.5 GPa and 1700 °C in a Walker-type multianvil apparatus [3]. According to the quantitative PEELS analysis, the oxygen-free grains of the product have a mean composition of B6N0.92, and its X-ray powder diffraction pattern seems to be similar to that of B6O and B4C. On this basis, the structure similar to that of other icosahedral boron-rich phases, more specifically to B6O, has been suggested. However, the X-ray powder diffraction data reported in Ref. [3] does not seem to be unambiguous. The lines at low angles, which are suppose to be intensive in the case of B6O- and B4C-like structures, are of very low intensity and only two lines at higher angles may be attributed to the B6N phase. At the same time, these two lines coincide with the 240 and 042 reflections of β-rhombohedral boron (β-rh B).
In Ref. [3], the samples have been powdered before taking the diffraction patterns; so, the absolute intensities of the lines should be close to the theoretical ones (how we see it for the patterns of B6O and B4C in Ref. [3]). Since preferred orientation is not expected for the B6N phase, such weak 101, 003 and 012 lines of ‘B6N’ observed in the pattern reported in Ref. [3] cannot be explained in the framework of the B6O- and B4C-like structures. Our crystallographic simulations using PowderCell program [4] have shown that neither nitrogen occupancy nor nitrogen/icosahedron positioning lead to such a strong decrease of intensities of all three lines in relation to the 104 and 021 reflections (Fig. 1). Therefore, the powder diffraction patterns reported in Ref. [3] can hardly belong to a B6N phase with B6O- or B4C-like structure.
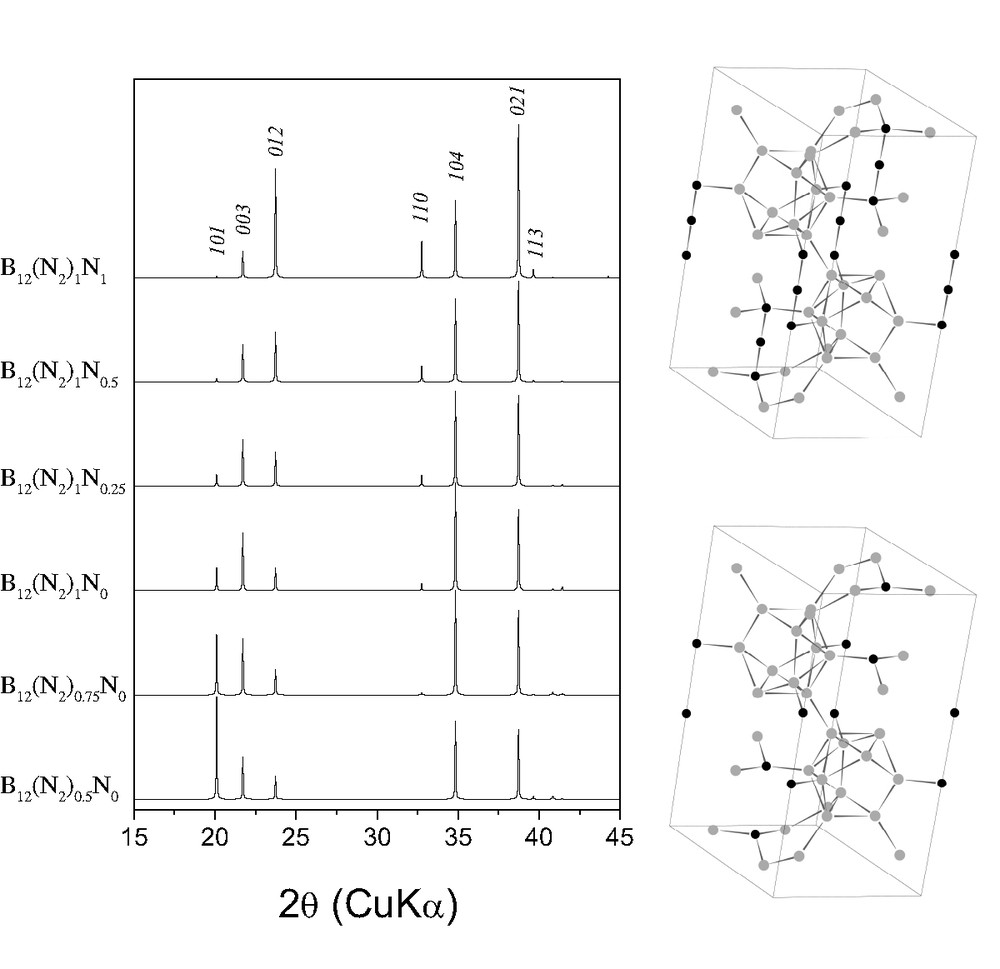
Crystallographic simulation of the powder diffraction patterns of boron subnitride with B6O-like (bottom) and B4C-like (top) structures (, a = 5.457 Å,c = 12.241 Å [3]). Three bottom patterns correspond to the B6O-like structure, while three top patterns represent the B4C-like structure). All patterns have different nitrogen content indicated in the legends.
Very recently we found that at high pressures and temperatures β-rh boron forms the bulks with strong texture [5]. Even after powdering between two WC discs (the same has been done in Ref. [3]), the X-ray diffraction patterns of the boron samples show unpredictable intensities of some reflections. Most probably, it is caused by the tendency of boron to form single crystals. Only a fine powdering in a hard alloy mortar has allowed us to observe the powder diffraction pattern of β-rh boron with reasonable line intensities. So, the possible explanation of the ‘B6N’ pattern reported in Ref. [3] is a strong preferred orientation of β-rh boron (or another boron phase that is stabilized by nitrogen impurity).
In order to clarify the situation, we have performed the experiments similar to that of Hubert et al. [3], but with an advantage of the in situ observation. The chemical interaction between boron and hBN at 7.4 GPa and 1700 °C have been studied using X-ray diffraction with synchrotron radiation at the European Synchrotron Radiation Facility. The experiments have been carried out using a new compact multianvil device combining a T-cup module with a Paris–Edinburgh press [6] at beamline ID27, ESRF. The mixture of β-rh boron2 and hBN (99.9%, Alfa) of the B6N stoichiometry has been subsequently loaded into a capsule of hBN and introduced into an octahedral cell assembly, which has been described in Ref. [6]. hBN has been used as an internal pressure calibrant [7]. Angle-dispersive diffraction patterns have been collected on an on-line MAR 345 image plate detector, which allows to record the high-quality data within less than a minute. High-brilliance synchrotron radiation from the ID27 two-phased undulator has been set to a wavelength of 0.3738 Å using an in-vacuum channel-cut Si (111) monochromator and collimated down to 100 × 30 μm2 (horizontal by vertical) by two sets of tungsten carbide slits. An oscillating multichannel radial collimator [8] has been used to avoid diffraction from the materials surrounding the sample. Exposure times have been between 60 and 120 s depending on the electron current.
Sample temperature has been measured with the W3%Re–W25%Re thermocouple without correction for the pressure effect on the thermocouple emf. If the thermocouple failed, the temperature was estimated by the power-temperature calibration curve. Also, pressures and temperatures have been independently determined by analytically cross-calibrating the equations of state of hBN and Re with the program PTX-cal [9] using the unit-cell volumes obtained by the least-squares fits of the diffraction patterns obtained in situ.
The precise calibration of the sample-to-detector distance (369.649 mm) has been made by measuring a LaB6 (99.999%) powder. Correction of the two-dimensional diffraction images for spatial distortions and integration of the Debye–Scherrer rings have been performed using the FIT2D software [10]. Lattice parameters of the crystal phases have been obtained by LeBail full profile refinement of integrated patterns using the GSAS [11] program package.
Fig. 2 shows the set of diffraction patterns obtained in situ during the heating of the boron–hBN mixture at 7.4 GPa. No interaction occurs in the mixture; only the random change in relative intensities of various boron reflections is observed. During the 30-min heating at 1700 °C, the boron lines do not disappear and the expected B6N reflections do not arise. In the powder diffraction pattern of the quenched sample (G3000 TEXT, INEL) we do not see the lines consistent with the pattern of B6N, although some reflections of boron have the same position as the reflections of hypothetical B6O-like boron subnitride.
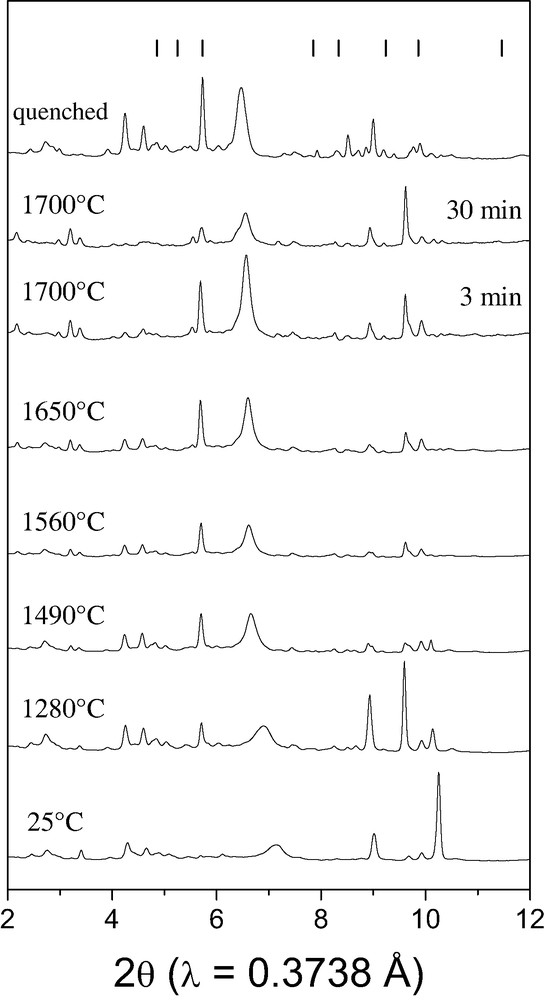
Sequence of the X-ray diffraction patterns collected in situ during the stepwise heating of the B–BN mixture at 7.4 GPa. The bars on the top represent the expected line positions for the hypothetical B6N phase with B6O-like structure (, a = 5.457 Å, c = 12.241 Å [3]).
Thus, the results of our in situ studies of the chemical interaction between boron and hBN at 7.4 GPa and 1700 °C has allowed us to conclude that the evidence for the solid-state synthesis of boron subnitride B6N with B6O-like structure reported in Ref. [3] is inconclusive.
Acknowledgements
We would like to thank our collaborators S. Klotz, G. Hamel, D. Martinez-Garcia, W.A. Crichton, and M. Mezouar for their contributions and valuable discussions. High-pressure experiments have been carried out during beamtime allocated to proposal HS-2532 at ID27, ESRF. This work has been supported by the ‘Agence nationale de la recherche’ Grant No. NT05-3_42601.