

1 Introduction
‘Ageing’ is a term used in many branches of polymer science and engineering when the properties of the polymer change over a period of time. The changes may be observed in engineering properties such as strength and toughness; in physical characteristics such as density; or in chemical characteristics such as reactivity towards aggressive chemicals. The origins of the changes may be independent of the surrounding environment and may be chemical, as in the case of the progressive cure of a thermosetting material, or physical, as in the case of a rapidly cooled polymer undergoing volumetric relaxation. In other cases the changes may be the result of interaction with the environment, such as when oxidation leads to chain scission. Sometimes a number of age-related phenomena operate simultaneously and/or interactively. The various kinds of ageing will be defined and discussed here and particular attention will be paid to interactive aspects.
The subject of ageing in polymers is vast and a comprehensive review would demand the collaboration of a large team of experts and would occupy several volumes. This paper presents a personal view and is based largely on studies conducted by the author and his co-workers. The major sections deal with Physical Ageing, Curing, Thermal Ageing and Weathering. These titles are chosen for convenience and some topics do not fall fully under a single heading.
2 Physical ageing
Physical ageing is probably the most common form of ageing. It often occurs alongside the other forms of ageing described below, and for that reason it is appropriate to deal with it first. Physical ageing occurs when a polymer is in a non-equilibrium state and is caused by molecular relaxations that are biased in the direction required to drive the material closer to equilibrium. This phenomenon is very common and is encountered in thermoplastics mouldings that have been cooled rapidly from an elevated temperature during the shaping operation. The material, in the form of a hot melt, is processed by passing it through a die (extrusion) or into a closed mould (injection moulding, blow moulding). After shaping, the material is cooled rapidly. In the case of amorphous polymers, the material is still cooling rapidly when the temperature drops below the glass transition temperature, Tg. Once below Tg the rate of molecular relaxation is too slow to keep pace with the changes required if the material is to remain at thermodynamic equilibrium. As a result, when the material reaches thermal equilibrium with the surroundings it is not at thermodynamic equilibrium. One symptom of this is the density of the material, because its volume is characteristic of a higher temperature. Consequently, after reaching thermal equilibrium with the surroundings, it will undergo volumetric relaxation, gradually increasing density during an extended ageing period. An example is given in Fig. 1 that is based on data obtained for injection moulded polystyrene by Iacopi and White [1]. Samples were taken, respectively, from the skin and the core of the moulded bar. The thermo-mechanical history of the two regions is quite different and this accounts for the difference in the density after the moulding operation is complete. Thereafter, the samples of material from the two locations appear to increase in density at fairly similar rates (Fig. 1). The rate of densification will depend on the departure from equilibrium at the ageing temperature, Ta, and on the difference between Ta and Tg. Ageing may be quite rapid at first if Ta is not too far below Tg but the approach to equilibrium will slow down and ageing effects may sometimes still be apparent after many years. If Ta is far below Tg the thermodynamic driving force will be large because the property (e.g. density) will be far from the thermodynamic equilibrium value and will favour change but the kinetics are determined by the difference between Ta and Tg, and this will limit the rate of change.
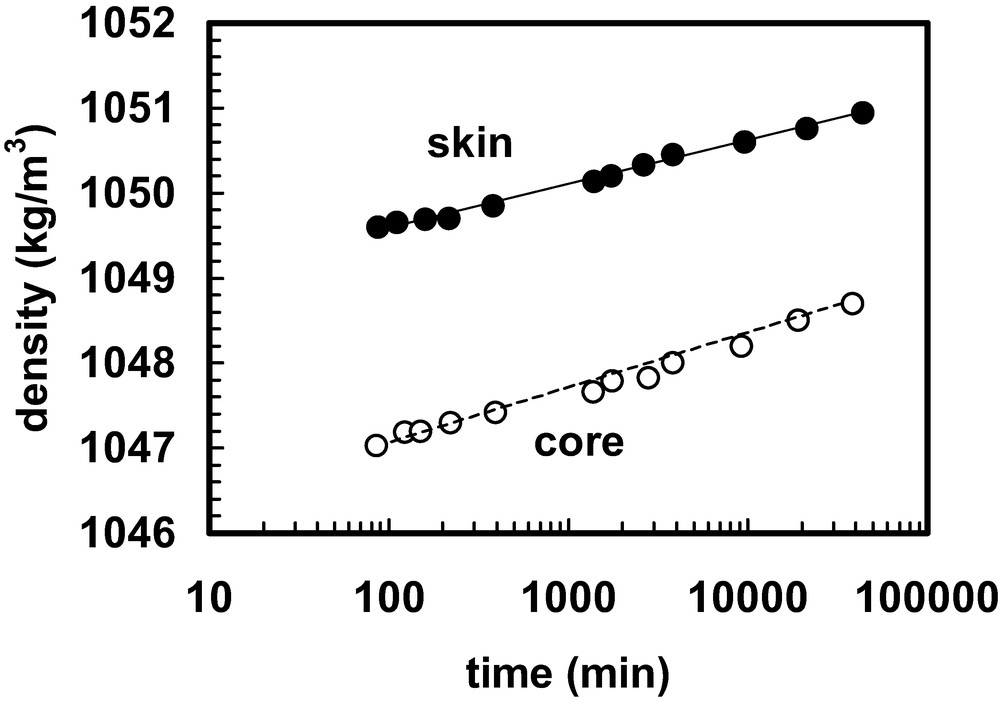
Density versus ageing time at room temperature of samples extracted, respectively, from the skin and the core of an injection moulded polystyrene bar. Ageing took place in a density column at 23 °C. Further experimental details and discussion of the interpretation of the data are given in Ref. [1].
Ageing of amorphous materials was studied extensively by Struik [2], and his book, published more than a quarter of a century ago, remains the foremost reference on physical ageing of polymers. The book is based largely on experimental data gathered in a meticulous fashion and included examination of the creep behaviour of materials in different states of ageing. Struik’s data and his analyses link closely to engineering properties. They also show close connection with the somewhat more fundamental approach to the understanding of the physical principles of polymer ageing followed by many others including Kovacs, Hutchinson, McCrum, McKenna and others [3–12]. Studies of inorganic glasses helped to set the agenda for the studies of these relaxation phenomena in the 1970s [13–15].
2.1 Relaxation behaviour and mathematical description of relaxation behaviour
Struik found that his creep data for amorphous polymers, obtained from tests on similar samples but commenced after various different ageing times, could be superimposed by applying a shift parallel to the time axis (where “time” was measured from the instant that the creep load was applied). A further discovery was that the relaxation behaviour of the polymers that he studied, specifically the onset of the glass–rubber transition, fitted a stretched exponential originally published by Kohlrausch and used also by Williams and Watt [16] (‘KWW function’) [2]:
| (1) |
J(t) is the creep compliance at time t; J0 is its value at t = 0; t0 is a characteristic time that depends on temperature and age; and m is a parameter that is typically ~1/3. This function and its application to relaxation phenomena is discussed further by Moynihan et al. [14,15], by Kovacs et al. [4,5] and by others [17–22].
The KWW ‘stretched exponential’ function gives a much better representation of relaxation behaviour than the single relaxation time function for the Maxwell body or the standard linear solid, but does not have a generally accepted physical basis. Another way to represent behaviour that appears to follow a stretched exponential form is to use a continuous distribution of relaxation times as in the following expression for the description of stress relaxation behaviour:
| (2) |
| (3) |
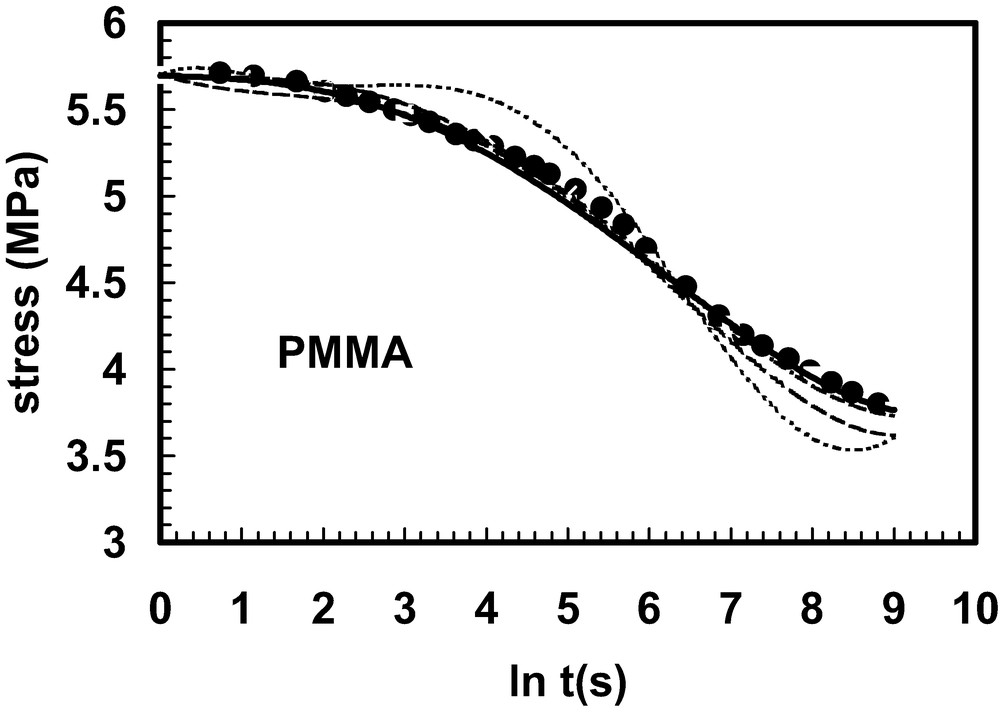
Stress relaxation of PMMA (data points) with fitting lines for:
(sum of exponentials) where τj = 33.3, 100, 333, 676, 103, 3333, 104 s;
(standard linear solid – single relaxation time);
(KWW function);
(power law).
(Adapted from Ref. [23]).
2.2 Ageing in crystallising polymers
Much of the research on polymer ageing has been concentrated on amorphous polymers. The development of an understanding of the observed phenomena in amorphous polymers is extremely challenging and this is one of its attractions to polymer physics researchers. Semi-crystalline polymers are even more complex, however, which may be why their ageing behaviour has attracted less attention from the polymer physics community. Although the crystal phase is relatively inert with respect to ageing, it restricts relaxation processes in the adjacent amorphous phase to which it is intimately connected. Its influence depends on the morphology and on the interaction between the crystal and amorphous regions. Therefore much of the property change that occurs on ageing a semi-crystalline polymer is due to relaxations occurring in the amorphous phase but it is controlled partly by the presence of the crystalline phase. To explain and/or predict the ageing behaviour of a semi-crystalline polymer therefore requires knowledge of the morphology with a level of detail rarely attained as well as of the ageing behaviour of the amorphous phase. Because of the widespread application of semi-crystalline polymers, it is important to know about their ageing behaviour but studies have concentrated more on the phenomenology as determined from the measurement of engineering property changes with ageing. A further complication is that the crystallinity may change during ageing. Crystallinity has a large influence over mechanical properties but it was discovered early on that changes in property on ageing could not be accounted for by changes in crystal characteristics as measured by the techniques available at the time and it was concluded that the amorphous phase must be playing a major role in the property changes [28,29].
When the physical ageing of crystallising thermoplastics is examined, it is usually found that the changes in property are at least as large as those observed with amorphous polymers. Physical ageing in the amorphous phase contributes significantly to the observed changes in properties. There are two additional potential sources of ageing. Firstly there is the possibility that secondary crystallisation may occur, causing the crystallinity of the material to increase [30,31]. Secondly there may be molecular re-arrangements in the amorphous phase of a different kind (or which simply have a different effect) to those that occur in totally amorphous polymers, such as rearrangement of tie molecules that control the interaction of the amorphous and crystalline phases [32].
The rate of crystallisation in a moulding operation is normally at its maximum at some temperature Tc,max between the melt temperature, Tm, and the mould temperature. The temperature falls rapidly through Tc,max, and the rate of crystallisation is often quite slow at Tm, as is also the case at the ambient temperature after de-moulding. Therefore the crystallinity is often incompletely developed. The molecular mobility is often sufficient to allow molecular segments in the amorphous phase to progressively realign and crystallise, either in the form of new crystals between the pre-existing lamellar crystals, or by adding to the pre-existing crystals. The mobility is particularly high in the case of crystallising polymers, such as polyethylene, that have Tg below ambient temperature and this secondary crystallisation may develop relatively quickly. Because of the enormous differences in property between the crystalline and amorphous phases, there can be very large property differences for even small changes in crystallinity (of the order of 1%). McCrum [33] considers the effect of crystallinity but Struik [34] claims that his ageing observations with polypropylene were not explained by secondary crystallisation. Struik [35–38] followed this study with a series of papers concerning physical ageing of semi-crystalline polymers in which the interpretation of the data again concentrated on the behaviour of the amorphous phase. He discusses the effect of the presence of the crystal units on molecular relaxation in the amorphous phase [36].
More recently, the current author has studied changes in crystallinity that occurred as the result of photo-ageing in polyethylene and polypropylene; this topic will be dealt with in Section 5.
2.3 Physical ageing at elevated temperature and the effect of large stresses
Physical ageing of a polymer is sometimes accelerated if the temperature is increased. Equilibrium is displaced at the higher temperature and the material may not finish in the same state as would have been achieved by a longer ageing period at a lower temperature. If the material is cooled rapidly from the elevated temperature a new ageing regime starts, especially if the elevated temperature is above Tg. The use of an elevated temperature is often referred to as “annealing” in analogy with the use of elevated temperature heat-treatments in metals. It has been claimed that apparent reversal of ageing can sometimes be provoked in polymers by the application of a large mechanical stress, a phenomenon sometimes referred to as “rejuvenation”. The observation of rejuvenation requires very careful experimentation and the interpretation of the results demands detailed consideration. McKenna [39] has provided a comprehensive review of this phenomenon and questions its existence, showing how a false impression can be obtained if the experimental design and execution of the measurements are not conducted adequately.
2.4 Residual stresses
The rapid cooling used in thermoplastics moulding processes, coupled with their low thermal conductivity, causes the formation of strong temperature gradients and, as a consequence, the development of residual stresses [40]. The stresses in the interior of mouldings made using conventional conditions are tensile in the interior and compressive near the surface. These residual stresses have a significant influence over behaviour. On extended ageing, they relax partially or completely, depending mainly on Tg in the case of amorphous polymers, and on the crystallinity in the case of semi-crystalline polymers. The effect of this change in residual stresses will also appear as an ageing phenomenon [1,41–48].
It is commonly observed that the residual stresses relax relatively rapidly at short ageing times. The rate of relaxation slows down progressively and the final residual stress levels after an extended period are often a significant fraction of the original values, immediately after moulding. This can be understood in terms of the stress-biased relaxation behaviour [49–52]. The ‘site-model theory’ [53] leads to the prediction that stress relaxation does not normally cause the stress in a polymeric body to reduce to zero [49], and residual stresses are therefore generally expected to equilibrate at a non-zero level [52]. The site-model theory predicts behaviour that is identical to that of a standard linear solid [50]. Although this has a single relaxation time, contrary to observed behaviour, combination of a series of standard linear solids in the manner shown in Eq. (2) (or Eq. (3)) resolves this. Elevated temperature changes not only the rate at which equilibrium is reached but the equilibrium state itself. An example of the change in residual stress distribution (in polystyrene) when ageing occurred at elevated temperature (83 °C) is given in Fig. 3; further examples are given in Refs. [42–45,48]. Rapid cooling after elevated temperature ageing will re-establish residual stresses that are attributable solely to this process.
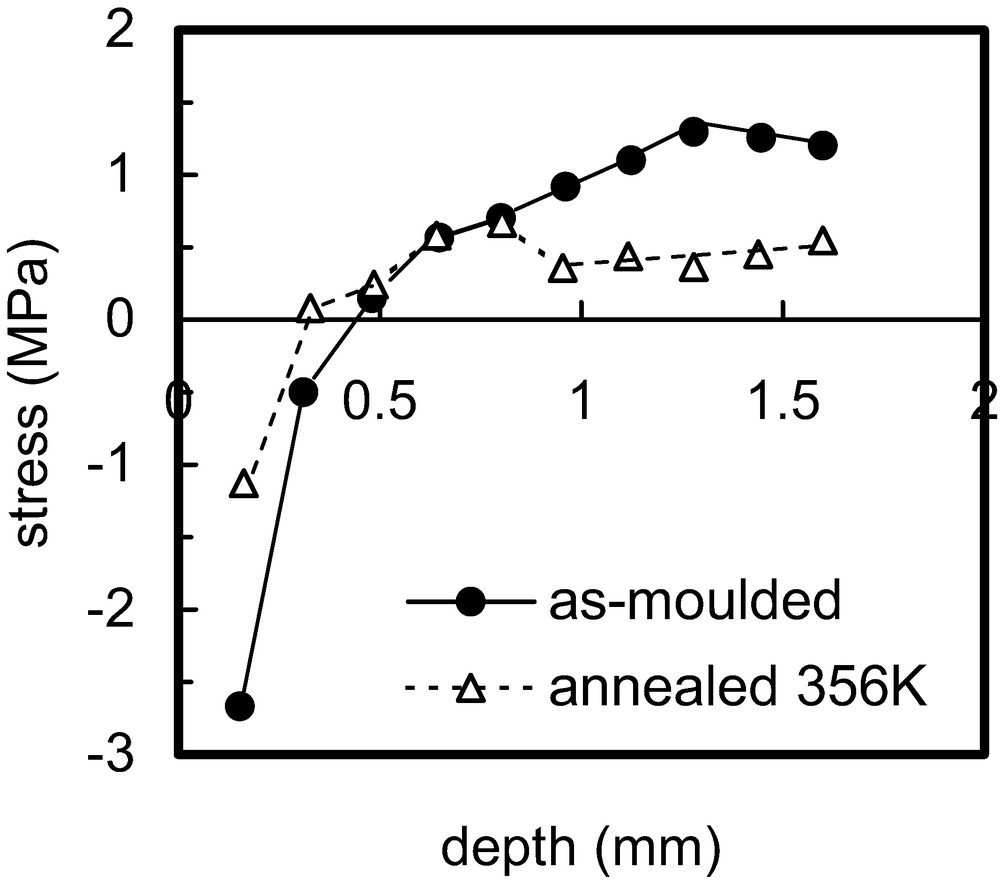
Residual stress distributions for polystyrene in the as-moulded state and after annealing at 356 K. Results are shown for half of the bar thickness (the moulding was ~3 mm thick). More details are given in Ref. [42].
Secondary crystallisation causes the material to shrink locally and if it occurs to a different extent at different positions within a solid polymer, residual stresses will develop. Thus if secondary crystallisation occurs in a depth-varying manner, residual stresses will develop or change through the depth of the polymer solid. This has been observed by Siegmann and Kenig [30] and analysed by White [31].
It is worth noting that the term ‘internal stress’ is often used instead of ‘residual stress’. This can sometimes be confusing because the term ‘internal stress’ is often used when molecular orientation is present in the cooled product, a practice discouraged by the present author [40,54]. Another use of the term ‘internal stress’ relates to the ‘back-stress’ reaction to applied stress that is a phenomenon that occurs also in metals and is explained by physical metallurgists using dislocation theory. The site-model theory can be invoked to explain the observed behaviour in polymers, in place of dislocation theory [40,54].
3 Curing
Curing of a thermoset is a very complex process. The starting state of a thermosetting resin is typically a viscous liquid in which fairly low molecular mass molecules are free to move. As crosslinking proceeds the molecules become larger and less mobile. When gelation (network formation) occurs the material effectively consists of two phases: the gel has a glass transition temperature that is higher than the glass transition temperature of the remaining ungelled fraction. Continued crosslinking causes the gel to grow at the expense of the ungelled material and the glass transition temperature climbs further; the maximum glass transition temperature is the glass–rubber transition of the fully cured resin. In a typical resin cure, the glass transition temperature of the gel starts out well below the temperature of the reagents but then climbs to finish above the temperature of the material. The progress of the changes depends on the mobility of the reactants and on their reactivity. It therefore depends on the temperature, particularly with relationship to the glass transition temperatures of the network and the ungelled material. Gillham [55] has developed a time–temperature–transformation diagram to formalise the description of thermoset curing. Continued crosslinking may continue for a very long time if unreacted groups remain and slowly diffuse to find partners with which to react. When this is the case, this phase of the curing process is sometimes termed ‘ageing’.
In addition to these changes, physical ageing occurs in thermosets; the effect is different if the ageing temperature is near the glass transition temperature than when it is well below it. Furthermore, both the progress of curing and the physical ageing may cause changes in residual stresses (often called “curing stresses” in thermosets). The stresses that develop during curing depend to a significant extent on the mechanical constraints that are applied during curing. The magnitudes of stresses that develop in thermosetting coatings applied to a deformable substrate are quite different from those that develop in coatings applied to a rigid substrate [56]. An example of this is given in Fig. 4 in which the internal stress in epoxy coatings applied to steel shims 0.1 mm thick and cured (aged) in dry air is plotted against ageing time. The sample that was free to bend during ageing developed larger stresses than the one that was on a substrate that was restrained during ageing (except for short periods when it was released to execute the stress measurement) [56]. In the case of epoxy resins the ageing behaviour is very sensitive to the humidity [57]. The effect of curing in air at different humidity levels is shown in Fig. 5. The results are for restrained-substrate ageing. Of special note is the fact that the samples that aged in moist air developed compressive stresses (plotted as negative values in Fig. 5). The same resin that was used to obtain the data in Fig. 4 was used for these series of measurements. Note that the stress magnitudes reported in Refs. [56,57] are in error: the correct values are given in Ref. [58] and have been used to construct Figs. 4 and 5. The highest stress levels (> 7 MPa for the material used as example here) represent a large fraction of the breaking strength of the coating. This will therefore contribute to cracking of the coating, whence it loses its barrier properties and the associated protective function. Alternatively, the residual stresses may cause the delamination of the coating.
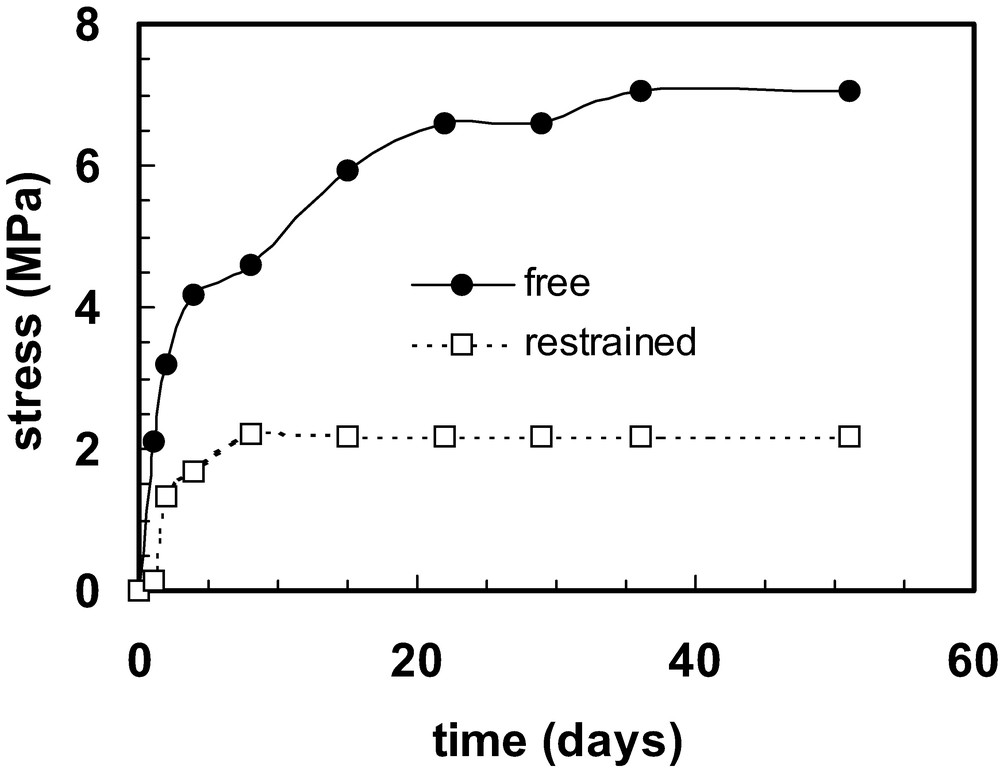
Internal stress development in an epoxy coating on a thin substrate as a function of curing (ageing) time in dry air at room temperature. Results are given for a sample on a substrate that was free to bend during the ageing process and for a sample on a substrate that was restrained from bending. Data are taken from Ref. [58].
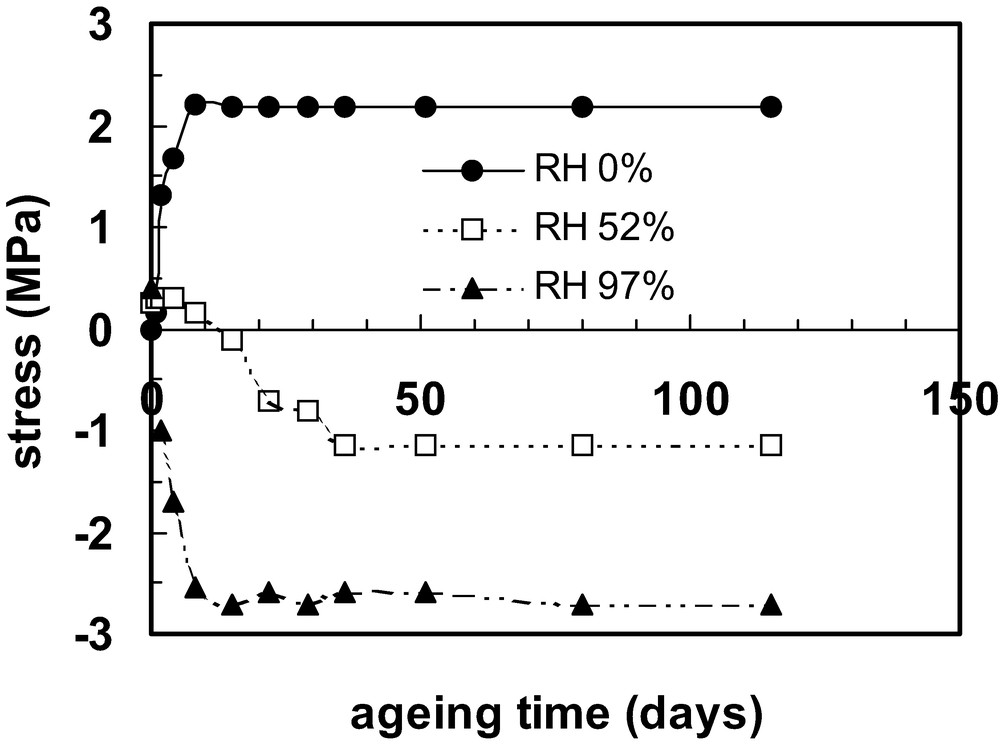
Internal stress development in an epoxy coating on a thin substrate as a function of curing (ageing) time at room temperature in (i) dry air; (ii) air at 52% RH; and (iii) air at 97% RH. All results were obtained using substrates that were restrained from bending during the ageing process. Data are taken from Ref. [58].
The term ‘ageing’ is sometimes used when coatings are conditioned under much more extreme conditions, for example when attempting to replicate the effects of a marine environment (and even when attempting to accelerate the changes in a coating that are provoked by the marine environment by the use of extreme temperatures and temperature cycling). Examples of the kinds of changes that can occur are given in Refs. [59,60] though the author does not advocate the use of ‘ageing’ to describe such conditioning procedures.
4 Thermal ageing
Some aspects of thermal ageing have already been dealt with in Section 2. In earlier section (Section 2), the acceleration of physical ageing caused by the application of an elevated temperature was discussed. No consideration was given to chemical reaction. Elevated temperature will cause acceleration of the completion of curing in a thermoset (‘post-cure’). If an elevated temperature is applied to a polymer in the presence of an aggressive chemical agent (often oxygen) then this may give rise to chemical reactions that may occur only very slowly, or not at all, at ambient temperature. The many changes that occur are collectively known as ‘thermal degradation’ and this is a highly studied aspect of polymer science, with a well-developed literature. It has importance even if the polymer is not destined to be exposed to elevated temperatures during its service life because the moulding process is often conducted at elevated temperature, for example, to melt the polymer if it is a thermoplastic. It is therefore commonplace to include a thermal stabiliser in moulding compounds, if only to protect the material from serious chemical degradation when it is fabricated into artefacts. Thermal stabilisation is another topic that has been studied extensively and has been the subject of numerous research papers. Tests to determine the effectiveness of stabilisers at temperatures well below the melt temperature are often referred to as ‘(thermal) ageing’.
A review of thermal oxidation of polymers has been provided recently by Pospíšil et al. [61], covering amorphous and semi-crystalline polymers, and discussing the physical and chemical changes that occur during thermal ageing, test procedures and with particular emphasis on the behaviour of stabilisers.
5 Weathering
Weathering trials are sometimes referred to as ‘natural ageing’. Polymers exposed outdoors can degrade through the action of several agents, including solar ultraviolet (UV) radiation; water; pollutants (in gaseous form or, more potently, as acid-rain); elevated temperature; and temperature changes. In a majority of cases, the main cause of property deterioration is photo-oxidation, which is initiated by UV irradiation and, as a consequence, much laboratory photo-ageing testing is conducted to determine the weatherability of polymers and to test the effectiveness of stabilisers introduced to improve their weather resistance. Several types of artificial UV sources can be used, the most popular being xenon lamps and fluorescent tubes. The spectral distribution can be adjusted by the use of appropriate filters in the case of xenon lamps or by appropriate phosphors in the case of fluorescent tubes. The spectral distributions of the sources are often designed to replicate the solar radiation distribution at terrestrial sites. Accelerated testing can be obtained by using (i) UV intensities higher than those normally encountered in service and/or (ii) lower wavelengths. The low wavelength option is discouraged by many, including the author, because it might provoke chemical reactions not present under natural conditions [62]. (A similar problem may arise if it is attempted to accelerate degradation by using an elevated temperature [63]). As with thermal degradation, a sequence of oxidative reactions follows in which both chain scission and crosslinking may occur; both of these molecular changes lead to embrittlement of the material. The reactions require oxygen, which diffuses in from the surface. The reaction can be very rapid in intense sunlight, even at ambient temperature. As a consequence, in a thick-sectioned article, the oxygen does not diffuse very far before it is consumed by reaction and the major part of the degradation is confined to a fairly thin layer near to the exposed surface [64–79]. Various characterisation procedures can be applied to investigate the depth profile, including determination of molecular weight, as shown in Figs. 6 and 7 [78,79]. The data given in Figs. 6 and 7 were obtained with 3 mm thick polypropylene injection moulded bars that were photo-aged in the laboratory at 30 °C using UVA-340 fluorescent tubes that simulate solar UV irradiation and do not emit any component with wavelength lower than the low wavelength limit in solar radiation at terrestrial sites. The weight average molecular weight prior to photo-ageing was 203 000 and it is evident that multiple scission events occurred, causing a very large reduction in molecular weight near the exposed surface. The apparent paradox that higher molecular weights are recorded in the interior after a longer exposure period is probably related to crosslinking and that the scission/crosslink ratio differs at different depths and changes as reaction proceeds [80].
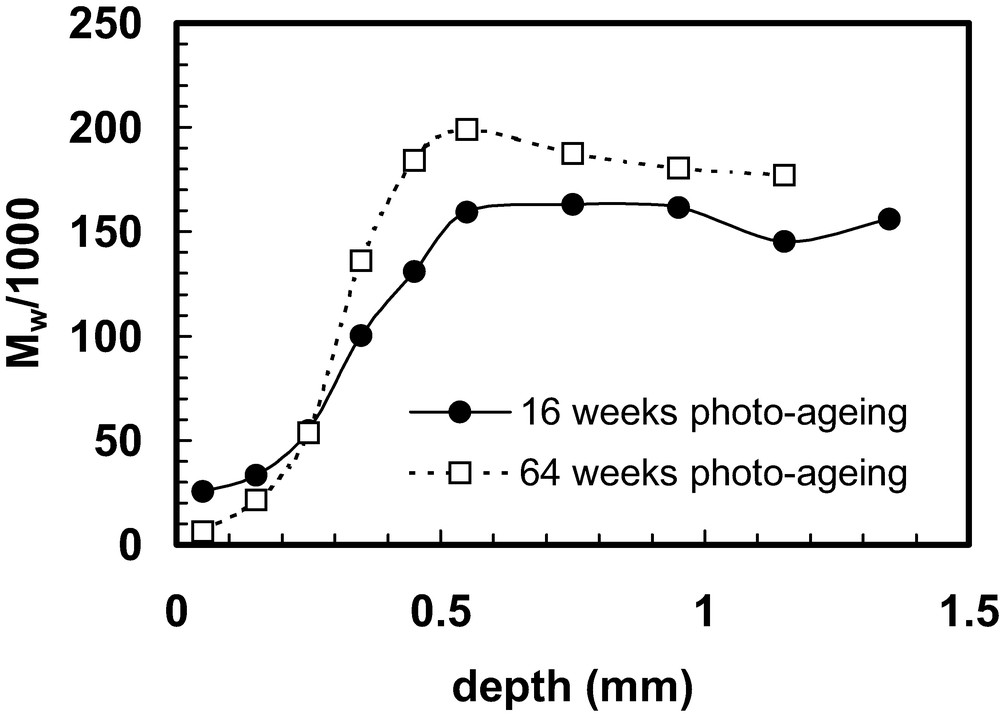
Weight average molecular weight data for polypropylene samples after photo-ageing. Samples, 0.1 mm deep, were extracted at different depths from the exposed surface: corresponding molecular weight plotted at layer centre (i.e. Mw for the layer 0.2–0.3 mm is plotted at 0.25 mm). The data are taken from a study described in Refs. [78,79] in which the experimental procedures are described in full.
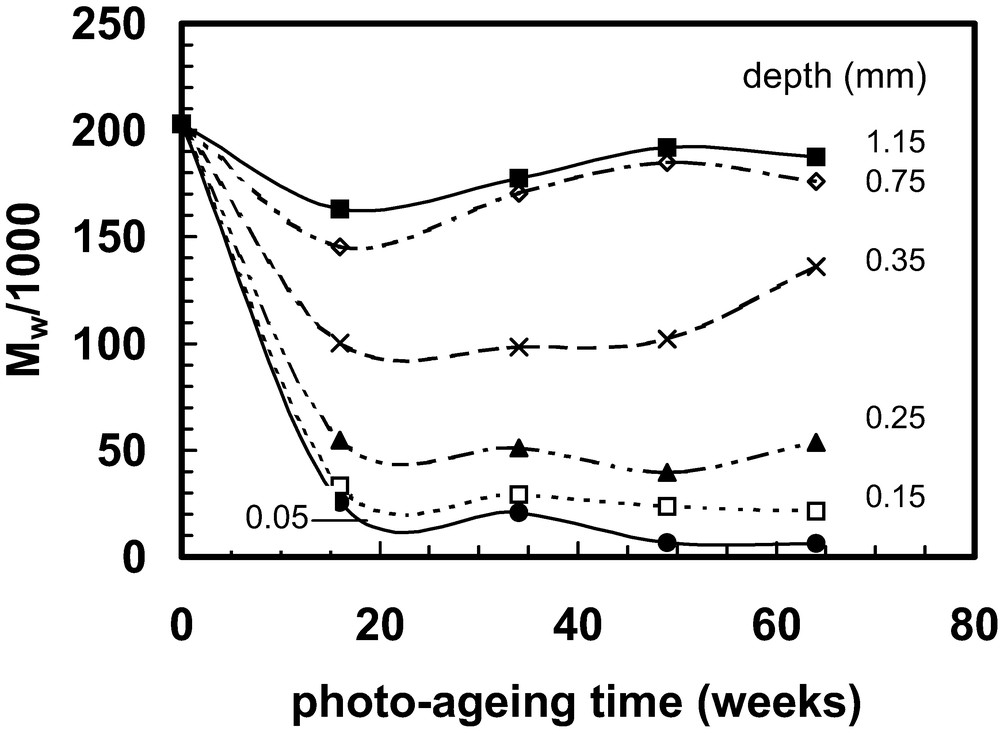
Molecular weight changes with photo-ageing time for six different depths from the exposed surface of injection moulded polypropylene (0.05, 0.15, 0.25, 0.35, 0.75 and 1.15 mm from the exposed surface). The data are taken from the study described in Refs. [78,79].
The molecular degradation in the form of chain scission and crosslinking caused by photoageing often leads to a very brittle layer that can break easily. For a ductile polymer that has a tendency to draw when subjected to a tensile deformation, the material beneath the embrittled layer remains ductile at short photo-ageing times and the cracks in the embrittled layer terminate when they reach the (relatively) undegraded zone (Fig. 8). The brittle layer then fragments and fissures form, allowing the underlying material to draw (Fig. 8). After longer photo-ageing times the degraded layer becomes thicker and the cracks that grow within it become deeper and therefore more potent from a fracture mechanics perspective. If the brittle layer is thick enough to produce a surface flaw of significant depth by this process and if the surface layer still retains sufficient mechanical integrity to transmit stresses to the relatively undegraded material in the interior, the whole section may be embrittled and fracture may follow rapidly when a stress is applied.

Scanning electron micrographs of moulded surface of a polypropylene injection moulded bar after 4 weeks photo-ageing followed by a tensile test. (a) General view near to a corner showing multiple fissures in the brittle surface layer. (b) Magnified image showing that the ductile material beneath the brittle degraded layer has drawn (more details in Ref. [76]).
On increasing the photo-ageing time still further, the degraded layer may become so fragile and have such poor mechanical contact with the underlying material that cracks formed in it no longer propagate into the interior (that has degraded much less because of oxygen diffusion limitation). Fig. 9 shows an example of this for a polypropylene sample in which the embrittled layer that had formed after 12 weeks photo-ageing was so fragile and so poorly adhered to the material in the interior that pieces fell out when it was subjected to a tensile test. Inspection of the fracture surface (at the top of Fig. 9) indicates that the brittle layer was thicker for this sample than for that photo-aged for only 4 weeks (Fig. 8). When this state is reached, recovery of mechanical strength may be observed [81–87]. This is illustrated in Fig. 10 for polypropylene prepared both by injection moulding and by compression moulding.

Moulded surface of polypropylene injection moulded bar after photo-ageing for 12 weeks then tensile tested. The fracture surface is visible at the top. The smooth fracture immediately behind the moulded surface indicates the extent of the severely degraded and embrittled zone (sample from the study reported in Ref. [76]).
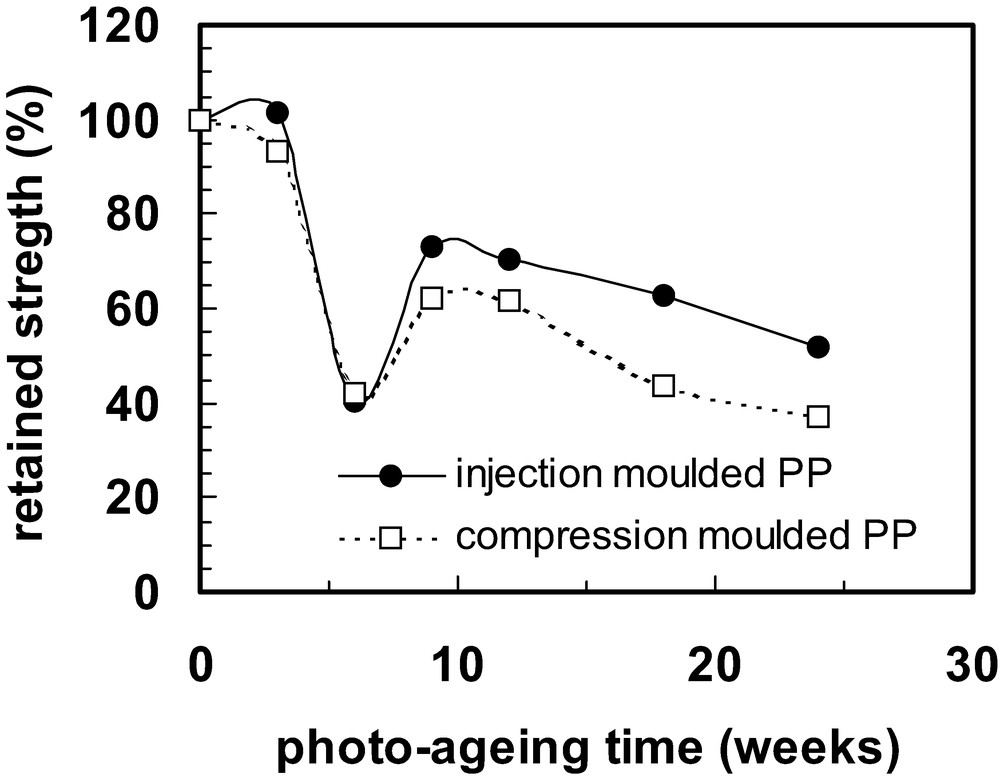
Retained strength versus photo-ageing time for (i) injection moulded polypropylene and (ii) compression moulded polypropylene, showing recovery behaviour. The standard deviations for four repeated tests were often smaller than the data decals except near to the recovery region and are omitted here for the sake of clarity. They can be checked in the original papers [85–87].
The embrittlement discussed above is not the only effect of photo-oxidation. In crystallising polymers, chain scission releases molecular segments that were immobilised by entanglements and allows secondary crystallisation in a manner that can be even more pronounced than that discussed in Section 2. In this case the secondary crystallisation is often referred to as ‘chemi-crystallisation’ [88–90] or sometimes ‘oxidative crystallisation’ [91]. Crystallinity changes caused by photo-ageing a polypropylene copolymer are given in Fig. 11. Measurements on 3 mm thick bars were made by differential scanning calorimetry (DSC) using layers 0.1 mm deep at the exposed surface (denoted ‘0.05’, for the mid-layer plane) and between 0.3–0.4 mm (‘0.35’). Measurements were also made using an X-ray diffractometer (XRD) at the exposed surface (‘0’) and at 0.3 mm (after milling away 0.3 mm from the exposed surface (‘0.3’) [92].
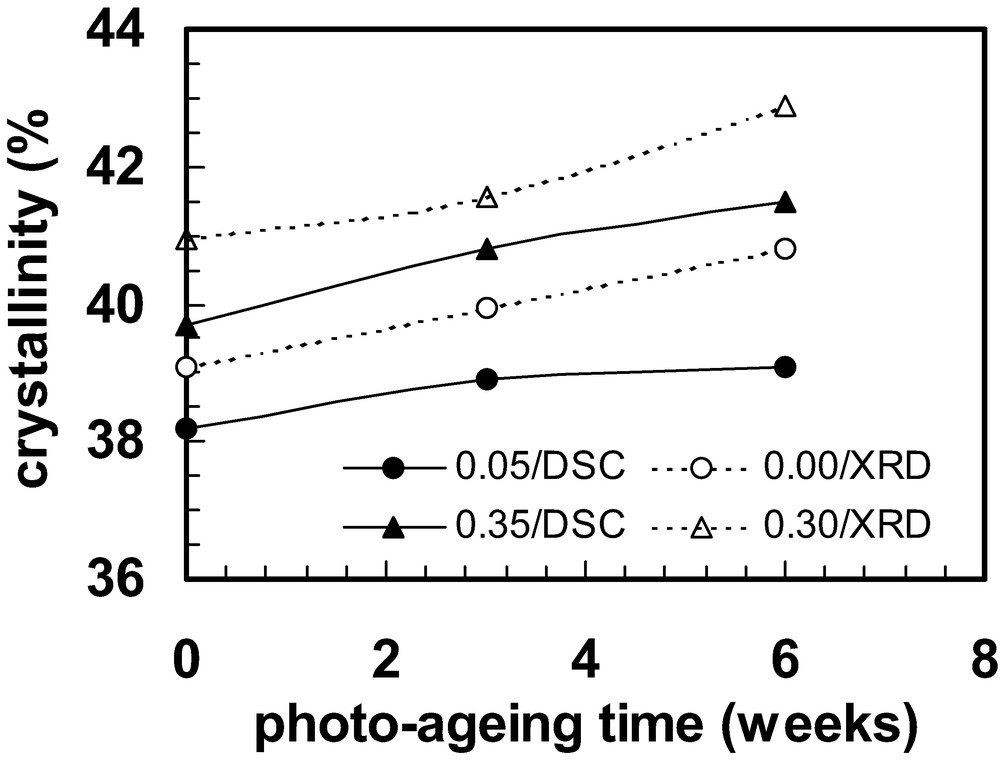
Crystallinity measurements at different depths within a 3-mm-thick bar of a polypropylene copolymer after different photo-ageing times. DSC measurements were made on samples removed by milling 0.1 mm layers at the surface (0–0.1 mm, denoted by layer midpoint ‘0.05’) and between 0.3 and 0.4 mm from the surface (‘0.35’). XRD measurements were made with a diffractometer using the exposed surface (‘0.00’) and the surface 0.3 mm beneath the exposed surface, revealed by milling away 0.3 mm (‘0.30’). These data were obtained in a study by Craig et al. [92].
Chemi-crystallisation often follows a sharply graded profile, with a large increase in crystallinity near the surface, where most of the photo-oxidation occurs, but very little change in the interior where oxygen starvation prevents extensive development of oxidation. When secondary crystallisation occurs, the material density increases because the molecules pack more efficiently in the crystals than in the amorphous phase. Therefore the material near the surface shrinks more than the material in the interior and this differential shrinkage causes residual stresses to develop [93,94]. The surface zone shrinks the most and tensile stresses develop here; this is where the material has embrittled, and both effects combine to weaken the material. Similar developments can be expected in thermal ageing if the temperature elevation is relatively small. At higher temperatures the oxygen diffusion rate is higher and depth-varying changes may not be as pronounced. Also, residual stresses will relax at elevated temperature.
Accelerated densification of photo-degraded amorphous thermoplastic has been observed, analogous to chemi-crystallisation (Fig. 12) [95]. This is believed to be the consequence of chain scission events releasing entanglements and allowing molecular segments to relax more readily to more favoured states, closer to equilibrium [95]. The relative changes in density at different depths will cause the development of residual stresses. The density changes recorded in Fig. 12 are of the correct order to account for the changes in residual stress observed with polystyrene photo-aged under similar conditions [95].
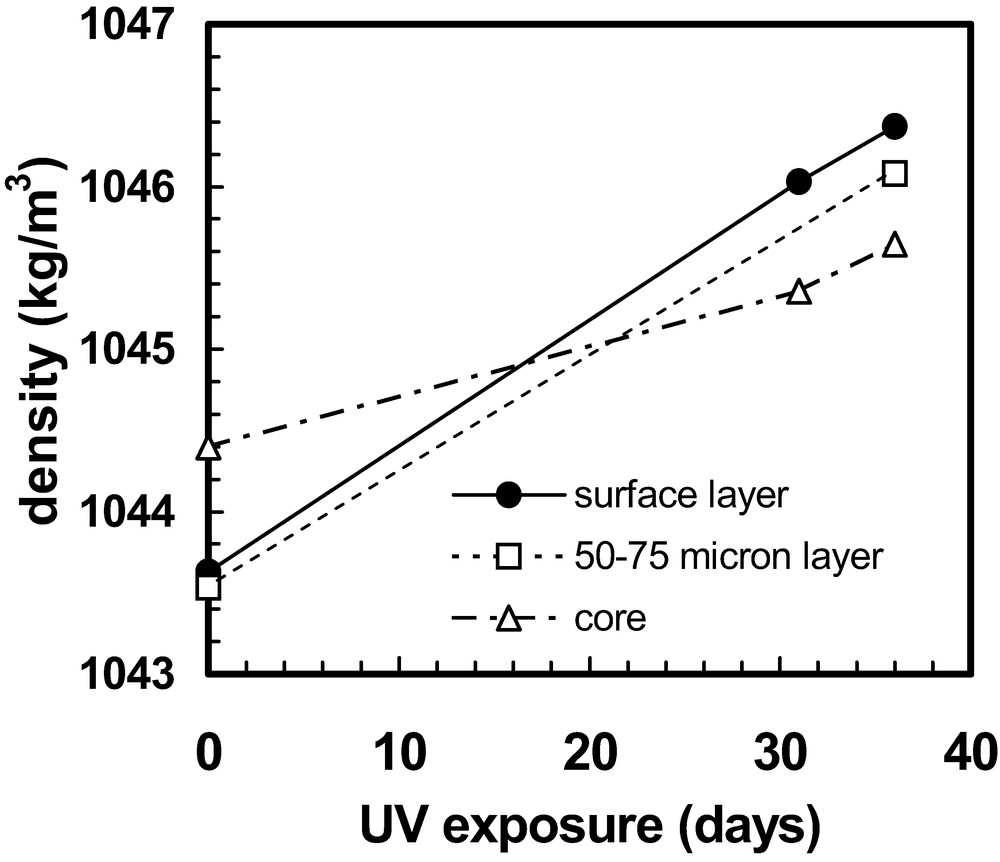
Density measurements for polystyrene samples extracted from different depths within photo-aged bars after different ageing times [95].
Most of the laboratory ageing experiments described above have been conducted with the objective of understanding the weathering degradation of polymers. To further this process it is necessary to conduct natural ageing trials outdoors. Acceleration can be achieved by exposing samples to extremely harsh climates, usually hot and with high UV levels but also involving extremes of other potential hazards such as high humidity and rainfall (e.g., Florida) or very dry (e.g., Arizona). Further acceleration can be achieved using sun-tracking sample holders to maximise the total dose received or mirrors that focus solar radiation onto the samples, so amplifying the intensity. As with artificial weathering, correlation of the results of an extreme climate with behaviour under service conditions may not be straightforward. Caution is always advised in standards and in the most authoritative texts [64,96,97].
Natural weathering studies showing the deterioration of engineering properties (strength, elongation to break, impact toughness etc.) are very common. Less common are attempts to relate the results of artificial weathering in the laboratory with observations made with natural ageing outdoors. The author and co-workers have attempted to do this in two important ways that relate to engineering properties and performance using samples exposed outdoors in Jeddah, Saudi Arabia, and in the laboratory using similar temperatures and UV spectral distributions and intensities. Firstly, failure analyses have been conducted on samples broken in tensile tests, and secondly residual stress analyses have been performed on samples after different weathering or laboratory photo-ageing exposures. Inspection of fracture surfaces by scanning electron microscopy confirmed that similar fracture mechanisms were provoked by natural and artificial photo-ageing. The following events have been observed in a range of thermoplastics after photo-ageing under both types of condition [76–79,84–87,93,94,98–103]:
- ● the formation of a brittle layer;
- ● multiple cracking of the brittle layer during tensile testing;
- ● drawing of ductile material under a multiply fractured brittle surface layer;
- ● development of a dominant surface flaw from which fracture developed over the whole sample section;
- ● occasional failure from flaws originating near to the surface that faced away from the UV source.
The last observation is perhaps counter-intuitive but may be associated with (i) the de-sensitisation of the exposed surface, as described in the discussion of the recovery phenomenon discussed above and illustrated by Fig. 10; or (ii) the development of a deeper flaw at the unexposed face because the lower UV intensity there leads to a lower reaction rate and a less steep degradation profile associated with oxygen-diffusion limited behaviour. Examples where failure was nucleated at the unexposed face have been observed with polypropylene exposed, respectively, in the laboratory and outdoors in Jeddah for similar periods [76,98,103].
Another characteristic shared by samples aged, respectively, in the laboratory and in natural sunlight is the observation that the residual stress distribution inverts on prolonged exposure. In the as-moulded state, tensile stresses are present in the interior of thermoplastics mouldings, balanced by compressive stresses near the surface. On photo-ageing the stresses first reduce in magnitude then eventually reverse, becoming tensile near to the surface and compressive in the interior. The shrinkage that accompanies photo-induced crystallisation and/or accelerated physical ageing occurs primarily near to the exposed surface and accounts for the development of tensile stresses. This has been found with laboratory photo-ageing [93,94] and outdoors weathering [98,99].
Although UV irradiation is generally regarded as the most aggressive element in natural weathering, exposure to water (rainfall, high humidity) can lead to significant changes. The effect of different humidity levels on thermoset curing was discussed in Section 3. Some thermoplastics are much modified by ageing in contact with water, especially at elevated temperatures. Some amorphous polymers such as polycarbonate that are normally very tough can be embrittled by the presence of water absorbed during prolonged exposure [104]. The water forms lacunae that the act as stress concentrators [104]. Some semi-crystalline polymers display secondary crystallisation when exposed to water. Water is absorbed, increasing the free volume and reducing the glass transition temperature, mobilising molecular segments in the amorphous region that then rearrange and crystallise. Nylon 6,6 shows this behaviour [105]. If only one surface is exposed to rainfall, water absorption may be unsymmetrical and this may lead to distortion [106]. The secondary crystallisation leads to volumetric changes that modify the residual stress distribution but additional changes in residual stresses occur because of swelling caused by water absorption and because of relaxation of residual stresses due to the reduced Tg. A further complication with nylon 6,6 is that the exposure to water causes a strong variation in Young’s modulus to develop and this has required the development of special procedures to deal with the analysis of the residual stress distribution [107,108].
In some climates, the temperature is high enough to cause changes in polymers that might normally be associated with thermal ageing. This is especially the case where the polymer contains an additive that absorbs solar radiation over a wide spectral range, heating the material to temperatures well above the air temperature. Temperatures well in excess of 60 °C are possible. The surface facing the sun is most affected and this may lead to further changes associated with a temperature gradient [44,104]. Differential thermal expansion and relaxation of residual stresses in the hottest parts of the artefact can lead to significant changes in the residual stress distribution, as verified by laboratory experiments [44,104].
6 Discussion and conclusions
Physical ageing of a semi-crystalline polymer involves molecular relaxations that can provide stress relief and can give rise to volumetric relaxation. Processed polymers are usually in a non-equilibrium state after rapid cooling at the end of the moulding or shaping operation and the density increases towards the equilibrium value on ageing. Physical ageing can be accelerated by elevating the temperature (but this also changes the equilibrium state) or by changing the chemical state of the material. During both thermal oxidation and photo-oxidation, chain scission occurs and this releases molecular chain segments from entanglements, facilitating conformational rearrangements. This allows the chain segments to take up lower energy states, characteristic of the equilibrium state for the material, so accelerating physical ageing. The material is changed irreversibly by the chain scission events; the physical ageing occurs alongside the chemical changes and it is not always obvious which has the controlling influence over the material properties.
Differential changes in volume result in the formation of residual stresses. This is how they form during processing, as the result of temperature gradients that exist during moulding operations. Residual stresses often have a strong influence over engineering performance. They may lead to warping and other dimensional defects, may influence the strength and toughness of the material, and may lead to environmental stress cracking in the presence of an aggressive chemical environment. Changes in residual stresses often occur after the moulding operation is complete and this can be regarded as a type of ageing process. The engineering properties change in response to the changes in residual stress as well as to any other ageing process that is operating concurrently. A most common example of this kind of change in residual stress occurs in photo-ageing where the molecular degradation depth profile falls steeply from the exposed surface. Accelerated volume relaxation near the surface results in shrinkage and gives rise to tensile residual stresses there. Therefore tensile stresses develop in the region that is embrittled by the molecular degradation and increases the likelihood of fracture.
With semi-crystalline polymers, secondary crystallisation may occur in addition to physical ageing in the amorphous phase. Chain scission caused by photo-ageing accelerates secondary crystallisation (‘chemi-crystallisation’) and the effect varies strongly with depth from the exposed surface. The large volumetric reduction that occurs on crystallisation, coupled with this depth profile, causes tensile residual stresses to form in the regions that display the greatest secondary crystallisation. As with amorphous polymers, the tensile stress near the surface increases the chance that fracture will occur. Secondary crystallisation can also be promoted by elevating the temperature or exposure to water, both of which mobilise molecule segments in the amorphous phase. All of these effects may be present when a polymer is aged in a hot climate.
Residual stresses in thermoplastics may be modified both by physical ageing and chemical ageing. They may influence other mechanical properties that are considered to follow physical ageing. The formation of curing stresses in thermosets is a familiar phenomenon, especially in coatings. Although the shrinkage that normally accompanies the crosslinking events that are at the heart of thermoset curing is a major contributor to the residual stress that is observed, physical ageing during post-cure can also have a significant influence. Thermoset coatings are often sensitive to the humidity in which they are aged and this can have a large influence over the sense and the magnitude of the stresses that develop.
Ageing of polymers is a highly complex process. Many studies have been conducted on aspects such as physical ageing in the amorphous phase and photo-induced secondary crystallisation that influence engineering performance using carefully controlled laboratory conditions but it is still difficult to predict the ageing behaviour under less well characterised conditions such as natural weathering because of the presence of multiple ageing processes and their interaction.