

1 Introduction
The magnetic properties of transition metal coordination compounds have attracted more and more the attention of chemists and physicists in the last two decades. The classical picture of long-range ordered magnetism in metals and oxides has been extended to the molecular level of oligo- and polynuclear transition metal coordination compounds exhibiting ferro- and antiferromagnetic interactions in 1D, 2D and 3D molecular lattices. Terms like molecular magnetism and molecule-based magnets have entered the literature [1,2], and the new facet of magnetic behaviour of materials has inspired chemists, physicists and theoreticians with increasing enthusiasm. International research programs supported e.g. by the European Community (TMR networks, Centre of Excellence) and the German National Science Foundation (Priority Program) have been initiated to bring researchers from all relevant disciplines together for extensive collaboration in a fascinating new field of solid-state research.
It has soon been realized that molecular magnetism should not only be confined to coordination compounds exhibiting magnetic exchange interactions between the electron spins of metal ions (or unpaired electrons in organic compounds) via suitable molecular orbital pathways set up by properly chosen bridging atoms or molecules. Dynamic electronic structure phenomena such as thermally induced spin transition (spin crossover) [3] and valence tautomerism [4] are known to show drastic changes in magnetic properties and have thus found a place in the category of molecular magnetism.
Spin crossover has turned out to be a particularly appealing phenomenon as it offers the possibility of bistability, i.e. that complex molecules may exist in two different electronic states at one and the same temperature. The reversible change between low-spin (LS) and high-spin (HS) states driven by variation of temperature and/or pressure or also by irradiation, mainly observed in pseudo-octahedral iron(II) coordination complexes, is up to now one of the best examples of molecular bistability [3,5,6]. At the molecular scale, spin crossover (SCO) in Fe(II) compounds corresponds to an intra-ionic transfer of two electrons between the t2g and eg orbitals, (t2g)4(eg)2 ↔ (t2g)6(eg)0, accompanied by a change of spin state, S = 2 ↔ S = 0. The 5T2g state corresponding to (t2g)4(eg)2 is the ground state only up to a critical value of the ligand-field strength equal to the spin pairing energy. Above this value, the 1A1g LS state corresponding to (t2g)6(eg)0 becomes lower in energy than the HS state [3]. In the HS state, the antibonding eg orbitals are doubly occupied, and consequently the Fe-donor atom bonds are longer than in the LS state by ca. 0.20 Å. This increase of the molecule size when passing from the LS to the HS state plays a crucial role in the cooperative mechanism of SCO giving eventually rise to abrupt transitions and hysteresis. Both features are important for eventual practical applications (sensors, optical devices). For instance, the magnetic and optical properties accompanying the spin transition may switch sharply in a very small range of temperature and/or pressure for cooperative transitions [6,7]. Due to this particularity the SCO phenomenon has been considered as one of the most interesting examples of molecular switching. The condition to accomplish in order to observe the phenomenon of spin transition is that the zero-point energy between the two states, ΔEHL0 = EHS0 − ELS0, has to be of the order of thermal energy, kBT. In this case, all molecules will be in the LS state at very low temperatures or higher pressures whereas at elevated temperatures or lower pressures, an entropy-driven almost quantitative population of the HS state will occur. In general, spin transition is a well-established phenomenon and many examples of SCO complexes exhibiting abrupt spin transitions at room temperature, with broad thermal hysteresis as well as an associated thermochromic effect (necessary conditions for display devices) have been reported up now [3].
The spin crossover phenomenon is very sensitive to pressure, as is expected from the fact that the molecular volume is larger in the HS state than in the LS state, the difference being of the order of 3–5%. A schematic representation of the pressure influence on the LS and HS potential wells of Fe(II) is shown in Fig. 1. Application of pressure increases the relative vertical displacement of the potential wells; the additional relative horizontal displacement due to a slight decrease in bond length accompanying an increase of pressure has been neglected. Increasing the pressure favours the LS state, thus shifting the spin transition to higher temperatures, because pressure increases the zero-point energy difference ΔEHL0 by the work term PΔVHL0 and decreases the activation energy ΔWHL0, favouring the LS state.
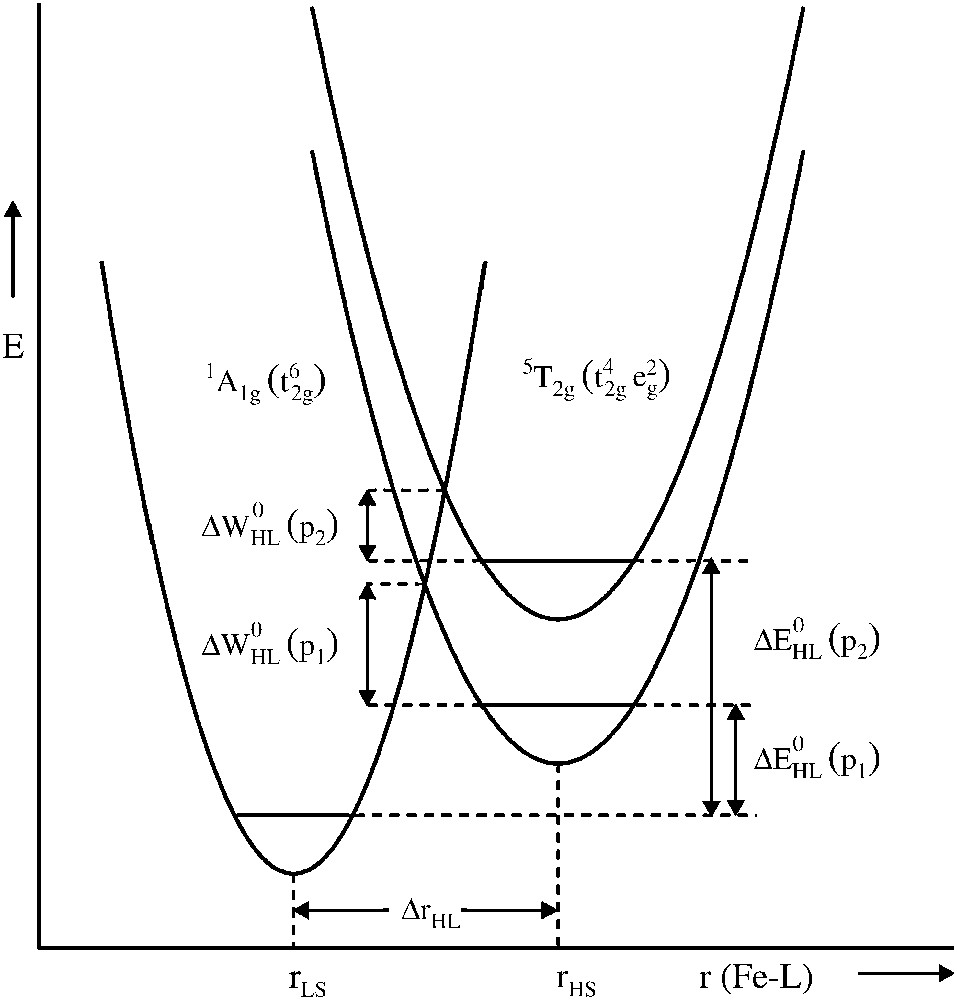
Schematic representation of the influence of pressure (P2 > P1) on the LS and HS potential wells of a Fe(II) spin crossover compound.
Barochromic properties of SCO materials have been recognized from the outset of SCO research, and yet pressure effect studies remained poorly explored mainly due to the experimental difficulties. There was a revival of interest in the effect of pressure on SCO complexes during the last decade involving studies using either hydrostatic cells adopted to magnetic susceptibility [7], Mössbauer [8], optical absorption [9] and reflectivity detection methods [10] or diamond anvil cells (DACs) in conjunction with methods of detection such as IR [11], EXAFS [12] or X-ray diffraction [13]. Both cells were also used with Raman spectroscopy [14]. The DAC technique is not well-adapted for studying the spin transition process because pressure loops are difficult to record in the most interesting pressure range (<1.0 GPa) and problems of non-hydrostaticity are likely to arise from pressure gradients in the cell. Hydrostatic pressure cells adapted for the other techniques have been used to study the effect of pressure on spin transition temperature, thermal and pressure-induced hysteresis cycles, the crystal structure, and relaxation processes.
The major part of the present article is devoted to the studies on the effect of pressure on SCO materials of different dimensionalities carried out in our laboratory using hydrostatic cells developed for magnetic susceptibility and Mössbauer measurements, illustrating how pressure effect studies can help in understanding the nature of cooperative interactions. Results from pressure effect studies on valence tautomerism in a cobalt catecholate complex and in a Prussian blue type system will also be presented. The article concludes with preliminary results from a pressure effect study of a molecule-based magnet.
2 Experimental section
2.1 Materials
The compounds under study have been synthesised and characterised according to procedures published in the original reports referred to in the following special sections.
2.2 Magnetic susceptibility measurements under hydrostatic pressure
The variable-temperature magnetic susceptibility measurements were performed using a PAR 151 Foner-type magnetometer equipped with a cryostat operating at 1 T in the temperature range 2–300 K and a Quantum Design MPMS2 SQUID susceptometer in a magnetic field of 1 T and a temperature range of 1.8–300 K. The hydrostatic pressure cell made of hardened beryllium bronze with silicon oil as the pressure transmitting medium operates in the pressure range of 1 bar < P < 1.3 GPa and has been described elsewhere [15]. Hydrostaticity was established during all our pressure effect studies. Cylindrically shaped powder sample holder dimensions are 1 mm in diameter and 5–7 mm in length. The pressure was measured using the pressure dependence of the superconducting transition temperature of a built-in pressure sensor made of high purity tin. Experimental data were corrected for diamagnetism using Pascal's constants.
2.3 Mössbauer spectroscopy under hydrostatic pressure
57Fe Mössbauer spectra were recorded using a conventional constant-acceleration spectrometer in conjunction with a helium bath cryostat for temperature dependent measurements down to liquid helium temperatures. Powder samples were measured in a Mössbauer pressure cell made of hardened beryllium bronze equipped with windows made of B4C and with silicon oil as the pressure transmitting medium. The construction enables hydrostatic pressure measurements to be carried out up to 1.5 GPa in the temperature range 2–350 K. The Mössbauer pressure cell was calibrated using FeF3 in accordance with published results [16]. The Recoil 1.02 Mössbauer Analysis Software was used to fit the experimental spectra [17]. Isomer shift values are quoted relative to α-Fe at 293 K.
3 Effect of pressure in Fe(II) spin crossover complexes
3.1 Mononuclear Fe(II) spin crossover complexes
The complexes of formula [Fe(L)2(NCS)2], where L stands for a bidentate α-diimine ligand, represent one of the most extensively studied class of SCO compounds [18–25]. They all have the FeN6 inner coordination sphere, by far the most preferred chromophore in iron(II) spin crossover complexes, in common, and display a wide range of SCO behaviour from gradual to abrupt transitions, even with broad thermal hysteresis loops. We shall illustrate the effect of pressure on the following examples of different SCO behaviours: an abrupt spin transition, a gradual one and a pressure-induced spin transition in a paramagnetic compound. We have chosen the SCO systems [Fe(phen)2(NCS)2] (phen = 1,10-phenanthroline) (1) [26], [Fe(PM-Aza)2(NCS)2] (PM-Aza = (N-(2′-pyridylmethylen)-4-azophenylaniline) (2) [7a] and [Fe(abpt)2(NCS)2] (abpt = 4-amino-3,5-bis(pyridin-2-yl)-1,2,4-triazole (3) [27] (Fig. 2). The two former compounds reveal an abrupt and a gradual spin transition, respectively, at ambient pressure, whereas the last one is in the HS state over the whole range of temperatures under study.

Molecular structures of the compounds [Fe(phen)2(NCS)2] (1) (left), [Fe(PM-Aza)2(NCS)2] (2) (centre), [Fe(abpt)2(NCS)2] (3) (right).
3.1.1 [Fe(phen)2(NCS)2] polymorph B (1)
The χMT vs. T plots of 1 at different pressures, χM being the molar magnetic susceptibility and T the temperature, are shown in Fig. 3. At ambient pressure the transition curve is extremely steep with T1/2 = 177 K. The presence of temperature hysteresis width ∼2 K and the value of the residual HS fraction (≅17%) are in agreement with published data [28]. As the pressure is increased, the transition curve moves upwards with an average rate of 220 K/GPa. At P = 0.17 GPa and higher pressures the hysteresis disappears and the transition curves become gradual. At P = 0.57 GPa the sample is mostly in the LS state; however, the residual HS fraction below the transition remains practically constant under pressure. This observation is in agreement with the results found in Ref. [18], where conservation of the space group during the SCO transition under pressure up to 1.0 GPa has been reported. A progressive diminishing of the pressure efficiency on the transition temperature (41.0 K/GPa at 0.17 GPa, 18.0 K/GPa at 0.34 GPa, 15.0 K/GPa at 0.57 GPa) calls for a sterical hindrance which plays a decisive role in preventing the complete HS → LS transformation in 1.
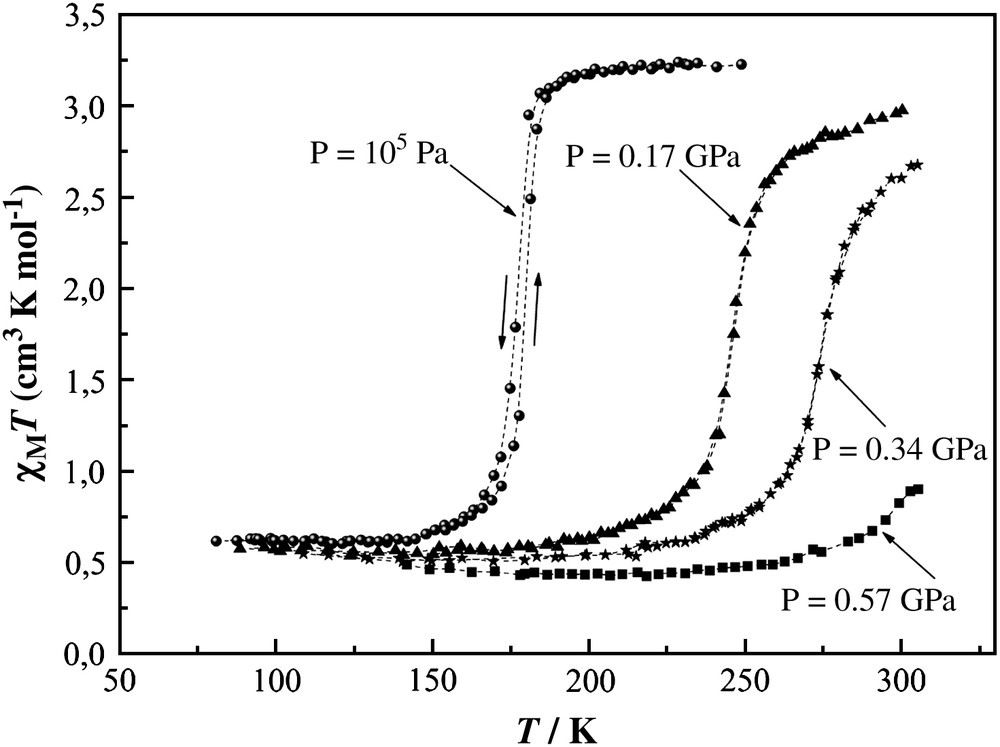
χMT vs. T plots of [Fe(phen)2(NCS)2] at different pressures up to 0.57 GPa.
3.1.2 [Fe(PM-Aza)2(NCS)2] (2)
The HS molar fraction γHS of the mononuclear compound 2 as a function of temperature decreases at ambient pressure continuously down to ca. 5% corresponding to a gradual and nearly complete spin transition. The conversion temperatures are T1/2↓ = 186 K and T1/2↑ = 192 K in the cooling and warming modes, respectively (Fig. 4). An increase of applied pressure shifts the transition temperature upwards and decreases the slope of the transition curve; at 0.25 GPa T1/2 is around 210 K, and at 1.08 GPa T1/2 is much above room temperature. Indeed, an increase of hydrostatic pressure stabilizes the LS state as expected due to the smaller volume of the LS as compared to the HS state. It should be noted as the linear behaviour of the pressure dependence of T1/2 for the spin conversion of 2. The slope of the T1/2 vs. P straight line, ∂T1/2/∂P = 16 K/GPa, is very close to that observed for the mononuclear compounds [Fe(2-pic)3]Cl2·EtOH (2-pic = 2-picolylamine) [29] and [Fe(abpt)2(NCS)2] [27] where ∂T1/2/∂P = 15 and 17.6 K/GPa, respectively. The type of pressure dependence as in this case appears to be favourable for pressure sensing in remote positions. To do so, a calibration graph is derived from the data in Fig. 4 by taking the γHS data at a given temperature, e.g. 295 K, and plotting them as a function of applied pressure. An optical light cable equipped with a suitable SCO material in the sensor head is transferred to the sensing position and indicates a change of colour from white (HS state) to red (LS state) resulting from the pressure-induced spin transition from HS to LS in the SCO material.

γHS vs. T plot of [Fe(PM-Aza)2(NCS)2] at different pressures up to 1.08 GPa.
3.1.3 [Fe(abpt)2(NCS)2] polymorph B (3)
Fig. 5 shows the temperature dependence of the χMT product for 3 at different pressures. At room temperature and at atmospheric pressure χMT is equal to 3.68 cm3 K mol−1 which is in the range of the values expected for an iron(II) compound in the HS state. As the temperature is lowered, χMT practically remains constant, the decrease of χMT at temperatures below 25 K is due to zero-field splitting of the HS iron(II) ions. This behaviour persists as pressure is increased up to 0.44 GPa, where an incomplete thermal SCO appears around T1/2 = 65 K. This T1/2 value is one of the lowest transition temperatures observed for an iron(II) SCO compound. Probably the low temperature slows down the kinetics of the HS ↔ LS equilibrium considerably and causes the incomplete spin transition observed for 3 at 0.44 GPa. A nearly complete and relatively sharp spin transition takes place at T1/2 = 106, 152 and 179 K, as the pressure attains 0.56, 0.86 and 1.05 GPa, serially. In the slow cooling and heating modes with the rate 0.1 K min−1 providing thermodynamic equilibrium conditions, the transitions are accompanied by a 2 K wide thermal hysteresis at all pressures studied. Noteworthy is the linear behaviour of the pressure dependence of T1/2 for the spin conversion of 3. The slope of the T1/2 vs. P straight line, ∂T1/2/∂P = 17.6 K/GPa, is very close to that observed for the mononuclear compound [Fe(2-pic)3]Cl2·EtOH (2-pic = 2-picolylamine) [29] where ∂T1/2/∂P = 17.6 K/GPa.

χMT vs. T plots of [Fe(abpt)2(NCS)2] at different pressures up to 1.05 GPa.
The above-described pressure influence on abrupt and gradual transitions can be qualitatively interpreted on the basis of a phenomenological theory of phase transitions in SCO systems [30]. It predicts that hydrostatic pressure transforms a discontinuous spin transition accompanied by hysteresis to a transition of continuous type. Indeed, we observed this behaviour in compound 1. In the frame of this theory the slope of the spin transition curve decreases under pressure and T1/2 shifts upwards. The thermal dependence of γHS under pressure for compound 2 demonstrates these features. The pressure studies on compound 3 demonstrate the principal possibility to “fine tune” the crystal field strength and thereby induce a thermal spin transition in a paramagnetic compound in a controlled way.
It is worth mentioning that not only the application of external pressure acting on an SCO compound influences the spin transition properties more or less dramatically. Embedding the SCO ions in the crystal lattice of a host matrix whose cationic complex volumes differ from those of the SCO cations creates “chemical pressure”, also called image pressure. Chemical pressure can be positive and negative, depending on whether the complex volumes of the host matrix are smaller or larger than those of the SCO cations. Extensive studies on the metal dilution effect have been reported and the results have served as a basis for the development of the model of elastic interactions and lattice expansion [3], which describes satisfactorily the metal dilution effect on the strength of the cooperative interactions in SCO systems. The effect of chemical pressure arising from embedding SCO complex ions into host lattices will not be pursued further in this article.
3.2 Dinuclear Fe(II) spin crossover complexes
The synthesis of dinuclear Fe(II) complexes exhibiting thermal spin transition reported by Real et al. [31] was not only the first step towards polynuclear SCO complexes as an alternative strategy to explore cooperativity [32], but also opened the path to a new class of SCO systems combining different electronic properties like magnetic exchange and spin transition in the same system. First efforts along this line began with the class of 2,2′-bipyrimidine (bpym)-bridged iron(II) dinuclear compounds.
The series of compounds {[Fe(L)(NCX)2]2(bpym)}, where L is bpym (2,2′-bipyrimidine) or bt (2,2′-bi-thiazoline) and X is S or Se, comprises four complexes, two of which, (bpym, S) and (bt, S), have been characterised by X-ray single crystal diffraction. The centrosymmetric dinuclear units {[Fe(L)(NCS)2]2(bpym)}, where L = bpym [31] or bt [33], are shown in Fig. 6. Each iron(II) atom is surrounded by two NCS− anions in cis positions, two nitrogen atoms of the bridging bpym ligand and the remaining positions are occupied by the peripheral bpym or bt ligands. The [FeN6] chromophore is rather distorted with Fe–N bond distances characteristic of an iron(II) ion in the HS state.
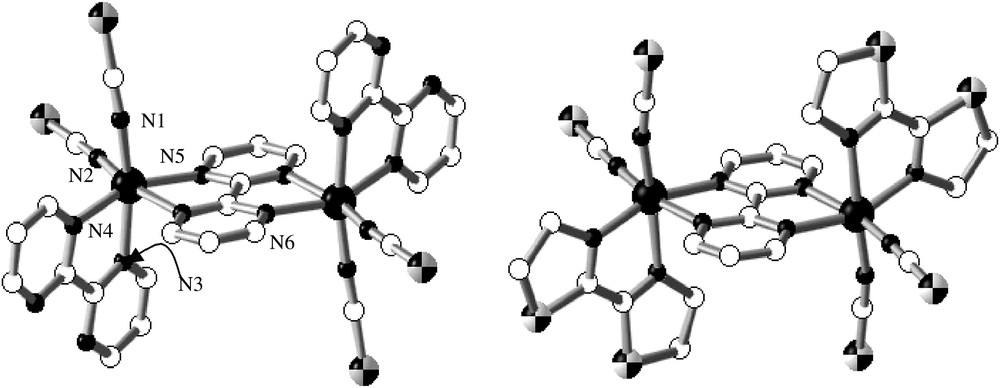
Molecular structure of {[Fe(bpym)(NCS)2]2(bpym)]} together with the corresponding atom numbering (a) and of {[Fe(bt)(NCS)2]2(bpym)]} (b).
No thermal spin transition is observed for the iron(II) complex denoted as (bpym, S) in the whole range of temperature (see next paragraph). At first sight this is rather unexpected as the iron(II) environment in the dinuclear compound is close to that in [Fe(bipy)2(NCS)2] [34]. However, the average Fe–N bond distances are noticeably greater for (bpym, S) which tends to favour the HS state. In contrast, the iron (II) complex denoted as (bt, S), which shows shorter Fe–N bond distances than (bpym, S), undergoes a complete spin transition [35]. The remaining members of this family, (bpym, Se) and (bt, Se), also undergo spin transition, but their crystal structures have not yet been solved. However, structural information on these compounds has been obtained using X-ray absorption techniques (EXAFS) at 300 and 77 K. The EXAFS data afforded a rather satisfactory description of the iron(II) coordination core both in the HS and in the LS states of these compounds [36].
The magnetic behaviour of this series at ambient pressure is depicted in Fig. 7. As stated before, (bpym, S) does not display thermally induced spin conversion, but exhibits intramolecular antiferromagnetic coupling between the two iron(II) ions through the bpym bridge (J = −4.1 cm−1, g = 2.18). When thiocyanate is replaced by selenocyanate the resulting (bpym, Se) derivative shows an abrupt spin transition in the 125–115 K temperature region with a small hysteresis loop of 2.5 K width (Fig. 7). Only 50% of the iron(II) ions undergo spin transition. The decrease of the χMT values at lower temperatures is due to zero-field splitting of the S = 2 state (see below). The magnetic properties of (bt, S) and (bt, Se) are similar to one another and show a complete spin transition with the remarkable feature that it takes place in two steps centred at 197 and 163 K for (bt, S) and at 265 and 223 K for (bt, Se). In both cases, the plateau corresponds approximately to 50% spin conversion.
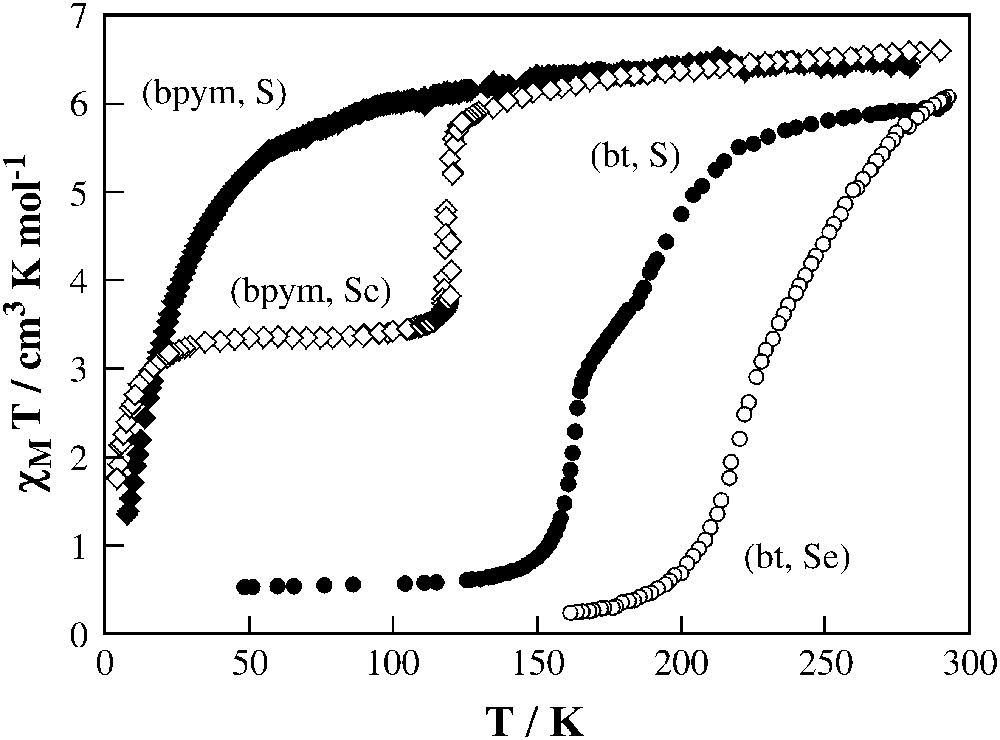
Temperature dependence of χMT for {[Fe(L)(NCX)2]2(bpym)]} (L = bpym and X = S (bpym, S) or Se (bpym, Se) and L = bt and X = S (bt, S) or Se (bt, Se)).
These observations, also confirmed by Mössbauer spectroscopy and calorimetric measurements, were interpreted in terms of a microscopic two-step transition between the three possible spin pairs of each individual dinuclear molecule [35]:
The stabilisation of the [HS–LS] mixed-spin pair results from a synergistic effect between intramolecular and cooperative intermolecular interactions (see below).
The pressure dependence of the thermal variation of χMT has proved to be a useful diagnostic probe to show that the formation of [HS–LS] spin pairs is not fortuitous but that they are the preferentially formed species in the dinuclear-type complexes [7b]. It is shown next that application of external hydrostatic pressure can help to unravel features of this whole class of compounds, which usually can be revealed by the variation of chemical composition.
It has already been shown that increase in hydrostatic pressure favours the LS state in mononuclear complexes, and there is no reason expect for a different behaviour for dinuclear systems. Two members of the {[Fe(L)(NCX)2]2bpym} family are particularly suitable candidates in this regard: (bpym, S) and (bpym, Se). Fig. 8 displays the thermal dependence of χMT at different pressures. At ambient pressure, and over the whole temperature range, (bpym, S) contains only the antiferromagnetically coupled [HS–HS] pairs (Fig. 8a). Occurrence of antiferromagnetic coupling and SCO in (bpym, S) clearly follows from magnetic susceptibility measurements at P = 0.63 GPa. When the pressure is increased to 0.63 GPa a partial conversion from 100% [HS–HS] to 55% [HS–LS] species takes place. The incompleteness of spin conversion is due to the fact that at low temperatures the spin conversion is so slow that the HS state becomes metastable. Thus antiferromagnetically coupled [HS–HS] pairs and [HS–LS] uncoupled pairs become co-existent in (bpym, S) at 0.63 GPa, as reflected in the thermal dependence of χMT. Finally, for P = 0.89 GPa the total conversion to [HS–LS] pairs is accomplished. It is worth noting that, at this pressure, (bpym, S) undergoes a similar [HS–HS] ↔ [HS–LS] spin transition at T1/2 ≈ 150 K as in (bpym, Se) at ambient pressure. The effect of pressure on the thermal dependence of the spin state of (bpym, Se) seems to be due to a weakening of the cooperativity, as can be concluded from the more gradual χMT function as compared to that under ambient pressure, and a shift of T1/2 towards higher temperatures for pressures up to 0.45 GPa (Fig. 8b). For even higher pressures, a second transition appears in addition to the former, due to the onset of thermal spin transition in the second metal centre. Between 0.72 and 1.03 GPa a two-step spin transition function is observed.
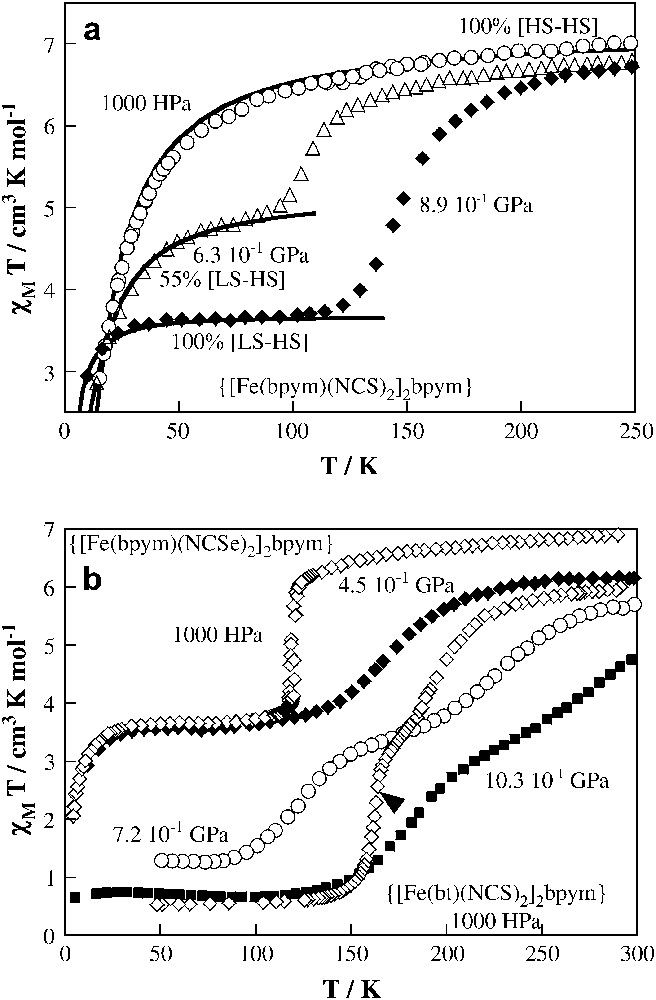
Temperature dependence of χMT for {[Fe(bpym)(NCS)2]2(bpym)} at different pressures (a). The solid lines, together with estimated concentrations of [HS–LS] and [HS–HS] species correspond to calculations using the appropriate Hamiltonian. Temperature dependence of χMT for {[Fe(bpym)(NCSe)2]2(bpym)} at different pressures (b). The magnetic behaviour of {[Fe(bt)(NCS)2]2(bpym)} at ambient pressure has also (1000 HPa) been included for comparison.
As mentioned above, a special feature of the dinuclear compounds is the appearance of a plateau in the spin transition curve. The results of the pressure experiments suggest that the plateau stems from successive spin transition in the two metal centres, leading first to the formation of relatively stable [HS–LS] pairs and then, above a critical pressure, to the formation of [LS–LS] pairs on further lowering of the temperature. The pressure-induced low-temperature state of (bpym, S) consisting almost entirely of the [HS–LS] units is stable at least up to 1.1 GPa. For (bpym, Se), a pressure of 0.45 GPa shifts T1/2 by ca. 50 K upwards without increasing the amount of the LS fraction. Only at higher pressures does the second step appear for this derivative. These experimental data underline the role of intermolecular interactions, particularly short range competing with omnipresent long-range interactions, in the stabilisation of the hypothetical “checkerboard-like” structure consisting of [HS–LS] units as proposed by Spiering et al. [37].
3.3 1D, 2D and 3D Fe(II) spin crossover coordination polymers
The number of polymeric iron(II) SCO compounds reported up to now is rather limited. Most of them incorporate multidentate N-donor heterocyclic bridging ligands such as 1,2,4-triazole, 1-R-tetrazole, polypyridine-like derivatives as well as tetra- or di-cyanometallate complex ligands [32,38]. These compounds generally exhibit abrupt spin transitions with hysteresis effects whose width strongly depends on the nature of the molecular bridge between iron(II) sites. In this section, we review the behaviour under pressure of the spin transition of a selection of 1D, 2D and 3D iron(II) coordination polymers.
3.3.1 1D chain compounds
4R-1,2,4-Triazole based polymeric chain compounds of iron(II) belong to the most investigated SCO compounds, presumably due to their potential for practical applications (memory devices, displays, sensors) [6]. [Fe(4-R-1,2,4-triazole)3](anion)2·nH2O are made up of linear chains in which the adjacent iron(II) ions are linked by three N1,N2-1,2,4-triazole ligands. The non-coordinated species such as counteranions and water molecules are localised between the chains. In these polymeric compounds, the molecular bridge is sufficiently rigid to allow an efficient transmission of cooperative effects. Consequently, abrupt spin transitions with broad thermal hysteresis loops around room temperature along with remarkable colour change from deep purple (LS) to white (HS) have been observed [6]. Several approaches aiming to tune the SCO behaviour in the room temperature regime have been followed including the use of external pressure [6,7f,39,40].
Fig. 9 shows the temperature dependence of the HS molar fraction for [Fe(hyptrz)3](4-chlorophenylsulfonate)2·H2O (hyptrz = 4-(3′-hydroxypropyl)-1,2,4-triazole) at different pressures up to nearly 0.6 GPa [7f]. At 105 Pa, a very steep and complete spin transition is observed with a hysteresis loop of width ∼5 K (T1/2↓ = 178 K and T1/2↑ = 183 K). As the pressure increases, the spin transition curves are shifted upwards to room temperature. The profiles of the curves remain essentially unchanged with the steepness retained at all pressures. The spin transition is observed at 260 K under 0.41 GPa, at 286 K under 0.5 GPa, at 301 K under 0.53 GPa, and at 324 K under 0.59 GPa. Interestingly, the hysteresis width reveals a non-monotonic character under pressure. It first diminishes and is no longer observed at 0.41 GPa before reappearing at a constant value of ∼5 K above 0.5 GPa. On release of the pressure, the same magnetic behaviour as observed at 105 Pa was obtained. Fig. 9 also shows the pressure dependence of the LS fraction, γLS, of [Fe(hyptrz)3](4-chlorophenylsulfonate)2·H2O. A very steep HS → LS transition is observed at room temperature around ∼0.6 GPa accompanied by a colour change from white to deep purple. This property is suitable for application such as pressure sensor or display [40].

Top: γHS vs. T plot for [Fe(hyptrz)3](4-chlorophenylsulfonate)2·H2O at different pressures (●, P = 105 Pa; ▪, P = 0.41 GPa; ▴, P = 0.5 GPa; ♦, P = 0.53 GPa; ▵, P = 0.59 GPa; ○, P = 105 Pa after releasing the pressure). Bottom: γLS vs. P plot for [Fe(hyptrz)3](4-chlorophenylsulfonate)2·H2O at 290 K.
This magnetic behaviour under pressure contrasts the one observed for mononuclear SCO compounds with a systematic flattening of the spin transition curves together with a variation in the hysteresis width with increasing pressure [7a,41]. This lends support to the assertion that cooperative interactions are confined within the Fe(II) triazole chain for this compound. Thus a change in the external pressure has an effect on the SCO behaviour comparable to a change in internal electrostatic pressure due to anion–cation interactions. Both lead to considerable shifts in transition temperatures without significant influence on the hysteresis width [7f]. This behaviour under pressure appears to be a general trend for 1D polymeric chain compounds with 4-R-1,2,4-triazole as ligands. Indeed, a similar shift of the hysteresis loop upwards to room temperature was observed for the polymeric chain compound [Fe(hyetrz)3](3-nitrophenylsulfonate)2 (hyetrz = 4-2′-hydroxyethyl-1,2,4-triazole) [42]. Several theoretical models have been developed to predict such SCO behaviour of 1D chain compounds under pressure [43–45].
The magnetic properties of the HS iron(II) chain compound [Fe(bpym)(NCS)2] have also been investigated under pressure. As a result, a spin transition was induced under ∼1.2 GPa involving about 50% of iron(II) ions as found for the dinuclear compound [Fe(bpm)(NCS)2]2bpym [7b].
3.3.2 2D and 3D coordination polymers
[Fe(btr)2(NCS)2]·H2O (btr = 4,4′-bis-1,2,4-triazole) is a 2D polymeric SCO compound [46] which is considered as a model material in SCO research. The crystal structure obtained at room temperature reveals that each iron ion is bridged by one N1,N1′ coordinating btr ligand defining an infinite stack of layered grids. Two thiocyanato anions in apical positions complete the coordination sphere of iron(II) (Fig. 10). Non-coordinated water molecules are linked by hydrogen bonding to the peripheral nitrogen atoms of the triazole. The layers are connected by means of van der Waals forces and weak hydrogen bond bridges involving the water molecules [46].
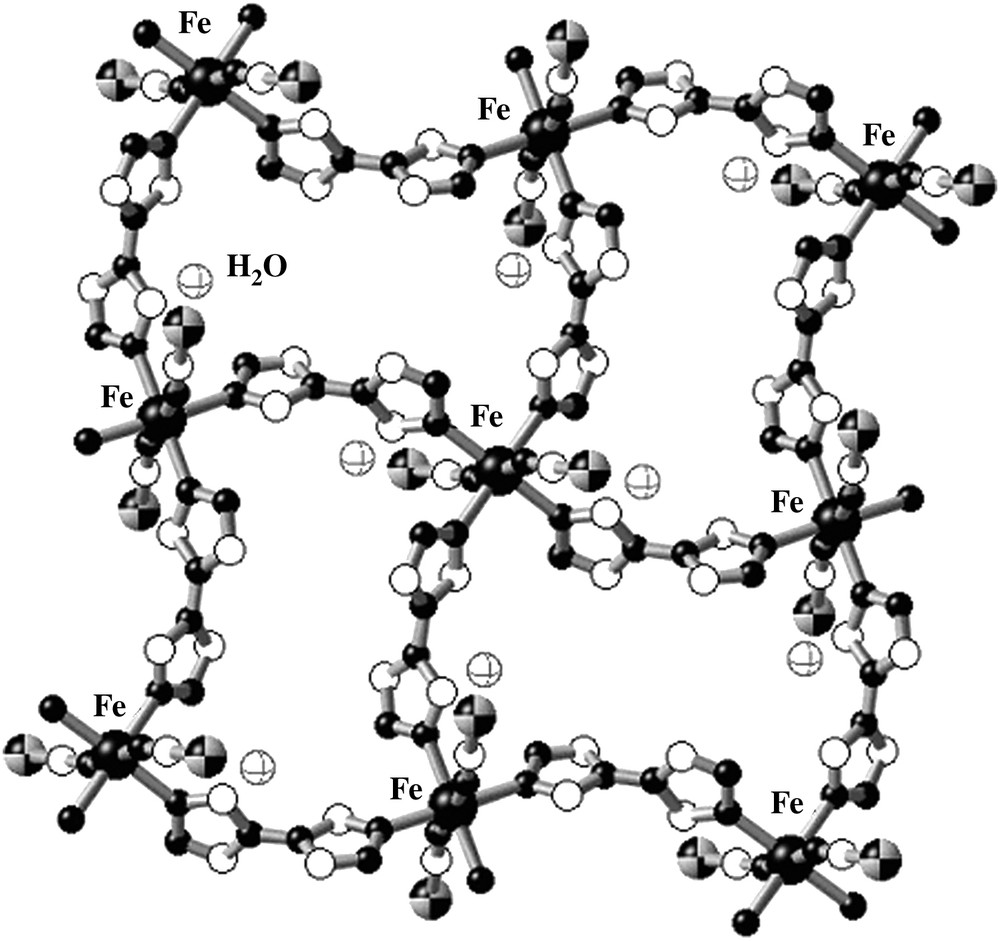
Representative fragment of the layered structure of [Fe(btr)2(NCS)2]·H2O.
[Fe(btr)2(NCS)2]·H2O undergoes a complete spin transition centred at ∼133 K with a hysteresis loop of width 23 K under ambient pressure with T1/2↓ = 121 K and T1/2↑ = 144 K (Fig. 11). At 0.08 GPa, the hysteresis loop broadens and becomes asymmetric. Interestingly, T1/2↓ is not modified, whereas T1/2↑ is slightly shifted to higher temperature. At 0.3 GPa the spin transition curve moves upwards and flattens, and the hysteresis width decreases. Also a noticeable fraction of iron(II) ions remaining in the HS state on the whole temperature range (∼8%) is detected. At 0.67 GPa, the hysteresis loop is now shifted to around 215 K and its width decreases to 19 K. A pronounced increase of the residual HS iron(II) sites is observed with ca. 50% of the molecules being in the HS state at low temperatures. When the pressure is further increased, the spin transition becomes increasingly gradual and incomplete. At 1.05 GPa, the spin transition is nearly quenched, the complex molecules are essentially all in the HS state. Thus, application of hydrostatic pressure surprisingly results in the stabilisation of the HS state, contrary to the normal expectation that pressure should stabilize the LS state due to its smaller volume. On release of the pressure, the HS state remains partially trapped. Indeed, an incomplete spin transition involving ∼50% of iron(II) ions which is shifted by 7 K to lower temperatures (T1/2↑ = 137 K and T1/2↓ = 105 K) and whose hysteresis becomes larger (32 K) compared to the ones obtained before applying pressure, is observed [7d].

χMT vs. T plots for [Fe(btr)2(NCS)2]·H2O under different pressures up to 1.05 GPa.
LIESST experiments have been performed on this compound at 10 K [7d]. These have shown that after thermal relaxation of the metastable HS state obtained by light switching, a pure LS state was observed in contrast to the pressure experiments. This different behaviour suggests that pressure leads to a structural modification that is presumably responsible for the pressure-induced HS state [7d]. Another possibility would be to consider that under pressure some water molecules enter the coordination sphere of iron(II) and thus establish a weaker ligand-field strength leading to the observation of the HS state. 57Fe Mössbauer spectroscopy under pressure and structural studies are necessary to clarify this unexpected magnetic behaviour.
The magnetic properties of the 3D SCO compounds {Fe(L)2[Ag(CN)2]2} with L = bipy = 4,4′-bipy and L = bpe = bis-pyridyl-ethylene have also been investigated under pressure [7e]. {Fe(bipy)2[Ag(CN)2]2} is HS over the whole temperature range under ambient pressure. Application of 0.48 GPa induces an incomplete SCO behaviour with T1/2 ∼ 150 K. At 0.7 GPa, the compound becomes essentially LS at room temperature. This spin transition induced by pressure at room temperature is found to be reversible. A similar magnetic behaviour under pressure is found for {Fe(bpe)2[Ag(CN)2]2} [7e]. Pressure investigation of the Hofmann like 3D SCO coordination polymer, {Fe(pz)[Ni(CN)4]}·2H2O (pz = pyrazine), by Raman spectroscopy, revealed an asymmetric piezo-hysteresis loop of width 0.07 GPa at room temperature [14]. Unfortunately, the spin transition was rather incomplete, since only 50% of the SCO iron(II) ions were involved in the spin transition process, and the hysteresis loop was strongly distorted. The observation of pressure tunable thermal hysteresis and piezo-hysteresis loop at room temperature of larger width (∼0.1 GPa) has been recently reported for the 3D coordination polymer {Fe(pmd)(H2O)[Ag(CN)2]2}·H2O (pmd = pyrimidine) [47]. In this material, pressure allows to place at will the hysteresis loop in a large range of temperatures, including room temperature, without losing its well defined square shape, which is a necessary requirement to achieve a reliable memory system. Furthermore, pressure also allows tuning of the thermal hysteresis width but this compound exhibits too an incomplete spin transition as a result of the presence of FeN4O2 sites [47].
4 Effect of pressure on the mononuclear spin crossover complex [CrI2(depe)2]
The phenomenon of thermal spin crossover is found most often in mononuclear compounds of iron(II), iron(III), cobalt(II), and is rather rare in complexes of other transition elements [48]. The first thermal SCO in chromium(II) compounds was reported by Halepoto et al. [49]. The ground state of the divalent chromium ion is 5D which splits into 5Eg and 5T2g states in a weak octahedral field yielding high-spin Cr(II) complexes with S = 2 ground states. In strong ligand-fields, the ground state will be 3T1g with LS behaviour and S = 1. The chromium(II) compound of the present study, bis[1,2-bis(diethylphosphino)ethane]di-iodochromium(II) (hereafter [CrI2(depe)2]), has a trans configuration (Fig. 12a) and at ambient pressure exhibits a very sharp spin transition with T1/2 = 169 K without noticeable thermal hysteresis [49]. A magnetic susceptibility study under pressure shows a progressive increase of T1/2 and a decrease of transition steepness with increasing pressure (Fig. 12b). Application of pressure of 0.8 GPa transforms the compound entirely to the LS state at room temperature. Qualitatively, one can interpret the behaviour of the transition curves under pressure in the approximation of mean-field theory [50], where the pressure dependence of the spin transition temperature obeys the Clausius–Clapeyron relation:
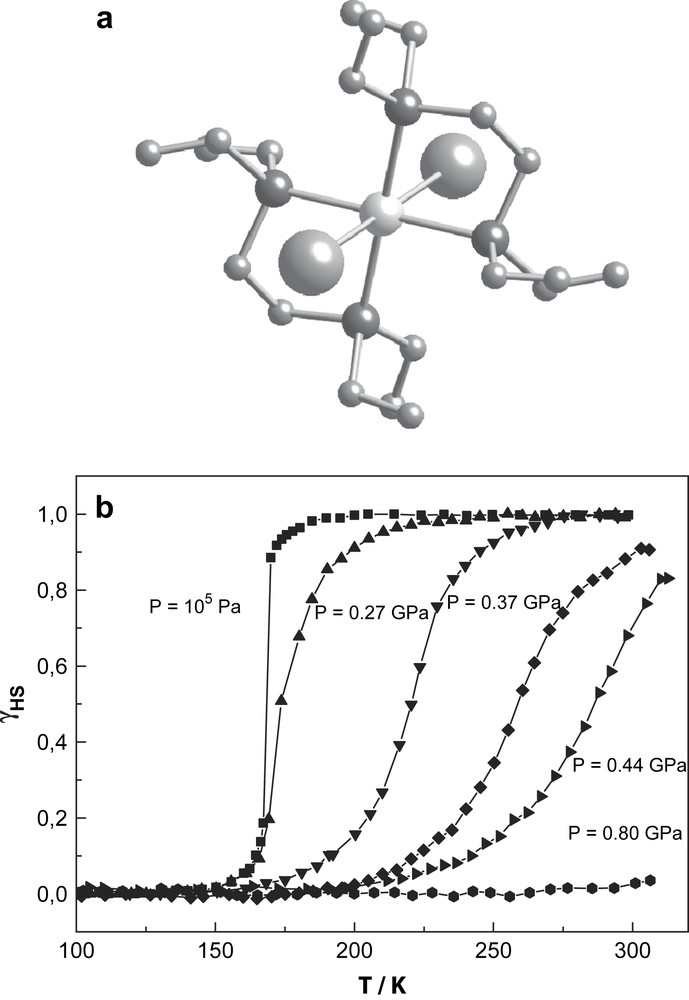
Molecular structure (a) and γHS vs. T plots at different pressures for the spin crossover complex [CrI2(depe)2] up to 0.8 GPa (b).
This relation reflects essentially the pressure dependence of the transition temperature T1/2 on the volume change. The pressure dependence of T1/2 for [CrI2(depe)2] shows strong nonlinearity (Fig. 13). It is expected that the iodine ions are more easily compressible than the phosphorus atoms that are of lower size, and, as a consequence, this should lead to an unisotropic volume change by application of pressure. A quantitative interpretation of the influence of pressure on the SCO behaviour of this chromium compound, particularly the nonlinear pressure dependence of the transition temperature T1/2 as indicated by the experimental data in Fig. 13, is only possible with a detailed crystallographic study of [CrI2(depe)2] under pressure and at variable temperatures.
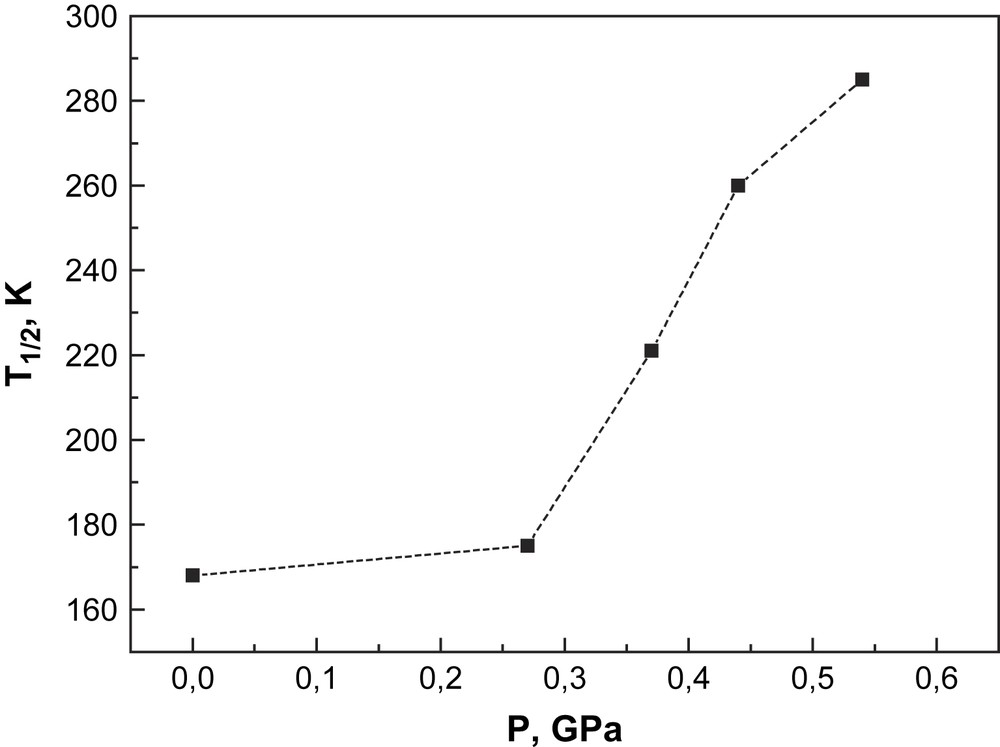
Pressure dependence of the spin transition temperature of [CrI2(depe)2].
5 Effect of pressure on valence tautomeric systems
5.1 O-Dioxolene adduct of a cobalt–tetraazamacrocycle complex
The phenomenon of temperature-induced valence tautomerism in cobalt complexes has been well-established in the literature [4,51]. In these systems, a thermally induced intramolecular one-electron transfer takes place between the catecholato ligand and the LS cobalt(III) acceptor with a spontaneous change in spin state from Co(III) (S = 0) to Co(II) (S = 3/2) at the cobalt centre, converting thereby the catecholato to the semiquinonato ligand with S = 1/2. The equilibrium between the two valence tautomers with different total spin states, viz. S = 0 and S = 2, respectively, can be easily followed by magnetic susceptibility measurements. The phenomenon resembles very much the thermal spin transition process in iron(II) compounds.
We have investigated the influence of pressure on the temperature dependence of the valence tautomeric interconversion between the catecholato (cat) and semiquinonato (sq) forms, [CoIII(L)(cat)]+ ↔ [CoII(L)(sq)]+, in the system [Co(cth)(phendiox)]PF6·H2O [52]. It has been inferred from crystal structure determination that the volume of the unit cell shrinks by more than 4% on going from the paramagnetic high-spin [CoII(L)(sq)]+ species to the diamagnetic low-spin [CoIII(L)(cat)]+ species, which is far more than can be accounted for by thermal contraction. Thus, it is clear that the magnetic properties of this valence tautomeric system should be pressure dependent. Indeed, as shown in Fig. 14, the transition curves are shifted to higher temperature and become more gradual with increasing pressure. When the pressure reaches the value of 0.74 GPa, the compound is practically diamagnetic at room temperature [52]. These findings are very similar to those obtained from pressure effect studies on SCO compounds as described above. After appropriate calibration, i.e. taking the χMT values for different pressure values at a given temperature, such valence tautomeric systems appear to be suited too for application in pressure sensors.

χMT vs. T plots at different pressures for [Co(cth)(phendiox)]PF6·1.5CH2Cl2 up to 0.74 GPa.
5.2 Pressure-induced electron transfer in ferrimagnetic Prussian blue analogues
In 1996, Hashimoto and co-workers found a photo-induced magnetization (PIM) effect in a cobalt–iron Prussian blue analogue [53]. This phenomenon was explained as being due to the presence of diamagnetic Co3+(LS)–NC–Fe2+(LS) pairs and a photo-induced electron transfer from Fe2+ to Co3+ through the cyanide bridge to produce Co2+(HS)–NC–Fe3+(LS) magnetic pairs [54]. Since the discovery of PIM, much effort has been devoted to the explanation of the appearance of diamagnetic pairs and their role in the PIM process [55]. Introduction of alkali metal cations into the tetrahedral sites of the fcc structure of Co4[Fe(CN)6]3 induces an electron transfer from cobalt(II) to iron(III) resulting in the stable diamagnetic Co3+(LS)–NC–Fe2+(LS) pairs. In studying K0.1Co4[Fe(CN)6]2.7·18H2O (hereafter K0.1Co4Fe2.7), which shows no spontaneous Co2+(S = 3/2)–Fe3+(S = 1/2) → Co3+(S = 0)–Fe2+(S = 0) process, we found a pressure-induced charge transfer taking place in the paramagnetic Co2+(HS)–NC–Fe3+(LS) units and leading to diamagnetic Co3+(LS)–NC–Fe2+(LS) units.
The χMT vs. T plots measured on K0.1Co4[Fe(CN)6]2.7·18H2O at ambient and under applied hydrostatic pressure are displayed in Fig. 15. This compound shows at ambient pressure antiferromagnetic interaction and a ferrimagnetic ordering below TC ≅ 16 K, which remains unaltered up to 0.3 GPa. Drastic changes are observed as the pressure increases to 0.4 GPa. In the temperature range 200 K < T < 300 K a strong decrease of the χMT product is observed and at low temperature the long-range magnetic ordering disappears. When the pressure is increased further, the pressure-induced feature in the magnetic behaviour above 200 K shifts to higher temperatures with a rate of ca. 170 K/GPa. At 1.02 GPa, the χMT value varies between 3 and 5 cm3 mol−1 K in the temperature range 4.2–300 K.

Temperature dependence of χMT for K0.1Co4[Fe(CN)6]2.7·18H2O at different pressures. Measurements at 105 Pa after release of pressure reveal a reversible behaviour in the samples.
57Fe Mössbauer spectroscopy confirmed the pressure-induced Co2+(S = 3/2)–Fe3+(S = 1/2) → Co3+(S = 0)–Fe2+(S = 0) charge transfer in the K0.1Co4Fe2.7 system. At ambient pressure and 4.2 K the magnetically split Mössbauer spectrum proves the existence of magnetic ordering in K0.1Co4Fe2.7 below 16 K (Fig. 16a). The spectrum with an average hyperfine field 〈H〉 = 164(2) kOe and isomer shift δ = −0.07(2) mm/s corresponds to 100% of Fe3+ in the S = 1/2 spin state. At a pressure of 0.3 GPa, a singlet with isomer shift δ = −0.015(2) mm/s indicative of a Fe2+ (S = 0) component appears in addition to the magnetically split spectrum (Fig. 16b). This is the only iron species present at pressures exceeding 0.4 GPa (Fig. 16c).
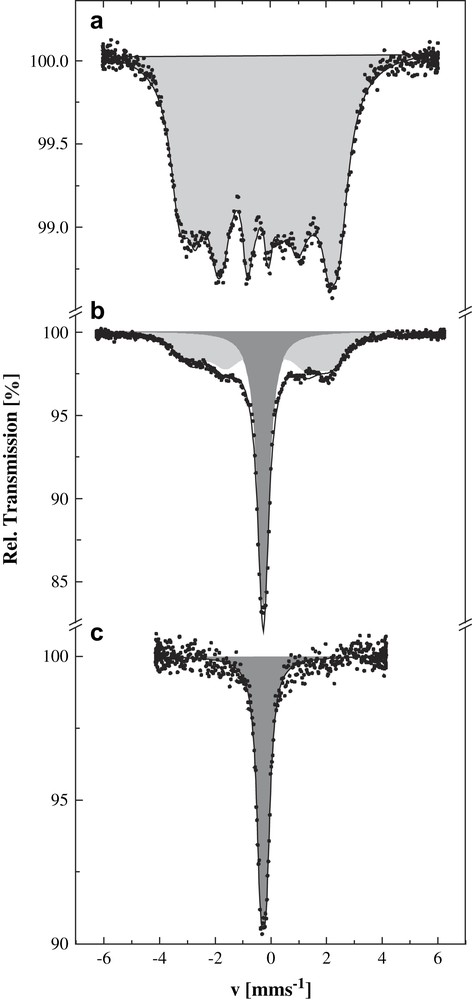
Mössbauer spectra of K0.1Co4[Fe(CN)6]2.7·18H2O recorded at 4.2 K and different pressures: (a) ambient pressure, (b) 0.3 GPa, (c) 0.4 GPa. Shaded subspectra correspond to Fe2+(S = 0) (dark grey), Fe3+(S = 1/2) (light grey).
The joint study of the magnetic properties and hyperfine interactions by Mössbauer spectroscopy under pressure in K0.1Co4Fe2.7 and other related Prussian blue analogues [56] gives clear evidence of pressure-induced electron transfer Co2+(S = 3/2)–NC–Fe3+(S = 1/2) → Co3+(S = 0)–NC–Fe2+(S = 0). The mechanism seems to be quite complicated. Recent investigations on other samples of this type of systems doped with alkali ions of different ionic radii and variable concentration lend support to the conclusion that it could be primarily the internal “chemical pressure” exerted on the iron chromophore sites leading to an increase of the ligand-field strength and thus favouring the formation of the diamagnetic Co3+(S = 0)–NC–Fe2+(S = 0) pairs, where both metal centres have smaller volumes than in the paramagnetic pairs [57]. Results from other experiments recently carried out, however, such as EXAFS studies, support another model where vacancies introduced into the lattice as a consequence of alkali ion doping are believed to play a decisive role. It is hoped to clarify this problem with further XRD and NIS (nuclear inelastic scattering of synchrotron radiation) studies in due course.
A pressure-induced Fe2+ SCO was recently reported for another Prussian blue analogue. The compound K0.4Fe4[Cr(CN)6]2.8·16H2O exhibits a reversible linkage isomerisation of the cyanide–metal bond from Cr3+(S = 3/2)–CN–Fe2+(S = 2) leading to Cr3+(S = 3/2)–NC–Fe2+(S = 0) as shown by a set of magnetic susceptibility, Raman and Mössbauer spectroscopic measurements under pressures up to 1.2 GPa [58].
6 Influence of pressure on the molecule-based magnet [FeII(TCNE)2]·xCH2Cl2
Among the molecule-based magnets possessing spins on organic radicals, the room temperature V(TCNE)x·y(solvent) magnet has the highest ordering temperature Tc ≈ 400 K [59]. In the related family of compounds [MII(TCNE)2·xCH2Cl2 (M = Fe, Mn, Co, Ni) the iron-based magnet [FeII(TCNE)2]·xCH2Cl2 possesses the next highest Curie temperature and orders ferromagnetically at ambient pressure at Tc ≈ 100 K, as follows from magnetic and Mössbauer measurements at ambient pressure [60]. Application of hydrostatic pressure provides new possibilities of modifying the properties of organometallic compounds by non-chemical methods [7c]. It is expected that application of pressure changes the interatomic distances and possibly the structure, which in turn may modify the magnetic interactions leading finally to changes of the macroscopic magnetic properties. We have chosen the magnetically well characterised compound [FeII(TCNE)2]·xCH2Cl2 to study such pressure-induced effects [61]. A sample of approximately 9 mg for magnetic measurements under pressure was packed into the high-pressure cell in a glove-box in anaerobic atmosphere. Dried and deoxygenated silicon oil used as a pressure transmitting medium in the high-pressure cell protected the sample against oxidation. The magnetic susceptibility was measured on polycrystalline samples in the temperature range 5–300 K with a PAR 151 Foner-type magnetometer, later on repeated with an SQUID magnetometer. The data were corrected for magnetization of the sample holder and for diamagnetic contributions.
Fig. 17 shows the temperature dependence of the magnetization at various pressures. The inset in Fig. 17 shows the magnetization at ambient pressure. It is obvious that the application of hydrostatic pressure (≥4 kbar) on the molecule-based magnet [FeII(TCNE)2]·xCH2Cl2 results in considerable improvement of its magnetic properties. The Curie temperature of the sample under pressure increased from Tc ≈ 100 K to Tc ≈ 160 K. Spontaneous magnetization in zero-field increased drastically from ≈30 emu G mol−1 up to ≈14 000 emu G mol−1. At the same time the strong increase of the coercive field of 8500 Oe at 10 K and 6000 Oe at 80 K was also a consequence of the pressure treatment. On release of the pressure, the new magnetic properties persisted at least for two weeks. This behaviour supports the assumption of a structural transformation leading to increased number of nearest neighbors of the iron atoms as the likely reason of the pressure-induced metastable state. Meanwhile, preliminary EXAFS measurements under hydrostatic pressure have confirmed the pressure-induced increase of the local coordination number of iron [62].
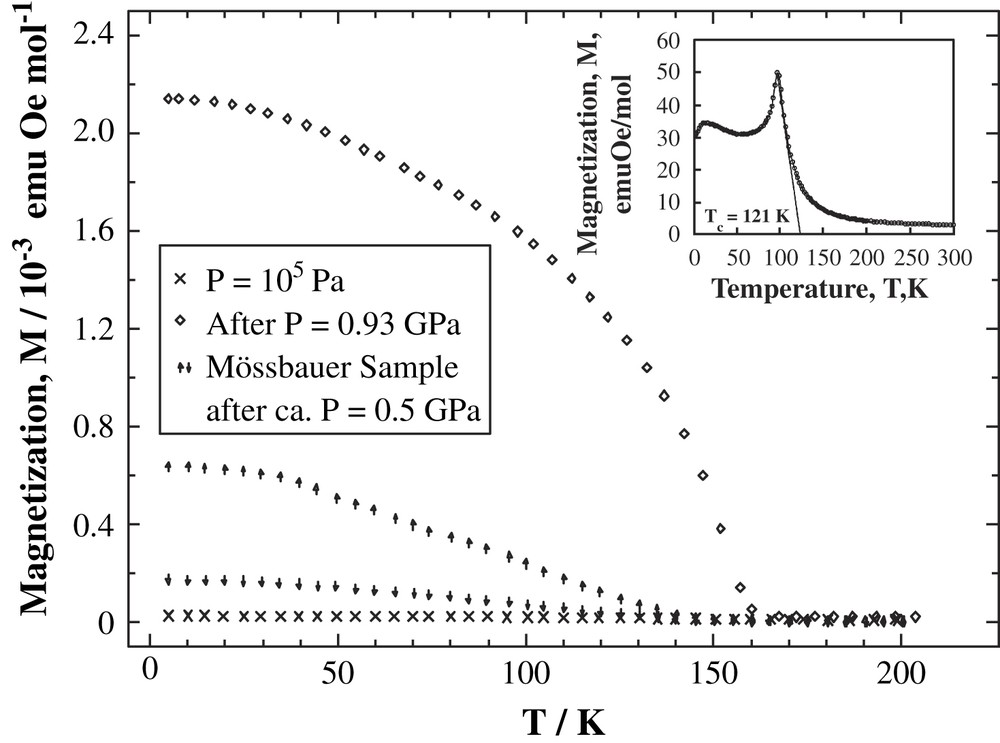
Pressure dependence of magnetization of [FeII(TCNE)2]·xCH2Cl2. The inset shows the magnetization at ambient pressure.
The oxidation and spin state of [FeII(TCNE)2]·xCH2C12 have been determined by 57Fe Mössbauer spectroscopy. Within the detection limits, only one iron site is observed. Above 95 K, the sample shows signals with large isomer shift [δ = 1.23(1) mm s−1] and quadrupole splitting [ΔEQ = 3.26(1) mm s−1] indicative of HS FeII ions. Below 95 K, the spectra become more complex due to magnetic splitting. The onset of the magnetic splitting with Hint = 229(1) kOe is in agreement with the critical temperature determined by magnetic measurements (≈100 K). The isomer shift and the quadrupole splitting derived from the magnetic hyperfine split spectra are δ = 1.24(1) mm s−1 and ΔEQ = 3.31(1) mm s−1, respectively.
7 Conclusion
Application of hydrostatic pressure in studies of molecular magnetism has proven to be a powerful technique. We have described pressure experiments on selected examples exhibiting thermal spin crossover, intramolecular magnetic coupling and valence tautomerism. The electronic structures in these systems change more or less dramatically on varying the temperature, observed e.g. by magnetic susceptibility and Mössbauer effect measurements. The electronic isomeric species involved in the transitions differ in their spin states, this in turn leads to differences in the molecular volumes. Logically, such phase transitions are also susceptible to pressure. The transition curves in terms of the product χMT vs. T derived from magnetic susceptibility measurements or in terms of the HS fraction γHS(T) as a function of temperature derived from Mössbauer effect measurements using custom-made hydrostatic pressure cells are strongly influenced under pressure. The reason is that the metal-donor atom distances decrease under pressure, this in turn increases the ligand-field strength at the transition metal centres and finally stabilizes the LS state by moving valence electrons from the antibonding eg∗ orbitals (in the HS state with the larger complex molecule) to the slightly bonding t2g orbitals (in the LS state with the smaller volume). As the changes of the molecular volumes play a significant role in the cooperative interactions in solid compounds showing these phenomena, it is obvious that application of pressure, can serve as a tool for changing the ligand-field strength in a controlled manner, an important method in studying the mechanism of dynamic electronic structure phenomena like spin crossover and valence tautomerism. Likewise, the magnetic paths in molecular systems with magnetic exchange interactions can be influenced strongly by pressure-induced changes of distances and angles with immediate consequences for magnetic coupling effects. As shown in the last example, desired magnetic properties can be improved considerably under pressure, like increase of magnetization, shifting upwards the magnetic ordering temperature, creation of new magnetic phases.
Acknowledgements
Financial support from the European Commission TMR_Network ‘Thermal and Optical switching of Spin States (TOSS)’, contract No. ERB-FMRX-CT98-0199EEC/TMR), the Deutsche Forschungsgemeinschaft (Priority Program 1137, ‘Molecular Magnetism’), the Fonds der Chemischen Industrie, the University of Mainz and the Fonds Special de Recherche of the University of Louvain is gratefully acknowledged. A.B.G. expresses her thanks to the Alexander von Humboldt Foundation for work-visiting fellowships and acknowledges the Spanish MEC for a research contract (Programa Ramón y Cajal) and for the project (CTQ 2004-03456/BQU).