

1 Introduction
Heme peroxidases are an important class of iron-porphyrin-containing enzymes which efficiently catalyze substrate oxidations using hydrogen peroxide [1]. The first step of the catalytic cycle of these enzymes consists in the coordination of H2O2 to the iron, followed by heterolytic cleavage of the O–O bond to produce a species commonly named compound I. In the classical peroxidases, such as horseradish peroxidase, this species is an iron-oxo species containing a porphyrin cation radical, +PorFe(IV)O. The reaction is facilitated by the proximal and distal environment of the heme through the cooperation of several amino acids with a ‘push–pull’ mechanism [2]. In the proximal site the axially bound histidine residue has partial anionic imidazolate character, due to hydrogen bonding to an aspartate, and acts as electron donor, thus stabilizing the high oxidation state of the iron in compound I (the push effect). In the distal site, an arginine residue polarizes the iron bound hydroperoxide, and the distal histidine acts as a shuttle for the proton transfer required for the release of a water molecule (pull effect) [3,4]. The global effect of the right positioning of the catalytic amino acids in the heme surrounding is a very efficient compound I formation from the resting enzyme. The rate constant for the hydrogen peroxide activation by peroxidase is in the order of 106–107 M−1 s−1 [5,6]. A single point mutation of a relevant amino acid gives rise to a marked drop of this rate constant, which can be reduced by orders of magnitude [7].
Other heme proteins, such as myoglobin, hemoglobin, etc., are capable, in the presence of hydrogen peroxide, of generating active species but with a much reduced activity (rate constant below 103 M−1 s−1) [8,9].
The peroxidase compound I species generally oxidizes monoelectronically a substrate molecule forming the compound II species, an iron-oxo species PorFe(IV)O, while the electron from the substrate is transferred to the porphyrin radical. Since delocalized π-type orbitals are involved in the electron transfer and since upon reduction the enzyme undergoes only small structural changes, the compound I to compound II reduction is really a fast process, usually the fastest within the enzyme catalytic cycle [1]. A further one-electron reduction of compound II restores the enzyme in the resting state and closes the cycle. Two protons are required to transform the iron-oxo to the iron(III) form of the protein, with the release of a water molecule. Furthermore, upon reduction the iron ion slightly shifts toward the proximal porphyrin site. The larger rearrangement required for compound II reduction contributes to the reduction of the electron transfer rate with respect to that involving compound I. The distal arginine in peroxidases acts a proton donor for the iron-oxo group, thus facilitating the process. In fact, removal of the distal arginine by site-directed mutagenesis gives rise to a huge reduction in the rate constant for compound II reduction [10].
The high activity shown by peroxidases, together with the wide range of substrates that can be oxidized in the presence of hydrogen peroxide, makes these enzymes very appealing for the removal of pollutants present, for instance, in industrial waste waters but also to be used as catalysts in fine chemical industries. At the same time, the use of peroxidases has some drawbacks. They are generally expensive enzymes obtainable in small amounts from natural sources. Often they are unstable during catalysis and after a few thousands of catalytic cycles their activity falls down, with the consequence of requiring renewal of these precious enzymes. Furthermore, their specificity is controlled by the charge at the substrate binding site and the accessibility of the binding site at the heme periphery, where the electron transfer can occur readily; but the molecule to be processed by the peroxidase catalysis may be a poor substrate of the enzyme and this will make the enzymatic process inconvenient.
One way to overcome, at least in part, the above problems is the preparation of heme derivatives with peroxidase-like activity. It is difficult to reproduce in a synthetic molecule the peculiar features of peroxidases, i.e. the high reactivity vs. both hydrogen peroxide and the substrates, and the stability during turnover due to the protective action given by the protein backbone to the heme active species. Though, often an increase in the activity toward the substrates is obtained using synthetic models exhibiting reduced rates in the reaction with hydrogen peroxide.
This review deals with the preparation of peroxidase model systems by using heme-peptide complexes, and it is mainly focused on the results obtained in our laboratory. In particular, our research efforts were developed through four different approaches, as summarized below.
- (a) In the first approach, peptide residues are covalently attached to hemin (or deuterohemin, derived from hemin upon removal of the vinyl groups). The amino acid residues of the peptide(s) interact with the substrates giving rise to some selectivity effect, thus partly mimicking the substrate recognition mechanism exhibited by the enzyme [11–13].
- (b) In the second approach the hemin is further modified by covalently linking to the propionate side chain a histidine residue or a peptide containing histidine that better models the effect of the proximal histidine in the enhancement of the heme reactivity vs. hydrogen peroxide [11,14–17].
- (c) The third approach is based on microperoxidases, the heme-peptide complexes obtained from proteolytic degradation of cytochrome c. They already contain a proximal histidine and, through further chemical modification, it is possible to introduce other polar residues which may contribute to hydrogen peroxide activation or substrate recognition [11,18–20].
- (d) The last approach utilizes a synthetic heme-peptide complex for the reconstitution of a heme protein. The globular protein myoglobin is used to provide the amino acid scaffold around the heme; the polypeptide chain is useful for substrate recognition and protects the porphyrin during catalysis. The peptide bound to the heme has the effect of opening the myoglobin active site allowing an easier access to the substrate and can fine tune substrate recognition [8,9,21–29].
2 Hemin/deuterohemin-peptide complexes
Synthetic and natural iron(III) porphyrins are able to generate highly reactive species upon reaction with hydrogen peroxide. These species are strong oxidizing agents which can react with substrates in one-electron or two-electron oxidation, often in catalytic processes. The activation of the metal catalyst by peroxide is generally a slow step; in fact, the synthetic systems usually lack the acid–base activation required for an efficient heterolytic cleavage of the bound peroxide in the first step of the catalytic cycle. Often this activation is given by the buffer, through general acid–base catalysis and is facilitated by basic conditions, where a larger fraction of peroxide exists in the deprotonated form [30–34]. In spite of that, the active species thus generated is highly reactive due to the easy approach of the substrate to the porphyrin plane and the iron-oxo group. The capability of the catalyst in discriminating between different substrates depends on the redox potentials of the active species and of the substrates. Another important aspect is the mode of interaction between the two molecules; in fact, in order to have an efficient oxidation process, the substrate must bind with high affinity to the iron-porphyrin complex and with a disposition which favors the electron transfer. In peroxidases, substrate discrimination is controlled by the protein backbone in the active site. In synthetic iron-porphyrins the accessible distal and proximal sites do not allow such a discrimination. In tetraaryl iron-porphyrin derivatives, it is possible to introduce charged or bulky groups on the phenyl rings in order to introduce some selectivity [35]. The two propionate substituents present in hemin and deuterohemin complexes may also exhibit some electrostatic interaction with charged substrates.
The preparation of synthetic catalysts with peroxidase-like activity which are capable of selectively oxidize a certain type of substrate is of particular interest. Such systems must have only a partly accessible porphyrin and a substrate binding site where, after binding, it can undergo oxidation by the catalyst active species. The iron-porphyrin-peptide complexes we used to mimic the environment of the heme present in the enzymes were obtained by covalently linking to one of the propionate side chains of deuterohemin a peptide [11–13]. Deuteroporphyrin was preferred to protoporphyrin IX as iron ligand because it does not contain the oxidation-sensitive vinyl groups and therefore it is supposed to be less prone to autoxidation. In the two heme-peptide complexes the peptides we prepared were a deca-l-alanine (DH-Ala10) and Ala-Phe-Ser-Phe-Glu-Ala-Gln-Gly-Gly-Leu-Ala-OH (DH-pep11), both bound through the amino terminal group (Fig. 1).
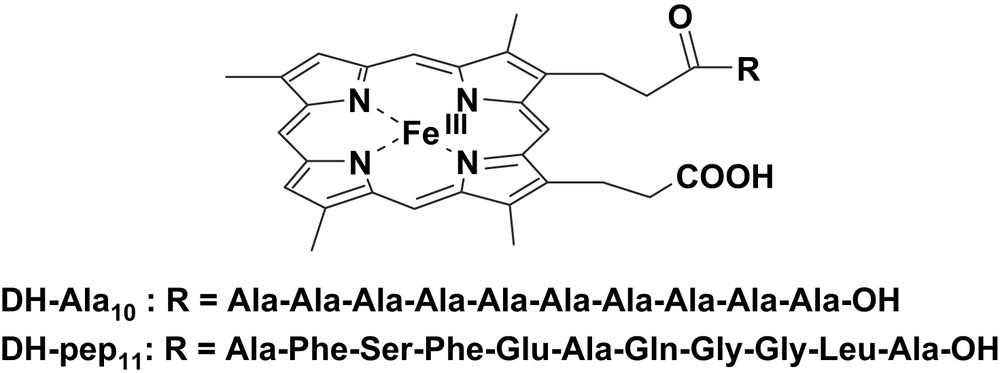
Structures of the deuterohemin derivatives DH-Ala10 and DG-pep11.
It should be noted that the synthesis of these complexes gives rise to an equimolar mixture of the two isomers carrying the peptide linked to each of the propionate side chains. The two isomers have very similar properties and this prevents their separation. Both DH-Ala10 and DH-pep11 derivatives catalyze the oxidation of l- and d-tyrosines to o,o′-dityrosine in the presence of hydrogen peroxide. It is worth noting that the dependence of the reaction rates on substrate concentration, under saturating peroxide concentration, follows a hyperbolic behavior, suggesting the need of binding the substrate at the catalyst active site during the catalytic cycle according to the following pre-equilibrium reaction scheme:
| Feox + substrate ⇄ [Feox(substrate)] → Fe3+ + product |
The degree of stereoselectivity obtainable with these deuterohemin complexes is limited (up to 50% preference for one enantiomer). This is probably connected to the flexibility of the peptide arm, which can span almost freely most of the space around deuterohemin, a behavior which is clearly different from the reduced mobility of the peptide backbone in the protein. Furthermore, the activity in the reaction with hydrogen peroxide, required for the formation of the active species, is low due to the lack of the ‘push–pull’ effect around the iron porphyrin.
3 Proximal histidine coordination
The easiest way to introduce the push effect by the proximal histidine of peroxidase in model systems in order to increase the activity vs. peroxides is the introduction of coordinating molecules in the solution. The buffer itself or an exogenous base ligand, like imidazole added to the solution, can act as iron ligand [30–34]. The problem of the simple addition of the ligand is the high tendency of iron-porphyrins to form the six-coordinated low-spin adduct. When the imidazole derivatives are kept at low concentration only a minor fraction of the iron is bound; raising the concentration of the ligand, the six-coordinated species which forms is catalytically inactive. As a result, the activation observed with the addition of free ligand is quite low [13].
In order to force the coordination of an axial imidazole, covalently modified protohemin or deuterohemin complexes, through the linkage of imidazole residues to the propionate side chains of the porphyrin, were synthesized [11,14–17]. Complexes of this type have been previously used to investigate the ligand binding properties of the iron center [36–39], and also as peroxidase mimics [30,31].
To combine the activation effect given by the axial imidazole and the substrate recognition given by an appended peptide chain, two model systems were studied by our group. These complexes contain a l-histidine methyl ester residue linked to one propionate group of deuterohemin and a poly-l-alanine peptide, containing a l-phenylalanine residue at 3- or 4-position, linked to the other propionate arm (Fig. 2) [15]. The chelated histidine residue improves the reactivity of the model systems; it also has the effect of forcing the peptide of the other arm to fold toward the opposite (distal) plane of the porphyrin. The catalytic oxidation of the two enantiomers of tyrosine methyl ester in the presence of tert-butyl hydroperoxide follows a Michaelis–Menten behavior indicating the formation of an intermediate complex between the catalyst active species and the complex. Interestingly, only the complex bearing the phenylalanine residue in the fourth position of the peptide chain exhibits enantioselectivity in the oxidation of the two isomers. This result has been explained considering that the chiral recognition required interaction with the polar groups of the peptide; when the interaction is mostly through hydrophobic ring stacking with the Phe residue, the substrate is not immobilized in the adduct and its large mobility prevents the observation of appreciable stereoselectivity effects.
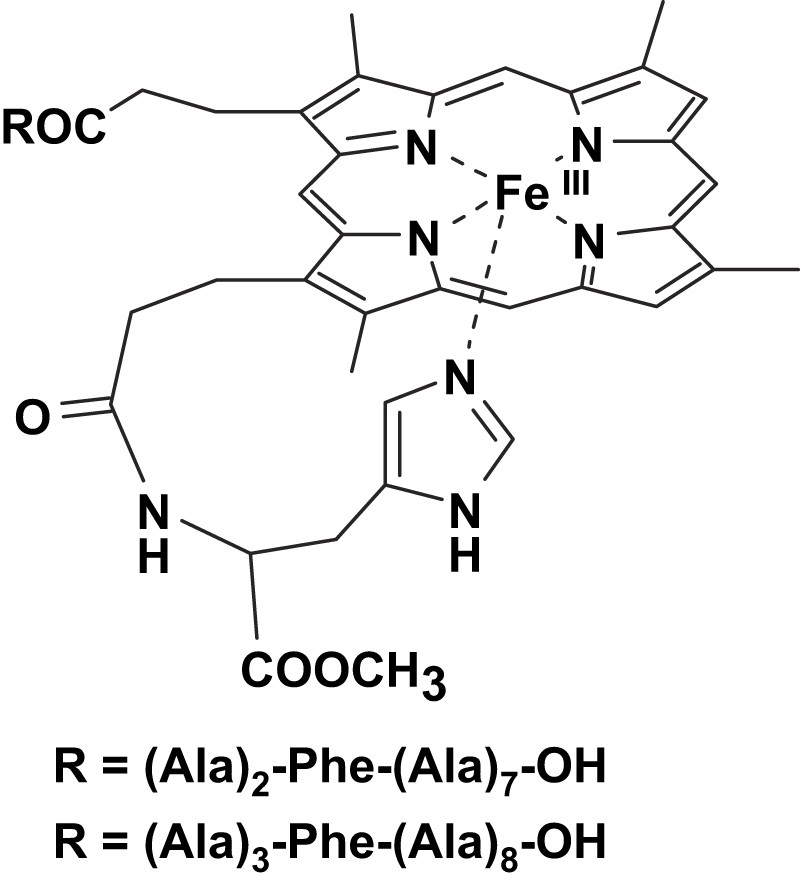
Structures of the deuterohemin derivatives modified by a decapeptide containing alanine residues and a phenylalanine at 3- or 4-position.
The strain in the coordination to the iron of the chelated imidazole alters the binding properties of the iron-porphyrins. For instance, it has been shown by Traylor et al. that this strain affects the CO dissociation in the iron(II) forms [39], but it also has strong effect on the catalytic activity of the complexes because it affects both the binding of ligands/substrates in the opposite plane of the porphyrin and the donor strength of the axial imidazole. Molecular mechanics and molecular dynamics calculations carried out on five-coordinated deuterohemin complexes bearing a histidine, or a short peptide containing a His residue, bound to one propionate chain have shown that strain in the coordination depends on the His position in the peptide sequence [14]. When His is the first amino acid and is directly bound to the propionate (as in deuterohemin-6(7)-l-histidine methyl ester, DH-His) (Fig. 3), the interaction with the iron center is weaker and the imidazole plane is not perpendicular to the porphyrin ring but with somewhat tilted. This strain is released when histidine is the second residue in the peptide, as in the complex containing Ala-His bound to the propionate (deuterohemin-6(7)-l-alanyl-l-histidine methyl ester, DH-Ala-His) (Fig. 3).
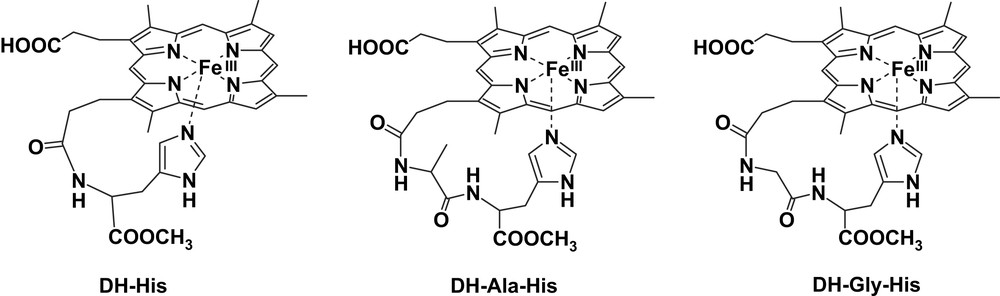
Structures of the deuterohemin derivatives DH-His, DH-Ala-His and DH-Gly-His.
The position of the histidine in the peptide has a marked influence in the affinity constant for imidazole binding to iron(III), with DH-His showing a 4-fold lower binding constant with respect to DH-Ala-His, but the difference is much larger when the binding is studied in an organic solvent [15].
Also the reactivity of the complex in the catalytic oxidation of N-acetyl-tyrosine is affected by the position of the histidine in the chelated complex. DH-Ala-His shows a higher reactivity with respect to DH-His both in terms of increased turnover number (larger kcat) and affinity vs. the substrate (lower KM).
The chelating arms containing the histidine residue in DH-His and in the complex with a Gly-His-OMe peptide covalently bound to deuterohemin, (deuterohemin-6(7)-glycyl-l-histidine methyl ester, DH-Gly-His) (Fig. 3), exhibit a further effect on the reactivity. These complexes show appreciable stereoselectivity in the oxidation of the l and d isomers of tyrosine and tyrosine methyl ester [16]. The selectivity can only be due to the interaction of the chiral substrate with the chelating arm containing the l-His residue. Analysis by molecular mechanics and molecular dynamics shows that, in the substrate–catalyst adduct, phenol interacts with the histidine bound to the iron and approaches the porphyrin edge from the proximal site of the molecule. The disposition of the two tyrosine isomers is different, thus justifying the catalyst stereoselection.
The effect of the position of the histidine residue on the catalytic activity of heme-peptide complexes has been observed by the Nordlander group with modified protohemins [17]. They synthesized the hemin modified by covalently linking to one propionate arm of the porphyrin a Gly-His-OMe (protohemin-6(7)-l-glycyl-histidine methyl ester, HGH) or a Gly-Gly-His-OMe (protohemin-6(7)-l-glycyl-glycyl-histidine methyl ester, HGGH) residue. The analysis of the catalytic activity of these complexes in the oxidation of p-cresol, l-tyrosine methyl ester, and ABTS in the presence of hydrogen peroxide indicates that the longer and less coordinatively strained heme-peptide complex (HGGH) exhibits higher reactivity.
4 Modified microperoxidases
Microperoxidases (MPs) are heme-containing peptides derived from proteolytic digestion of cytochrome c, in which the heme is covalently linked to the peptide chain through two thioether bonds [40–42]. The peptide size and sequence in MPs can be modified by changing the source of the starting cytochrome c or the type of proteolytic enzyme (i.e., trypsin, pepsin or both of them one after the other). In all MPs the histidine residue, acting as iron proximal ligand in cytochrome c, is maintained while the second axial ligand (Met80 in cytochrome c) is removed by proteolysis; this leaves the iron(III) center weakly bound to a water molecule, allowing the coordination of hydrogen peroxide or other exogenous ligands. The best known among MPs is MP8 (Fig. 4), which is obtained from the peptic and tryptic digestion of horse heart cytochrome c; it retains the amino acid residues 14–21 of the starting protein. Treatment of the protein with pepsin alone yields MP11, which contains residues 11–21; instead, using trypsin proteolysis alone generates MP9, containing residues 14–22. In the presence of hydrogen peroxide, all these MPs form active species which are able to oxidize several substrates, thus acting as models for peroxidases [19].
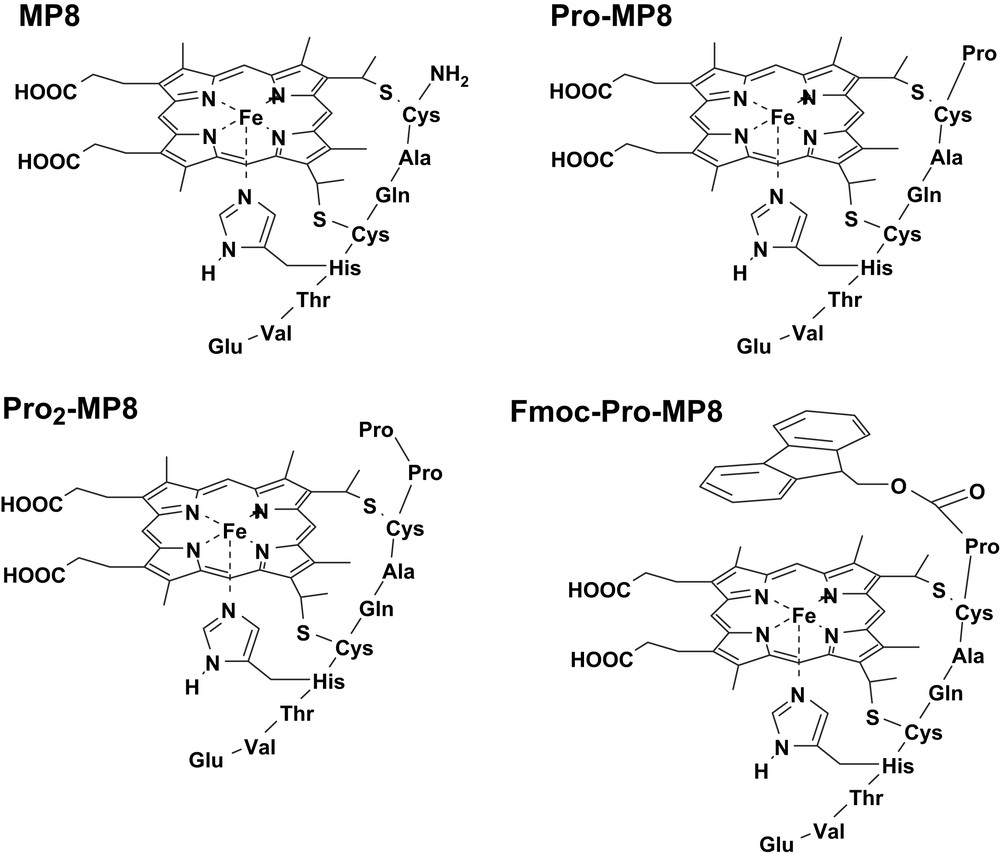
Structures of the microperoxidase derivatives MP8, Pro-MP8, Pro2-MP8 and Fmoc-Pro-MP8.
Microperoxidases are highly soluble in water medium and can take advantage of the activating ‘push’ effect due to the presence of a suitably positioned axial histidine. Therefore, MPs are good starting points for the building up of heme-peptide catalysts with enhanced activity and selectivity. At the same time, the presence of a rigid proximal site makes the modified MPs good probes for investigating the effect of modifications at the distal site on the reactivity of the complex. MP8 is characterized by an open distal site, which is freely accessible to both hydrogen peroxide and substrate molecules. The reduced substrate selectivity observed in its reactions is due to electrostatic interaction with the protonated N-terminal amino group. The lack of charged residues at the distal site, that could reproduce the effect of distal histidine and arginine in peroxidases, makes the reactivity of MP8 toward H2O2 orders of magnitude lower than that of the enzyme.
To investigate the effect of distal residues on the formation of the active species upon reaction with H2O2, some MP8 derivatives were synthesized by covalent linkage to the N-terminal amino group of one (Pro-MP8) or two (Pro2-MP8) proline residues, or a single proline N-protected by an aromatic fluorenyl group (Fmoc-Pro-MP8) (Fig. 4) [18]. Table 1 shows the second order rate constants for the reaction with hydrogen peroxide of these MP8 derivatives at two pH values. The data show that the reactivity is controlled by the polarity around the iron. In Fmoc-Pro-MP8, the amine protecting group reduces the polarity and introduces some steric hindrance; as a consequence, the k1 value drops down with respect to the value observed for MP8. In Pro-MP8, the protonated proline group acts as an internal acid–base catalyst in the heterolytic cleavage of the heme-bound peroxide, thus increasing the activity of the catalyst. The addition of a second Pro residue further reduces the distance between the proline N atom and the peroxide oxygen atom, increasing the k1 value for Pro2-MP8. The second order rate constants observed at pH 9 are higher than those at pH 5. This is because at basic pH a larger fraction of anionic peroxide is present in solution, facilitating its coordination to iron; at the same time, the acid–base catalysis by OH− from the buffer further activates the complexes.
Second order kinetic constants for oxidation of p-cresol by MP8 derivatives
| Catalyst | k1 (M−1 s−1), pH 5 | k1 (M−1 s−1), pH 9 |
| MP8 | 680 ± 10 | 13 300 ± 350 |
| Fmoc-Pro-MP8 | 390 ± 10 | 5300 ± 150 |
| Pro-MP8 | 1100 ± 20 | 13 500 ± 400 |
| Pro2-MP8 | 2000 ± 50 | 17 500 ± 400 |
The analysis of the reactivity of MP active species toward the substrates is complicated by the complete accessibility of the porphyrin ring of MPs by exogenous molecules. Therefore MPs substrate oxidation is extremely rapid compared to the formation of the active species and usually occurs as a fast step in the catalytic cycle. But operating at high peroxide concentration and analyzing only the initial phase of the reaction (where catalyst degradation is negligible), the substrate oxidation reaction becomes the rate determining step of the turnover process. It is thought that MPs react with a mechanism similar to that of peroxidases, i.e.:
| MP + H2O2 ⇄ [MP/H2O2] → MPox1 + H2O | (1) |
| MPox1 + SH → MPox2 + S + H+ (fast) | (2) |
| MPox2 + SH + H+ → MP + S + H2O | (3) |
The activity of several MP derivatives in the oxidation of two representative phenols, 4-(2-aminoethyl)-phenol (tyramine) and 3-(4-hydroxyphenyl)propionic acid (HPA), by hydrogen peroxide has been investigated. The MP derivatives used were MP8, MP11, AcMP8 (obtained from MP8 after acetylation of the terminal amino group), MP′11 (obtained from baker's yeast cytochrome c), and the synthetic N-Fmoc-Arg(Mtr) MP8 (FM-R-MP8) (Fig. 5) [20]. The latter compound was obtained from MP8 by linking to the N-terminal amino group a double protected arginine residue, by 9-fluorenylmethoxycarbonyl (Fmoc) at Nα and by a 4-methoxy-2,3,6-trimethylbenzenesulfonyl at Nγ. The kinetic parameters for the activity of these MPs are reported in Table 2. The data show that the residues on the distal MP site alter the reactivity of the active species by controlling both the substrate approach and affinity (KM), and the electron transfer rate (kcat), which is modulated by the phenol disposition at the active site. An indication of the substrate disposition in the adduct with the catalyst is given by the iron–substrate proton distances evaluated from the paramagnetic effect of the metal ion on the 1H NMR relaxation rates. It is interesting to note that the binding of HPA and tyramine to MP complexes occurs with iron–proton distances in the range of 7.4–9.5 Å, which is the same range commonly observed for substrate binding to peroxidases [43–45]. The collective data show that the difference in reactivity between tyramine and HPA is little dependent on their redox potential; this is consistent with a high redox potential for the MP active species. Comparing the data for MP8 and AcMP8 clearly indicates that the protonated amino group of the former complex exhibits substrate recognition, because of their different charge; its acetylation leads to loss of selectivity in the oxidation of the two phenolic substrates. The electrostatic interaction between substrate and MP controls substrate binding but stacking π–π interactions also seem to play an important role. This is evident with FM-R-MP8 where, besides the lack of the protonated amino group, the two aromatic protective groups make the distal site of the complex much more hydrophobic. It binds the substrates with high affinity, as shown by the low KM values, but the binding site is quite far from the iron and this leads to strong reduction in the reaction rates, since it is known [46] that for an efficient electron transfer between the substrate and the catalyst active species a good overlap of the orbitals is required. Table 2 also shows that polar residues near the heme facilitate the oxidation of the substrate. This is particularly evident with MP11 and MP′11 where in some cases the substrate oxidation rates are so high that only a guess for the lower limit of kcat is possible. The notably high electron transfer rates observed with MP11 and MP′11 is probably connected to the presence of protonated residues which can approach the iron-oxo group. This may favor the protonation of the ferryl oxygen required for the transformation of the active species into the iron(III) form. Therefore, these charged residues mimic the effect of the distal arginine in peroxidases in the activation of compound II reduction [10]. The efficiency of this process is so high in MP11 and MP′11 that their reactivity becomes comparable with that of HRP, and even larger than that in the case of tyramine oxidation. To our knowledge this is the highest peroxidase-like activity ever reported for a heme model system.
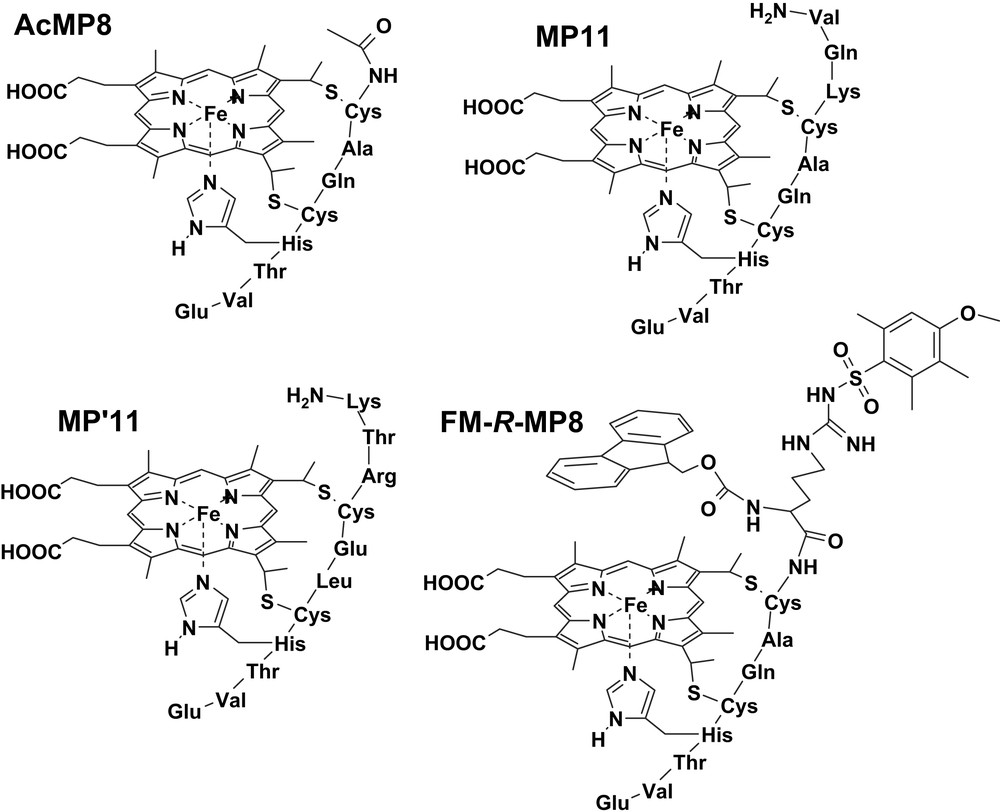
Structures of the microperoxidase derivatives AcMP8, MP11, MP′11 and FM-R-MP8.
Kinetic parameters for the catalytic activity of MPs in the oxidation of HPA and tyramine by hydrogen peroxide
| kcat (s−1) | KM (M) | kcat/KM (M−1 s−1) | References | ||
| MP8 | HPA | 26.9 ± 1.6 | 0.0053 ± 0.0010 | 5050 ± 750 | [19] |
| Tyramine | 22.9 ± 1.3 | 0.0119 ± 0.0015 | 1920 ± 150 | [19] | |
| AcMP8 | HPA | 32 ± 2 | 0.0071 ± 0.0001 | 4200 ± 600 | [20] |
| Tyramine | 31 ± 1 | 0.0053 ± 0.0005 | 5800 ± 400 | [20] | |
| FM-R-MP8 | HPA | 0.80 ± 0.02 | 3.4 × 10−4 ± 4 × 10−5 | 2300 ± 200 | [20] |
| Tyramine | 1.18 ± 0.07 | 6 × 10−4 ± 1 × 10−4 | 1900 ± 300 | [20] | |
| MP′11 | HPA | >800 | n.d. | n.d. | [20] |
| Tyramine | >800 | >0.030 | 25 700 ± 500 | [20] | |
| MP11 | HPA | >800 | n.d. | n.d. | [20] |
| Tyramine | 257 ± 8 | 0.015 ± 0.001 | 16 700 ± 700 | [20] |
The emerging picture from the activity of covalently modified MPs is that to have an efficient catalyst the distal site must contain positively charged residues which can approach the iron; their presence increases both the reactivity toward hydrogen peroxide and the substrates. The selectivity for different substrates can take advantage of the addition of other charged residues which do not necessarily have to approach closely the iron, since the electron transfer occurs through the porphyrin plane. Aromatic residues can be used to increase the substrate affinity through stacking π–π interactions. To keep a good activity, the reduction of the polarity around the iron by the aromatic groups should be prevented; moreover, the substrate binding site must be kept close to the porphyrin edge.
5 Reconstitution of myoglobin with heme peptides
A main goal of protein engineering is the construction of new enzymes which are capable of enhancing the catalytic activity and/or the specificity toward a substrate. In particular, efforts to introduce new functions into the heme enzymes and other heme proteins such as myoglobin (Mb) have been increasingly reported in the recent years. Moreover, protein modification is useful to get a fuller understanding of both the enzymatic reaction mechanism and physiological function, including structure–function relationships. Myoglobin, the protein of storage and intracellular transfer of molecular oxygen [47], has a potential for catalytic peroxide-dependent one-electron oxidation reactions, such as the oxidation [28,48] and nitration [49,50] of phenolic substrates, and for reactions involving formal two-electron oxidation of substrates, such as the sulfoxidation of organic sulfides and the epoxidation of alkenes [51]. Even if the peroxidase activity of Mb is much lower than that of the peroxidases, it can be improved by protein modification.
Different approaches can be explored to convert a protein into a peroxidase: rational design of heme enzymes, direct evolution of heme enzymes, exon shuffling, and modification of the heme prosthetic group [52]. Regarding the peroxidase activity, the rate of substrate oxidation depends on two main factors: the activation of hydrogen peroxide and the rate of electron transfer from the substrate to the iron active species, which is controlled by the disposition of the substrate in the active site, with respect to the heme. Site-directed mutagenesis enables to improve activation of H2O2 but does not represent the best route for the construction of a specific receptor site for a particular type of substrate. On the other hand, the heme reconstitution method is a powerful way to enhance the selectivity of Mb. An interesting possibility is the engineering of Mb by combination of the genetic and reconstitution approaches, to potentiate the different effects on the enhancement of the catalytic activity [28].
The chemical modification of the protein cofactor as a new strategy for introducing new chemical functions into the protein emerged in the eighties [53]. The most interesting improvements in the peroxidase activity of Mb by a nongenetic method where a functionalized heme is inserted into apoMb were reported by the group of Hayashi and by our group. The work of Hayashi focused on the construction of a binding interface for substrates through the functionalization of the two heme propionates [21]. It is well known that these groups play a role in fixing the heme in the protein pocket by electrostatic interactions with Lys-45 and Ser-92 [54,55]. Moreover, to evaluate the specific role of the heme-6- and heme-7-propionate side chain in horse heart Mb, Hayashi reconstituted the protein with hemin derivatives methylated at the 6- and 7-position [22]. The results indicate that the elimination of the salt bridge between the 6-propionate side chain and Lys-45, which further makes a network with His-64 via one water molecule, might affect the physiological binding of molecular oxygen. Differently, the lack of heme-7-propionate has an influence on the proximal site since it regulates the hydrogen-bonding network between Ser-92 and His-93, perturbing the coordination of the latter residue to the heme iron.
The chemical reactivity of Mb can be improved by setting a substrate binding site on the protein surface, near the entrance of the heme pocket. With the aim of creating such an artificial binding domain, Hayashi reconstituted Mb with two different types of chemically modified cofactors: an anionic heme with four or eight carboxylates at the terminal of the two propionate side chains [23,24] and a cationic heme with four amino groups at the end of the two propionate chains [25]. These modified Mbs form stable complexes both by electrostatic interactions with substrates carrying an opposite charge and by hydrophobic contacts [25]. Moreover, the proteins functionalized with the binding domain are useful models to clarify the behavior of the protein–protein recognition and interprotein ET (with cytochrome c as the cationic partner of Mb reconstituted with anionic heme) [26]. The findings of Hayashi on the reactivity of the Mb intermediate compound II indicate that modification of the heme propionates creates a binding site with specificity for some substrates: with guaiacol, for example, the peroxidase activity of the reconstituted Mb was 14-fold higher than that of the native protein [8].
The interest of our group focused on the peroxidase-like activity of Mb: the combination of site-directed mutagenesis and prosthetic group modification allowed us to develop a powerful method to enhance the reactivity and selectivity of Mb derivatives. The active site similarities between Mb and peroxidases make the changes to be introduced in the protein of relatively limited entity, compared to those eventually necessary to attempt to simulate the activities of other enzymes using Mb. The scope of our modifications of Mb aimed at introducing into the protein the amino acid residues that are essential for the activation of hydrogen peroxide (a distal arginine and a proximal aspartate) and a specific cleft for substrate binding near the heme. In our initial attempts, horse heart apoMb was reconstituted with hemin modified by attachment of an Arg-Ala residue to one of the two propionate side chains (Mb-RA) (Fig. 6) [9]. As we have seen before, insertion of a modified hemin into Mb affects the pre-existing network of hydrogen bonds between the propionate groups of the cofactor and side chain residues of the protein. In the case of Mb-RA this had a deleterious effect, as the key peptide segment of Mb Phe-43-Lys-47 acquired sufficient mobility to allow collapse of the distal helix and binding of distal His to the sixth coordination position of the iron [9]. The presence of distal His as the sixth ligand hindered the binding of hydrogen peroxide to the iron and reduced the activity of the reconstituted protein. On the contrary, when the heme cofactor was modified by attachment of a histidine residue (heme-His), the reconstituted Mb (Mb-H) contained a significant portion of high spin form that reacted normally with hydrogen peroxide (Fig. 6). Since the His imidazole group of the modified prosthetic group is partially protonated at physiological pH, it could play a role similar to that of distal arginine in peroxidases. Moreover, mobilization of the peptide chain around the heme in the reconstituted Mbs increases the accessibility of the substrates to the active site facilitating the electron transfer in the peroxidase-type oxidations.
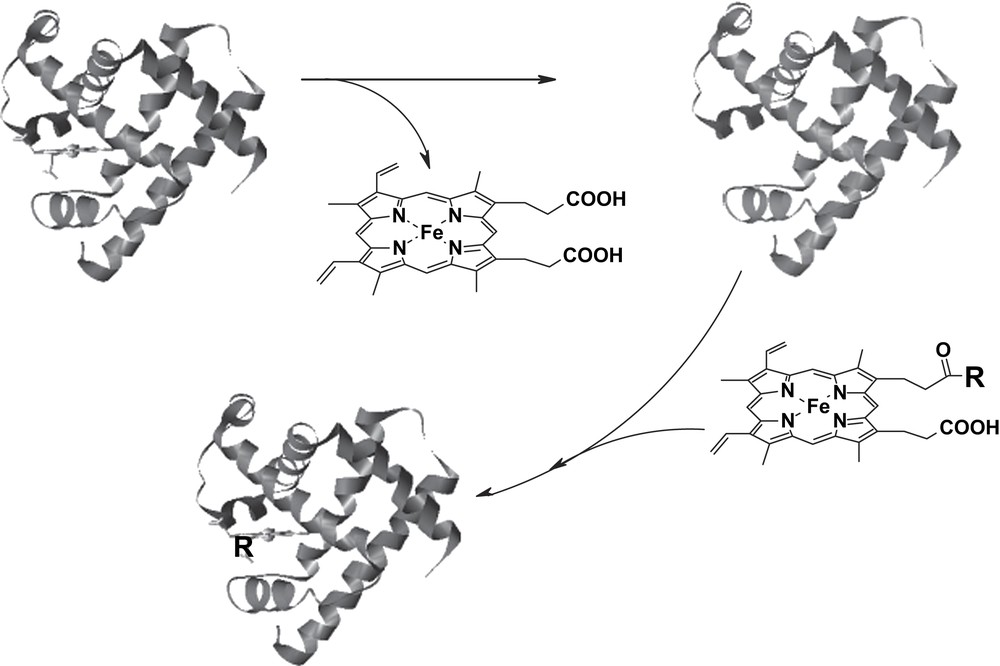
Schematic representation of the reconstitution of Mb with hemin derivatives. In the case of Mb-RA and Mb-H, R represents the Arg-Ala-OH dipeptide and the protected amino acid His-OMe, respectively.
It should be noted that upon reconstitution of Mb with monosubstituted protohemins, four reconstituted protein isomers are obtained, depending on the position (6 or 7 of the porphyrin ring) of the covalent modification and the mode of incorporation (‘up’ or ‘down’) of the prosthetic group into the protein. The ratio of these protein isomers is dictated by their relative stability and generally produces one major and one minor isomer for both 6- and 7-substituted derivatives, as it can be assessed by NMR [9].
In further developments, we produced site-directed mutants of sperm whale Mb containing those amino acid residues critical for peroxide activation in peroxidases. In particular, we replaced the distal Thr-67 with either Arg (T67R Mb) [56] or the more flexible Lys residue (T67K Mb) [29], and obtained a double mutant where, in addition to the Thr-67-Arg mutation, the proximal Ser-92 was substituted with an Asp residue (T67R/S92D Mb) [27]. The reactivity toward H2O2 is measured by the k1 rate constant for the formation of the intermediate compound I-like species. Such a parameter shows an appreciable increase for the modified Mbs (from the value of 760 ± 10 M−1 s−1 for wild type Mb up to 2100 ± 40 M−1 s−1 for the T67R/S92D Mb mutant) [27,29,56], indicating that the positively charged residue introduced into the modified Mbs favor the heterolytic cleavage of the bound hydroperoxide by a polarization effect analogous to that promoted by distal Arg in peroxidases. A greater increase in reactivity toward H2O2 was indeed obtained by Watanabe repositioning the distal His of Mb as it is in peroxidases [57], though further greater enhancement was observed for a quadruple variant of Mb generated through random mutagenesis (with a 24.7-fold increase in the k1 rate constant) [58]. In the latter case, the mutations were all peripheral and far from the heme pocket, showing that protein reactivity is also controlled by still incompletely understood factors associated with the folding and local flexibility of the polypeptide backbone.
We also reconstituted the mutants T67K Mb and T67R/S92D Mb with heme-His (obtaining the T67K Mb-H and T67R/S92D Mb-H protein derivatives, respectively), while with T67R Mb we could not obtain a stable reconstituted derivative. The kinetic studies on the catalytic oxidation of a representative phenolic substrate such as HPA by the various Mb derivatives demonstrated that the catalytic performance of Mb is enhanced in an additive fashion by engineering and cofactor reconstitution (Table 3) [28]. The electron transfer rate from the substrate to the protein intermediate compound II, for a given substrate, depends on (i) the redox potential of the ferryl species, which is likely increased by introduction into Mb of positively charged residues, and (ii) the mode of substrate interaction to the protein, which allows a closer approach to the heme especially in the case of the reconstituted proteins. The anionic substrate HPA carries a side chain with a charge complementary to that of the residues introduced in the heme environment of Mb and is therefore favored from the point of view of protein–substrate interaction. Among the Mb derivatives, the highest catalytic activity is exhibited by T67K Mb-H and T67R/S92D Mb-H (with the latter protein being the most efficient so far reported for any Mb derivative) because with these reconstituted proteins the substrate can reach the closest distance to the iron center and the most favorable arrangement for the electron transfer process [27,29].
Kinetic parameters for the catalytic oxidation of p-hydroxyphenyl propionic acid by hydrogen peroxide in the presence of the Mb derivatives
| Protein | kcat (s−1) | KM (mM) | kcat/KM (M−1 s−1) | References |
| WT Mb | 1.0 ± 0.1 | 72 ± 5 | 14 ± 0.4 | [56] |
| WT Mb-H | a | a | a | [29] |
| T67K Mb | 2.3 ± 0.2 | 28 ± 5 | 83 ± 9 | [29] |
| T67K Mb-H | 6.7 ± 0.4 | 26 ± 3 | 255 ± 18 | [29] |
| T67R Mb | 2.3 ± 0.1 | 12 ± 2 | 190 ± 25 | [56] |
| T67R/S92D Mb | 1.5 ± 0.6 | 20 ± 2 | 80 ± 6 | [27] |
| T67R/S92D Mb-H | 6.6 ± 0.3 | 20 ± 2 | 330 ± 20 | [27] |
a Biphasic kinetics.
The fact that active site engineering and cofactor modification are additive in enhancing the reactivity of heme proteins like Mb is promising in a broader perspective, particularly because the reconstitution approach can be extensively elaborated to introduce more sophisticated substrate recognition sites into the proteins.
Acknowledgements
This work was supported by the Italian MIUR, through a PRIN project, and the University of Pavia, through FAR.