

1 Introduction
A series of basidiomycete genomes have been completed during last years corresponding to plant and animal pathogens, and soil and mycorrhizal species, together with wood-rotting fungi. The latter have raised much interest in the search for peroxidases with industrial interest in bulk and fine chemistry, food and other sectors. They include the genomes of Phanerochaete chrysosporium [1] (the first basidiomycete genome to be sequenced) and Postia placenta [2], the recently published genome of Schizophyllum commune [3], and many others that are in different phases of the sequencing process (up to 53 only at the Joint Genome Institute, JGI, of the US Department of Energy, DOE) including those of Pleurotus ostreatus and Ceriporiopsis subvermispora (both to be published soon), Pycnoporus cinnabarinus (to be released) and Phlebiopsis gigantea.
Among the above basidiomycetes, several white-rot fungi from the genus Pleurotus have been described as being able to degrade lignin selectively [4]. The limited attack to cellulose makes them very interesting in different biotechnological applications related to the use of plant biomass, including the integrated lignocellulose biorefineries for the future production of chemicals, materials and biofuels. This fact, together with the understanding of the regulation of the ligninolytic system constituted by heme peroxidases and other enzymes [5], as well as the increasing interest in Pleurotus as an edible mushroom (with a world production near that of Agaricus bisporus [6]) were the main reasons to include a representative of this genus among the organisms to be sequenced by the DOE, in a project coordinated by Dr A.G. Pisabarro from the Public University of Navarre (Spain).
Ligninolytic peroxidases from P. ostreatus and other white-rot fungi have been widely studied over the last years [7–9]. However, only in the case of P. chrysosporium a complete catalog of these enzymes can be obtained from its genome sequence. On the other hand, new superfamilies of heme peroxidases, namely heme-thiolate peroxidases and dye-decolorizing peroxidases, exhibiting very interesting catalytic properties have been recently described in basidiomycetes [10]. Although their role in nature is still unknown, they have a very promising future as industrial biocatalysts due to the wide variety of aliphatic and aromatic compounds they are able to oxidize and oxygenate.
Herein we show the result of our participation in the search for genes encoding peroxidases in the genome of P. ostreatus. Different heme peroxidase genes models were identified, which could be grouped into different “classical” and “new” families and superfamilies of interest from a biotechnological point of view. At present, these heme peroxidases are being expressed in Escherichia coli and their stability and catalytic properties studied with the aim of determining their substrate specificity and real biotechnological potential.
2 Materials and methods
2.1 Fungal strains and P. ostreatus genome sequence
The JGI (DOE, http://www.jgi.doe.gov) has sequenced the genome of P. ostreatus (dikaryotic strain N001 deposited in the Spanish Type Culture Collection with the accession number CECT20600). The strategy followed consisted of sequencing the DNA of two monokaryons (PC9 and PC15 deposited in the aforementioned Type Culture Collection with the accession numbers CECT20311 and CECT20312, respectively) which had been previously obtained from the dikaryotic strain N001 [11]. The sequences of both monokaryons have been released to different collaborators after assembly by the Stanford Human Genome Center and subsequent annotation by JGI using custom analyses and the automated JGI Annotation Pipeline (genome assemblies v1.0 for PC9 and v1.0 and v2.0 for PC15). The 35.6 (PC9 v1.0) Mbp and 34.3 (PC15 v2.0) Mbp assemblies contain 572 and 12 nuclear scaffolds, and are predicted to have approximately 12206 and 12330 gene models, respectively. The significant differences in the number of scaffolds between both assemblies are a consequence of the sequencing method used for each monokaryon. The traditional Sanger sequencing technology was used for PC15, whereas a Sanger/454 pyrosequencing hybrid approach was used for PC9.
2.2 Screening and analysis of the heme peroxidase models
The search for heme peroxidase gene models in the genome sequence of both monokaryotic strains was performed at the Centro de Investigaciones Biológicas (CIB-CSIC). The process carried out to obtain the complete heme peroxidase inventory consisted of the following stages: (1) screening the automatically-annotated genome of P. ostreatus, monokaryons PC9 and PC15, using the Advanced Search option (“peroxidase” as search term) at the JGI web-site; (2) revising and manual curating (when necessary) the precise positions of introns, strongly affecting the deduced protein sequence and C-termini of the selected models; (3) comparing the amino acid sequence identities; and (4) confirming the presence of characteristic residues at the active center and substrate oxidation sites after homology modeling using crystal structures of reference proteins, deposited in the RCSB Protein Data Bank (PDB, http://www.pdb.org), as templates. The allelic variants of one dye-decolorizing peroxidase and three ligninolytic peroxidases genes previously cloned from P. ostreatus [12–15] were identified following this strategy, and used as references to validate the screening and analysis process. Programs used in this process were: BLAST (Basic Local Alignment Tool) at National Center for Biotechnology Information (NCBI) for searching nucleotide and protein databases; MEGA4 [16] for conducting automatic and manual sequence alignment during the heme peroxidase gene model search and curation process, and also for inferring phylogenetic trees; and SignalP 3.0 [17] for predicting the presence and location of signal peptides and cleavage sites. Theoretical molecular models of heme peroxidases were generated by using the programs implemented by the automated protein homology-modeling server SWISS-MODEL [18].
3 Results and discussion
3.1 Identification of heme peroxidase gene models in the P. ostreatus genome
Twenty-eight gene models were initially identified in the genome sequence of the P. ostreatus monokaryotic strain PC15 v1.0 (the first assembly released to collaborators) derived from the parental dikaryotic strain N001. However, eleven of these models were subsequently excluded by two main reasons: (i) seven of them (with identification numbers, ID#, 30148, 52526, 62301, 160045, 166150, 170674 and 173675) most probably do not correspond to heme protein genes, although they were automatically annotated as such due to the presence of short sequences reminiscent of heme protein conserved motifs; and (ii) four ones (IDs# 30776, 41714, 154272 and 170654) could correspond to heme proteins but they are not related to fungal peroxidase genes (the highest sequence identities were with oxygenases and cytochrome P450-type genes). The automatic annotation of the remaining seventeen heme peroxidase gene models was analyzed and subsequently their sequence manually improved in the PC9 v1.0 and PC15 v2.0 assemblies by modifying the intron positions and the deduced protein sequence and C-termini (affected by the intron changes) when errors were detected. Multiple nucleotide (DNA and cDNA) and amino acid sequence alignments with seventy basidiomycete and two ascomycete heme peroxidases were used for this task (protein sequence entries for the seventy peroxidases are given in the dendrogram of peroxidase evolutionary relationships present in Ruiz-Dueñas et al. [19]). The seventeen heme peroxidase gene models identified have the corresponding alleles in PC9 and PC15. All of them are summarized in Table 1, but only those corresponding to PC9 are hereafter cited in the text for simplifying the reading.
Summary of heme peroxidase gene models in the genome of P. ostreatus. Reference (ID #), scaffold number (Sc.), and position of the manually curated/annotated sequences in monokaryons PC9 (v1.0) and PC15 (v2.0); and model analysis including: (i) amino acid sequence identity of PC9 and PC15 alleles (Ide-PC9/PC15, %); (ii) name of best hit protein (after species abbreviation) and whole protein identity (Ide-Best hit, %); (iii) presence of ligninolytic peroxidase catalytic residues (Wc, Ea, Eb and Dc); and (iv) peroxidase type based on structural-functional classification.
| PC9 (v1.0) | PC15 (v2.0) | Analysis of the heme peroxidase models | ||||||||||||
| ID# | Sc. | Position | ID# | Sc. | Position | Ide-PC9-PC15 | Best hit | Ide-Best hit | Wc | Ea | Eb | Dc | Type | |
| 1. | 77045 | 3 | 583621-585384 | 1096819 | 5 | 734156-735921 | 99.7 | CC-CCP | 71 | − | − | − | − | CCP |
| 2. | 114464 | 1 | 3116892-3118095 | 1110336 | 1 | 1483346-1484549 | 99.7 | AB-CPO | 55 | − | − | − | − | CPO/APO |
| 3. | 123372 | 2 | 3213565-3214450 | 1111884 | 4 | 262016-262902 | 99.2 | AB-CPO | 27 | − | − | − | − | CPO/APO |
| 4. | 127284 | 7 | 560895-561684 | 1108212 | 9 | 535259-536048 | 98.8 | AB-CPO | 33 | − | − | − | − | CPO/APO |
| 5. | 87639§ | 7 | 191407-193574 | 62271§ | 9 | 213879-216064 | 98.6 | PO-DyP | 97 | − | − | − | − | DyP |
| 6. | 115057 | 2 | 2850334-2852370 | 1092668 | 4 | 657069-659105 | 99.8 | PO-DyP | 64 | − | − | − | − | DyP |
| 7. | 97865 | 6 | 2056440-2058411 | 52170 | 7 | 3196067-3198039 | 98.7 | PO-DyP | 62 | − | − | − | − | DyP |
| 8. | 117204 | 12 | 215278-217337 | 1069077 | 11 | 2524348-2526415 | 98.2 | BA-DyP | 44 | − | − | − | − | DyP |
| 9. | 137740§ | 3 | 1767677-1769279 | 1089546§ | 5 | 1981555-1983678 | 99.7 | PO-MnP | 99 | − | + | + | + | MnP |
| 10. | 137765 | 3 | 1625766-1627664 | 199511 | 5 | 1840285-1842183 | 99.7 | PSA-VPL | 76 | − | + | + | + | MnP |
| 11. | 137764 | 9 | 32682-34394 | 199510 | 2 | 1135915-1137627 | 100 | TV-MRP | 63 | − | + | + | + | MnP |
| 12. | 51713 | 12 | 2544215-2545903 | 1041740 | 5 | 2768778-2770466 | 100 | TV-MRP | 64 | − | + | + | + | MnP |
| 13. | 121638 | 1 | 2703196-2704913 | 1099081 | 1 | 1827850-1829567 | 100 | TV-MRP | 62 | − | + | + | + | MnP |
| 14. | 137757 | 8 | 536529-538627 | 1089895 | 6 | 1701105-1704309 | 100 | PSA-VPL | 98 | + | + | + | + | VPL |
| 15. | 137760§ | 2 | 3291830-3293748 | 1096331§ | 4 | 186162-188472 | 100 | PO-MnP | 99 | + | + | + | + | VP |
| 16. | 123383 | 2 | 3246008-3247917 | 156336 | 4 | 212622-214530 | 99.7 | PO-MnP | 78 | + | + | + | + | VP |
| 17. | 137766§ | 8 | 635476-637382 | 199491§ | 6 | 1618102-1620009 | 100 | PO-MnP | 99 | + | + | + | + | VPS |
During the automatic annotation process, the programs used by JGI identified canonical GT-AG splice sites (GT-AG introns), and only one noncanonical GC-AG site (GC-AG introns) which was located in the sequence of the putative dye-decolorizing peroxidase gene 87639 (see below for more details about this peroxidase superfamily). Additionally, two noncanonical GC-AG splice sites could be also manually predicted in the genomic sequence of both alleles of a putative manganese peroxidase (MnP) (137764) during the curation process after comparing the deduced cDNA sequences of all the heme peroxidase genes identified. This is not the first time this type of low frequent introns containing GC instead of GT at the 5’ splice site is identified in fungal genes. Their existence have been previously reported for both ascomycete and basidiomycete genes [20,21], including those encoding two oxalate-degrading enzymes from the white-rot fungi C. subvermispora and Dichomitus squalens [22,23]. On the other hand, the presence of a high number of introns (among 10 and 15) and the small size of some exons, several of them encoding for only three amino acid residues, resulted in very significant differences after manual curation of six of the nine sequences corresponding to ligninolytic peroxidases (sequences 9 to 17 in Table 1) (see below for more details about these sequences) compared with those initially included by JGI in the automatically annotated P. ostreatus gene catalog.
Once the sequences were manually curated, subsequent functional classification of the heme peroxidases was based on: (i) the use of BLAST scores (best hit proteins and whole sequence identities are shown in Table 1); and (ii) the 3D-modeling results. In order to define the gene family these heme peroxidases belong to, a structural-functional analysis was conducted. Identification of the gene family was based on the presence of conserved (active site and substrate oxidation sites) residues in molecular models built by homology modeling of the curated protein sequences. The structural models used as templates during this process were selected in function of the crystal structure availability of related heme peroxidases (see next subsections for each particular heme peroxidase). This approach allowed us to make a more precise definition of some of the automatic models, such as shown in the last column of Table 1 (e.g. several enzymes initially identified as MnP according to the best Blast hit score are putative versatile peroxidases (VP) and, conversely, a VP is in fact a MnP). After manual curation and annotation, new protein identification numbers were automatically assigned to the gene models by JGI (Table 1).
The seventeen heme peroxidase models from each monokaryon could be classified into four different groups, corresponding to classical families and new recently described superfamilies [10], as follows: (i) cytochrome c peroxidase (CCP) (1 model); (ii) ligninolytic peroxidases (9 models), including five MnPs and four VPs; (iii) heme-thiolate peroxidases (CPO/APO-type) (3 models); and (iv) dye-decolorizing peroxidases (DyP) (4 models). A dendrogram showing sequence relationships between these enzymes, and structural-functional classification of the ligninolytic peroxidases according to the presence of different catalytic sites in their theoretical molecular structure is shown in Fig. 1. Finally, an important question related to the presence or absence of lignin peroxidase (LiP) genes in the P. ostreatus genome has been solved. Their absence is definitively confirmed, as expected according to previous expression/production studies [24] and DNA hybridization analysis [25]. The heme peroxidase models identified in the P. ostreatus genome are described below considering the family they belong to, and some interesting aspects of these enzymes are discussed.
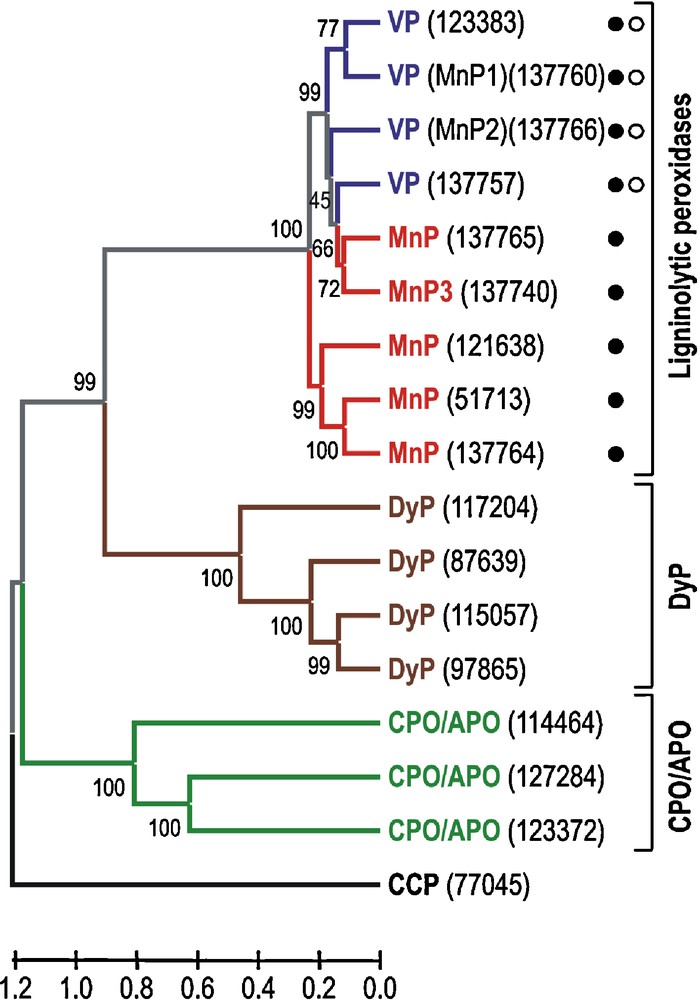
Dendrogram of seventeen deduced heme peroxidase sequences (accession numbers in parentheses) from the P. ostreatus genome (monokaryon PC9, v1.0) showing sequence relationships and structural-functional classification of the ligninolytic peroxidases (filled circle, Mn2+-oxidation site; and open circle, catalytic tryptophan). Sequence comparison as Poisson distances, and clustering using the UPGMA method and “pairwise deletion” option of MEGA4. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The corresponding allelic variants can be identified in the sequence of monokaryon PC15, v2.0 (Table 1).
3.2 Cytochrome c peroxidase (CCP)
CCP is a Class I low redox-potential enzyme of the superfamily of prokaryotic, fungal and plant peroxidases [26] with a His residue acting as the fith ligand of the heme iron. The only model of this family identified in P. ostreatus (77045) (Table 1) shows the highest amino acid sequence identity (71% of whole protein sequence) with the recently reported Coprinopsis cinerea CCP (NCBI XP_001830809) [27]. However, the theoretical structural model for this mitochondrial enzyme was obtained using the Saccharomyces cerevisiae CCP crystal structure (PDB entry 1CCP) as template, since this is the enzyme of this family exhibiting the highest amino-acid sequence identity (41%) whose crystal structure is available. Homology and crystal models of P. ostreatus and S. cerevisiae were superimposed and the rms distance between Cα measured. The low value obtained (0.26 Å, 256 residues computed) evidenced a high structural similarity between both proteins. Structurally, CCP is characterized, among others, by the heme distal and proximal residues (located above and below of the heme plane, respectively) similar to those found in ligninolytic peroxidases but showing two tryptophan, instead of phenylalanine, residues at both sides of the heme (see Fig. 2 for a comparison between CCP and ligninolytic peroxidases heme environments). As observed in Fig. 2A, the heme environment is completely conserved in the P. ostreatus CCP homology model. This fact strongly suggests a CCP-type activity for this enzyme. Among the different conserved amino acids, the proximal tryptophan has been described as a catalytically relevant residue in CCP. One of the two oxidizing equivalents of CCP compound I (generated after H2O2 enzyme activation) is stored in an intermediate protein radical centered on this amino acid residue [28] which would be involved in the electron transfer between the natural substrate cytochrome c and the enzyme [29].
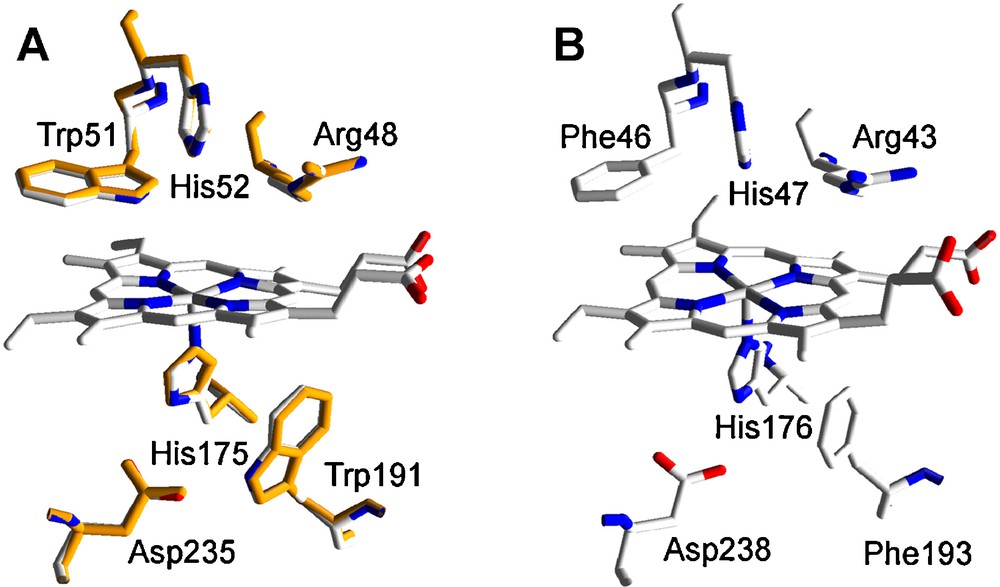
Heme environment in the homology model of P. ostreatus CCP obtained using S. cerevisiae CCP crystal structure (PDB entry 1CCP) as template: A. Structural alignment of the model obtained (orange, 77045) and 1CCP (CPK colors) (numbers refer to the amino acid residues in 1CCP). B. Equivalent residues in P. chrysosporium LiP (PDB entry 1LLP), as a model ligninolytic enzyme (MnP and VP present the same residues, here shown, in their molecular structure).
3.3 Ligninolytic peroxidases
Ligninolytic peroxidases are Class II high redox-potential enzymes of the above-mentioned superfamily of prokaryotic, fungal and plant peroxidases. They are secretion enzymes able to oxidize, directly or through mediators, the different units forming the lignin aromatic polymer as well as other aromatic compounds (such as polycyclic aromatic hydrocarbons, pesticides and dyes) [5,30]. Members of the three families of ligninolytic peroxidases (LiP, MnP, and VP) share an almost identical heme environment (Fig. 2B), responsible among others for their high redox potential [31,32], but differ in the substrates they can oxidize as consequence of the presence of different catalytic sites in their molecular structures. So, LiP oxidizes non-phenolic lignin model compounds in direct contact with a tryptophan radical exposed to the solvent [33,34], whereas MnP is very specific oxidizing Mn2+ in a small channel formed by three acidic residues and located directly on the heme internal propionate [35]. VP shares catalytic and structural properties with LiP and MnP. Furthermore, it is able to directly oxidize low redox potential substrates, including phenols, as well as high redox potential dyes that are oxidized by LiP only in the presence of some mediators [9].
Most changes with respect to the best gene model offered by the automatic annotation of the P. ostreatus genome by JGI corresponded to the nine ligninolytic peroxidase (five MnPs and four VPs, Table 1) models identified due, among other reasons, to the abundant information available on structure-function and molecular biology of these peroxidases of biotechnological interest [5,7–9,36,37]. Analyzing in detail the MnP models, it was observed that all of them correspond to the subfamily of “short” MnPs, recently described for the first time in Phlebia radiata [38], in comparison to the typical “long” MnPs (including an extra C-terminal extension in their amino acid sequence) from the model ligninolytic fungus P. chrysosporium. “Long” MnPs are specific for Mn2+, whereas “short” MnPs are also able to oxidize phenols, amines and ABTS in the absence of Mn2+. This fact suggests relative wide substrate specificity for the five MnPs identified in the P. ostreatus genome. Among these, three models (137764, 51713 and 121638) show the highest amino acid sequence identity (62–64% of the whole protein) with the so-called manganese-repressed peroxidase from Trametes versicolor (GenBank AAB63460) and are grouped together in the dendrogram of Fig. 1. By contrast, the other two MnP models (137740 and 137765) are clustered with VP models. One of them (137740) seems to be an allelic variant (differing in only two amino acid residues, one of them in the signal peptide) of the already described and cloned P. ostreatus MnP3 (GenBank BAA33449) [14], whereas the other one (137765) has the highest amino acid sequence identity (76% of the whole protein) with the unpublished VP (putative VPL isoenzyme) of Pleurotus sapidus (GenBank CAJ01576) although the catalytic tryptophan is absent and, therefore, it is classified as MnP.
Regarding VP, two models (137760 and 137766) seem to be allelic variants (differing in only 1-2 amino acids of the whole sequence) of the previously cloned and described P. ostreatus MnP1 (GenBank AAA84396) [12] and MnP2 (GenBank CAB51617) [13], respectively. The first one is a putative VP due to the presence of the catalytic tryptophan, whereas the second one has already been demonstrated to be a VP by its ability to act on Mn2+, and directly oxidize 3,4-dimethoxybenzyl (veratryl) alcohol and polymeric azo dyes at an exposed tryptophan [39]. A third model (123383) shows the highest amino acid sequence identity (78% of the whole protein sequence) with P. ostreatus MnP1 (the putative VP mentioned above), whereas the fourth model (137757) has high identity (98% of the whole protein sequence) with the aforementioned unpublished VP of P. sapidus. The allelic variant of this model in PC15 v2.0 (1089895) presents a stop codon in the middle of the sequence, suggesting it is a putative VP pseudogene. A sequencing mistake should be ruled out considering this codon appears in the two PC15 assemblies (v1.0 and v2.0). The amino acid sequence analysis of the above four sequences in PC9, three in PC15 due to the putative pseudogen, revealed the presence of both residues forming the manganese binding site and the catalytic tryptophan, typical from VP [9].
The subsequent analysis of the theoretical structural models of P. ostreatus MnP and VP isoenzymes, built using the wild type P. chrysosporium MnP and P. eryngii VP crystal structures (PDB entries 1YYD and 3FJW, respectively) as templates, revealed that these peroxidases maintain a well conserved tertiary structure. The homology models not only confirmed the typical predominant α-helical structure of the superfamily of plant, fungal, and bacterial peroxidases, but also the location of key amino acid residues. So, MnP isoenzymes exhibit the characteristic Mn2+ binding site formed by one aspartic and two glutamic acids (Fig. 3A), responsible for Mn2+ oxidation, whereas VP theoretical models include both the Mn2+ binding site and the tryptophan residue exposed to the solvent at the same position of that involved in oxidation of high redox potential substrates by P. eryngii VP (Fig. 3B) [9].

Axial view of the heme region in nine P. ostreatus ligninolytic peroxidase homology models obtained using P. eryngii VP and P. chrysosporium MnP crystal structures (PDB entries 3FJW and 1YYD, respectively) as templates: A. MnP-type models including a putative Mn2+-oxidation site (right) formed by two glutamic and one aspartic acidic residues (grey, 1YYD, E35/E39/D179; pink, 137764, E36/E40/D181; light blue, 51713, E36/E40/D181; red, 121638, E36/E40/D181; orange, 137765, E36/E40/D175; and green, 137740, E36/E40/D175). B. VP-type models including an exposed tryptophan (oxidizing high redox-potential substrates) in addition to the Mn2+-oxidation site described in A (grey, 3FJW, W164/E36/E40/D175; dark blue, 137766, W170/E36/E40/D181; light blue, 123383, W164/E36/E40/D175; green, 137760, W165/E36/E40/D176; and orange, 137757, W164/E36/E40/D175). The amino acid numbering showed above refers to putative mature sequences.
3.4 Heme-thiolate peroxidases (CPO/APO-type)
Heme-thiolate peroxidases have been described as secretion enzymes characterized by the presence of a cysteine residue acting as the proximal axial ligand of heme. CPO from the ascomycete Leptoxyphium fumago has been the only heme-thiolate peroxidase known, and exhaustively characterized, since its discovery in 1961 [40] until recently. Lately a large amount of sequences for putative heme-thiolate chloroperoxidases have been accumulated from the analysis of ascomycete and basidiomycete genome sequences, many of them being included in PeroxiBase (http://peroxibase.toulouse.inra.fr) [41]. Recently, a second heme-thiolate peroxidase described as an aromatic peroxygenase (APO) has been reported in agaric basidiomycetes, the APO from Agrocybe aegerita being the best characterized [42,43]. The CPO and APO wide substrate specificity, catalyzing halogenation reactions and sharing catalytic properties with peroxidases, catalases and cytochrome P450 monooxygenases (APO even oxygenating aromatic substrates), make these enzymes very interesting from a biotechnological point of view [44]. Amino acid sequence, catalytic activity and structure of these two heme-thiolate peroxidases differ from those of classical heme peroxidases, and they have been lately considered as members of the new heme-thiolate peroxidase superfamily [10].
Three heme-thiolate peroxidase gene models were identified in the P. ostreatus genome (sequences 2 to 4 in Table 1), being the first time this type of heme peroxidases is described in a fungus from the genus Pleurotus. All of them show the highest amino acid sequence identity with the unpublished putative CPO of A. bisporus (GenBank CAC03461). The identity values range from 55% of model 114464 (290 amino acids long) to 27–33% of models 123372 (255 amino acids long) and 127284 (242 amino acids long) for the whole protein sequence. Unlike the intron-less CPO gene of L. fumago, the three P. ostreatus gene models contain introns (3, 2 and 1 introns, respectively), as also observed for the sixteen genes encoding putative CPOs (fifteen genes containing four introns and one gene containing only one intron) in the recently sequenced genome of A. bisporus var bisporus (H97) v2.0 (JGI, DOE) and the apo1 gene (five introns) encoding the A. aegerita peroxygenase [45]. Surprisingly, only one (123372) of the three models identified in P. ostreatus seems to be clearly a secretory protein (signal peptide probability 0.987 for the allelic variant 1111884 in PC15) with a putative 23 amino acid signal peptide and the corresponding cleavage site, according to the scores reported by SignalP 3.0. The probability of the presence of a signal sequence in model 114464 is very low (0.334) and zero for model 127284. These data suggest a different cellular localization or secretion pathway for these three P. ostreatus heme-thiolate peroxidases, which would be also independent of a C-terminal propeptide (absent in their amino acid sequences), unlike what happen with L. fumago CPO, where a 21 amino acid N-terminal signal peptide and a 52 amino acid C-terminal propeptide, the latter probably having a chaperone-like function, have been identified [46].
Although the amino acid sequence identities with two model enzymes of this new superfamily, A. aegerita APO [45] and L. fumago CPO [47], were very low (only around 20% in both cases), theoretical molecular models for the three putative P. ostreatus heme-thiolate peroxidases were obtained using the crystal structure of the last one (PDB entry 1CPO) as template. This was because CPO of L. fumago is the only heme-thiolate peroxidase whose molecular structure has been experimentally solved [48] and it is available in PDB. Crystallization of A. aegerita APO has been recently reported [49] but its structure has not been yet published nor deposited in the PDB. Molecular models built for heme-thiolate peroxidases 123372 and 127284 fitted quite well with the whole crystal model of L. fumago CPO, and only the 10-11 N-terminal and 15-26 C-terminal amino acids, respectively, were not included in the final theoretical models. Both homology models were superimposed on the crystal model of L. fumago CPO. The rms distances between Cα were 0.37 Å with 123372 (194 amino acid residues computed of a total of 232 corresponding to the predicted mature protein) and 0.35 Å with 127284 (195 residues computed of a total of 242). This indicates high structural affinity taking into account, for example, that the rms distance between LiP isoenzymes H2 and H8 of P. chrysosporium is 1.1 Å [50]. By contrast, the structural homology model obtained for 114464 excluded not only the first 39 amino acid residues of the sequence but also the C-terminal 145 residues representing more than half of the amino acid sequence of the whole protein. These amino acid residues could not be automatically modeled and they were not considered in the final model. The modeled sequence of the remaining 102 amino acids fitted well with the equivalent region of L. fumago CPO, the rms distances between Cα being 0.32 Å. This region contains both the most relevant residues of the proximal side of the heme group, including the cysteine residue acting as the fifth ligand of the heme iron, as well as ligands for a Mn2+ ion described in the L. fumago CPO crystal structure. These data suggest that a structural “core” containing the heme active site is conserved in the three P. ostreatus heme-thiolate peroxidases and L. fumago CPO, even though the low percentage of amino acid sequence identity presented.
A detail of the heme environment in the three P. ostreatus heme-thiolate peroxidase homology models and the crystallographic model of CPO from L. fumago is shown in Fig. 4. Cysteine serving as axial heme ligand and surrounding residues stabilizing the cysteine-ligand loop by hydrogen-bonding are conserved at the proximal side. The only significant difference among them is the presence of an Ala and a Cys residues contiguous to the axial Cys in 114464, which could be reinforcing the hydrogen-bonds structure at this site. On the other hand, the theoretical models show the same three amino acid residues (Glu, Ser and His) responsible for cation coordination in CPO, and also include those present at the distal heme pocket. Differences at this region between the three models, and also with CPO, suggest different catalytic properties (activation rate and mechanism, substrate specificity, etc.) for these P. ostreatus hemo-thiolate peroxidases, since residues at this side in CPO have been involved in both enzyme activation by H2O2 (Glu183 acting as acid-base catalyst in the peroxide O-O bond cleavage mechanism, assisted by His105 and Asp106) and substrate binding (including, among others, Phe103, Phe186 and Asn74) [48,51,52]. Although catalytic differences can be suggested from this in silico analysis, these are only speculations. Homologous or heterologous expression of these P. ostreatus heme-thiolate peroxidases is necessary to determine their final catalytic properties, and definitively identify the heme-thiolate peroxidase family (CPO or APO) they belong to.
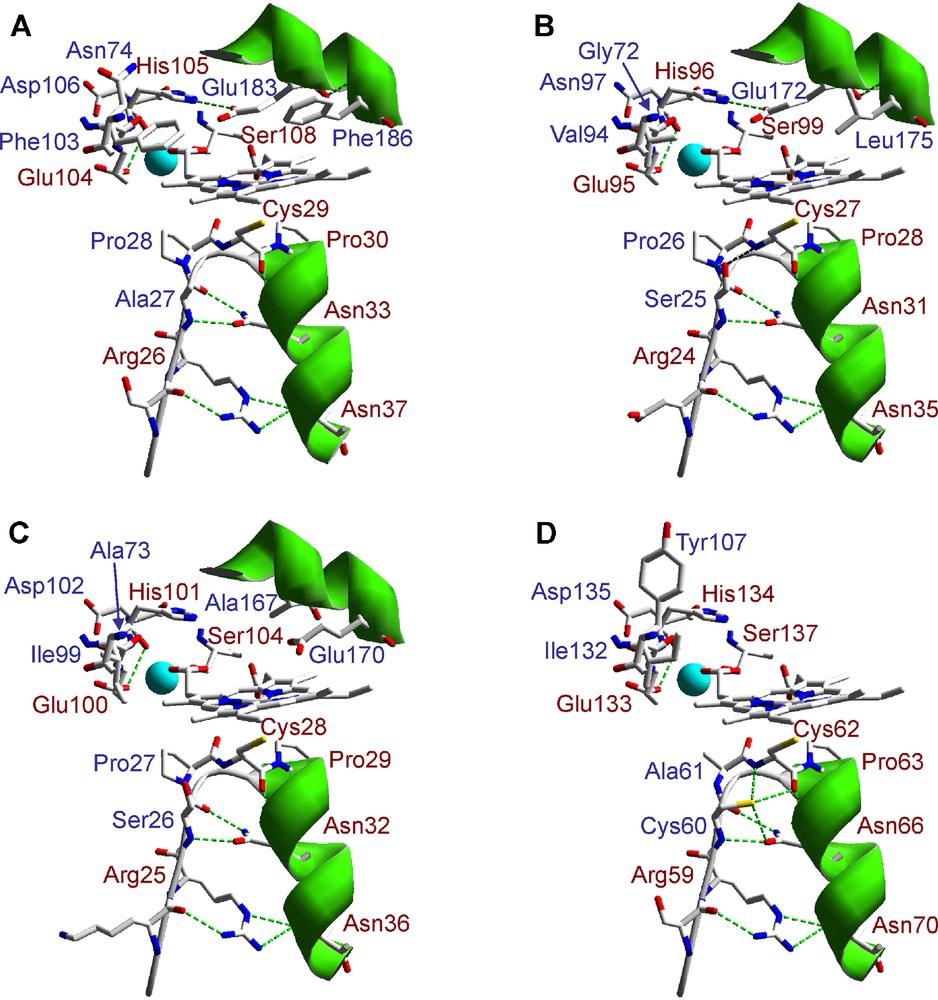
Lateral view of the heme region in three P. ostreatus heme-thiolate (CPO/APO) peroxidase homology models obtained using L. fumago CPO crystal structure (PDB entry 1CPO) as template. Conserved amino acid residues in the crystal and homology models at the heme proximal and distal sides, including those involved in the cation (Mn2+) (light blue sphere) binding, are shown in red color (A: 1CPO; B: 123372; C: 127284; and D: 114464). The amino acid numbering refers to putative mature sequences.
3.5 Dye-decolorizing peroxidases (DyPs)
DyP-type peroxidases constitute a new superfamily of heme peroxidases widespread in fungi and bacteria [53]. The first peroxidase giving name to this superfamilly was purified and characterized from cultures of the white-rot basidiomycete Bjerkandera adusta [54], although it appears in the bibliography as Thanatephorus cucumeris Dec 1 DyP due to a molecular misidentification.1 As previously described for ligninolytic and heme-thiolate peroxidases, fungal DyPs are enzymes with a high biotechnological interest. They exhibit a significant catalytic versatility due to its ability to bleach different dyes, including high redox potential anthraquinone derivatives not oxidized by other peroxidases, oxidize phenols and cleave carotenoids [53,57,58]. Additionally, DyPs have demonstrated to be very robust enzymes. So, isoform AjPI of Auricularia auricula-judae maintains 100% activity at pH 2.5 for 4 h [59] and MsP1 of Marasmius scorodonius is able to withstand high temperatures (up to 70 °C, with the optimum between 55 and 60 °C) and works at high pressure, even better than at atmospheric pressure (2-fold activity increase at 500 bar) [60]. Although the role of these enzymes in nature is still unknown, it has been suggested that some DyPs, e.g. those from A. auricula-judae, could be involved in lignin biodegradation according to their ability to oxidize methoxylated aromatics and lignin model compounds, as veratryl alcohol and a β-O-4 nonphenolic dimer representative of the most frequent substructure present in the lignin polymer [59]. The same authors have suggested that DyPs may supplement or replace the classical high-redox potential ligninolytic peroxidases (LiP and VP). In favour of the idea of peroxidase complementarity, Sugano et al. [61] have reported complete decolourization of an anthraquinone dye as result of a concerted action of DyP and VP from the above B. adusta strain (that they misidentified as T. cucumeris).
Up to four different models of this superfamily were identified in the genome sequence of P. ostreatus (Table 1). Model 87639 from PC9 v1.0 and the corresponding 62271 from PC15 v2.0 seem to be alleles (98.4% and 98.6% nucleotide sequence identities, respectively) of the already cloned and described DyP from P. ostreatus (GenBank CAK55151) [15]. The amino acid sequences of these three allelic variants are highly similar, that of 62271 differing in only one residue (located in the signal peptide) from the previously published sequence. Two more models (115057 and 97865) have the highest amino acid sequence identity (67–69% for the whole protein) with model 87639 mentioned above, all of them containing nine introns. By contrast, model 117204 has the highest identity (only 38% of the whole protein sequence) with DyP (GenBank BAA77283) cloned from a B. adusta strain (formerly misidentified as T. cucumeris), and differs significantly from the others from P. ostreatus in intron number (only seven compared with the nine introns observed in the other three DyP models) and position. Surprisingly, models 97865 and 117204 do not contain signal sequences guiding these peroxidases in their transit through endoplasmic reticulum and Golgi apparatus suggesting, as in the case of the heme-thiolate peroxidases mentioned above, different cellular locations or secretion pathways for these peroxidases.
Homology models for DyPs of P. ostreatus were built using the B. adusta DyP (reported as T. cucumeris DyP) crystal structure (PDB entry 2D3Q) as template. The rms distance between Cα of the crystal model and the four homology models (ranging from 0.34 to 0.44 Å and covering from 83 to 90% of the amino acid sequences) revealed a high structural similarity among them, as in the case of the other peroxidases previously analyzed. The theoretical models present characteristic β-barrel tertiary structure (Fig. 5A) and heme environment (Fig. 5B). This includes proximal histidine acting as the fifth heme iron ligand, and conserved arginine and aspartate residues involved in enzyme activation by H2O2 at the distal side of heme, instead of typical histidine and arginine residues present in classical fungal heme peroxidases (Fig. 2). Unfortunately, not much more about structure-function relationships of these peroxidases is known.
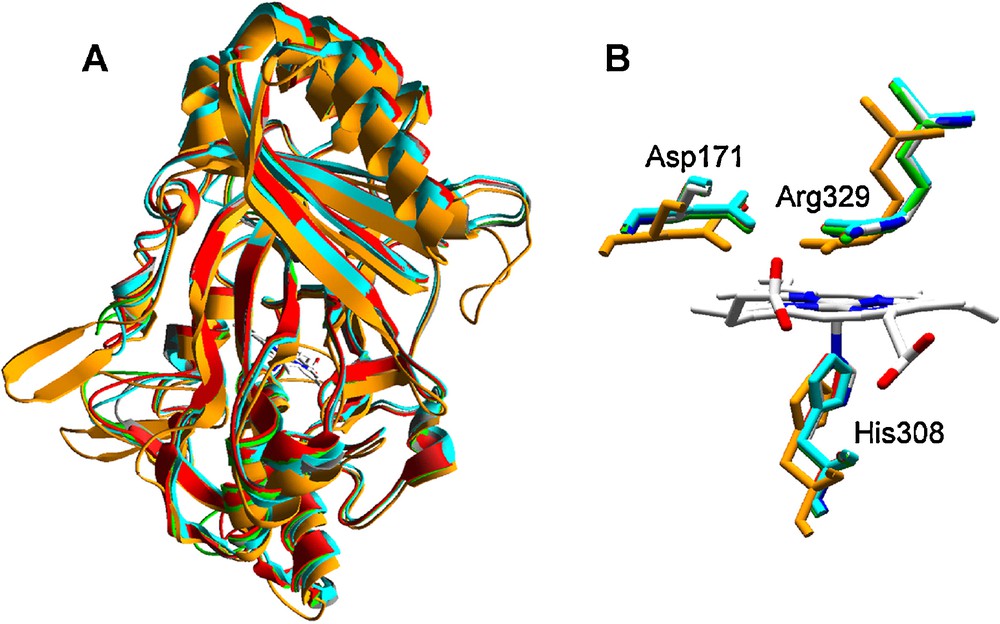
Homology models of four P. ostreatus DyP-type peroxidases obtained with B. adusta DyP (reported as T. cucumeris DyP) crystal structure (PDB entry 2D3Q) as template: A. Superimposition of ribbon-type molecular models (grey, 2D3Q; green, 87639; red, 115057; light blue, 97865; and orange, 117204) with the central heme as CPK bars. B. Detail of the heme region (lateral view from the propionate side) showing conserved histidine (heme axial ligand at the proximal side) and aspartic acid and arginine residues at the heme distal side (colors as in A but 2D3Q in CPK colors). The numbers of the conserved histidine, aspartic acid and arginine residues in 2D3Q (shown in figure) and the putative mature sequences of the different P. ostreatus models (2D3Q/87639/115057/97865/117204) are the following: H308/H356/H351/H343/H334, D171/D213/D208/D200/D196, and R329/R377/R372/R364/R360.
4 Conclusion
As a result of the present work, a complete inventory of ligninolytic peroxidase, including VP and MnP, genes is provided and the absence of lip genes in the genome of P. ostreatus is definitively demonstrated. Heme-thiolate peroxidase genes initially represented by the L. fumago CPO and related enzymes, but also including APO recently described in several agaric basidiomycetes with very interesting catalytic properties (e.g. aromatic oxygenation), are described for the first time in Pleurotus. Moreover, one already identified DyP-type peroxidase is confirmed and more genes of this family are reported. On the other hand, the analysis of the amino acid sequences and structural homology models obtained for these enzymes reveal certain differences, not only among the members of different peroxidase super/families, but also among the members of the same super/family. In this sense, cellular localization and secretion pathways seem to be different among CPO/APO-type peroxidases and also among DyP-type peroxidases. Moreover, small sequence and structural details could determine differences in the catalytic properties of these heme peroxidases. A good example of this refers to the ligninolytic peroxidases, since it is possible to distinguish among LiP, MnP and VP on the basis of the presence or absence of only one or a few amino acid residues. After publishing the APO crystal structure it will be possible to determine differences between this and CPO, and then determine whether the P. ostreatus heme-thiolate peroxidases correspond to the CPO or APO type enzymes.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
This study has been supported by the EU projects BIORENEW (NMP2-CT-2006-026456) and PEROXICATS (KBBE-2010-4-265397) and the Spanish project BIO2008-01533. The work conducted by the US Department of Energy Joint Genome Institute is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231. Antonio G. Pisabarro (Public University of Navarre) is acknowledged for coordinating the P. ostreatus genome project, and Yasushi Sugano (Tokyo Institute of Technology) and Walter Buzina (Medical University of Graz) for their collaboration in the reappraisal of the molecular identification of the DyP-producing B. adusta strain (formerly reported as T. cucumeris Dec 1). E.F. thanks CSIC for a JAE fellowship and F.J.R.-D. thanks the Spanish MICINN for a Ramón y Cajal contract.
1 This strain was first described as Geotrichum candidum [55] and reidentified as Thanathephorus cucumeris (the sexual state of Rhizoctonia solanii) based on ITS-5.8S rDNA sequence identity with GenBank AF455461 (Y. Sugano, personal communication). The latter corresponds to a medical strain that had been identified by ITS sequence identity with AJ000198 (W. Buzina, personal communication). GenBank AJ000198 was at the origin of the above and other misidentifications (over 20 rDNA GenBank entries) of B. adusta strains as T. cucumeris (or R. solanii). It corresponds to a fungus that had been misidentified as R. solanii by classical methods and, although the comparison of rDNA sequences showed that it did not cluster with R. solanii and related species [56], its rDNA sequence was deposited in GenBank with the erroneous name. However, this fungus, as well as the above Dec 1 and medical strains, show high rDNA sequence identity with authentic B. adusta cultures (such as GenBank Y089741) indicating that the DyP-producing fungus, initially described as T. cuccumeris, must be referred in the future as B. adusta.