

1 Introduction
Several oxidases and oxygenases contain two cooperating copper ions within their active site [1]. The two proximate metal ions serve to activate the kinetically inert O2, and their combined redox power is used to mediate and to control the multi-electron redox reactions. Among the most prominent examples of these so-called type 3 copper centers is catechol oxidase, which catalyzes the two-electron oxidation of ortho-diphenols to the corresponding quinones [2]. The X-ray crystal structure analyses of catechol oxidase from sweet potatoes revealed details of the enzyme structure in different redox states and in its complex with bound inhibitor, with both copper ions found in N-donor (histidine) ligation [3]. Based on these structural findings as well as on previous spectroscopic and biochemical evidences, a plausible mechanism has been proposed for the catecholase activity [4] that includes activation of O2 as a peroxide at the dicopper site. The two-electron substrate oxidation occurs by means of the cooperative action of the two copper ions, as each individual copper ion undergoes a one-electron redox shuttle between the +I and +II states during turnover. However, details of the mechanism are still far from clear, and several different scenarios are discussed in the literature [4–7].
In view of the great importance of oxidation reactions for industrial and synthetic processes, and in view of the ongoing search for new and efficient oxidation catalysts, it is of paramount interest to unravel the basic functional principles that govern such bimetallic reactivity of natural enzymes and their synthetic analogues [8]. Various mono- and dinuclear copper coordination compounds have thus been investigated as biomimetic catalysts for catechol oxidation [6,9]. Parameters that determine the activity of the synthetic analogues comprise, inter alia, the metal–metal separation [10–12], the redox properties [13,14], the identity of exogenous bridging ligands [15], the structure of the dinucleating ligand scaffold [12,16,14,17], and the pH [13,18–20]. No clear relation between the catalytic activity and the redox potential of the copper species has yet emerged, which may be explained by the manifold steps that are involved in the overall catalytic cycle [21]. Dinuclear copper complexes are generally found to be more reactive than mononuclear compounds, and a steric match between the dicopper site and the substrate is assumed to be advantageous [11,12,22]. Within a series of O-atom bridged dicopper(II) complexes, those complexes with a metal–metal distance closest to that of the met form of the enzyme (2.9 Å) displayed the best catalytic performance [11]. In addition, some correlations between the observed rates of reactivity and ligand architecture, copper coordination environment or preorganization of the metal ions have been observed for particular classes of complexes [6]. However, a more general understanding of structure–reactivity patterns that might allow for a rational design of catalytically most active copper complexes is still lacking. Further studies of well defined and tunable dicopper complexes are obviously needed to gain deeper insight into these copper-mediated substrate oxidations and to finally unravel the parameters that determine bimetallic reactivity both in natural metalloenzymes and in small synthetic catalysts.
In this contribution we extend our investigations of the catecholase activity of highly preorganized pyrazolate-based dicopper complexes [12]. The primary intention is to elucidate trends in reactivity that might be valuable for the use of this class of complexes in oxidation catalysis in a broader sense. To this end the pyrazolate-derived compartmental ligand scaffolds offer the attractive option of targeted modulation of electronic and geometric characteristics of the bimetallic core by changes of the ligand side arms attached to the 3- and 5-positions of the heterocycle. Important variations comprise the type and number of side arm donor atoms as well as the lengths of the chelate arms. While the former allows to modify coordination numbers and electronic structures of the metal ions, the latter controls the properties of the bimetallic pocket, including the metal–metal distance [23–25].
An assortment of four dicopper complexes 1–4 (Scheme 1) had previously been evaluated in the catecholase reaction using 3,5-di-tert-butylcatechol (DTBC) as the test substrate, and drastic differences in activity became apparent from the kinetic data [12]. Among the set of complexes, those with shorter Cu⋯Cu distances (1 and 2) showed much higher activity than the systems with larger Cu⋯Cu separations (3 and 4), although other parameters are certainly relevant as well.
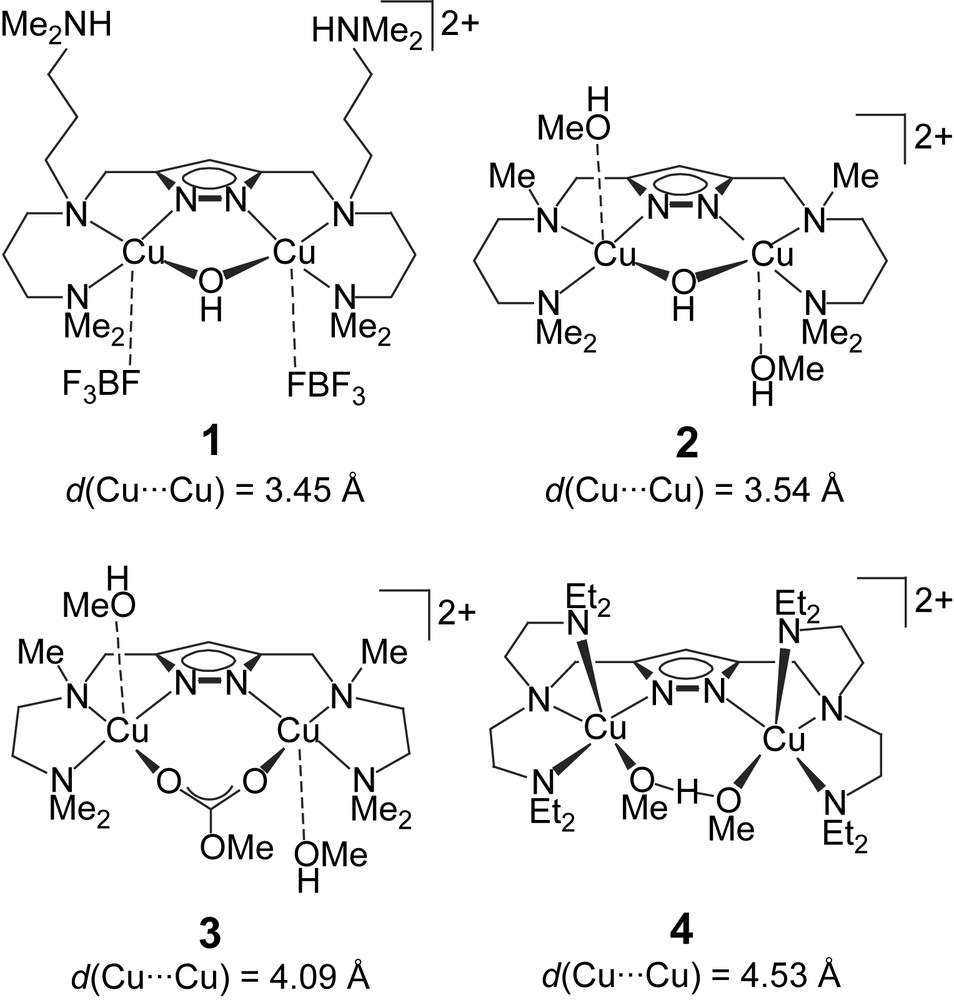
Dicopper(II) complexes tested previously as functional models for the active site of catechol oxidase [12].
We now include some new dicopper(II) complexes of pyrazole-derived ligands HL1–HL4 that provide either pyridyl-containing or triazacyclononane (tacn) side arms (Scheme 2) [24,25]. While the metal–metal distance in 1–4 is controlled by the length of the spacer between the side arms N-donors, a similar effect can be achieved by modifications of the spacer between the pyrazole ring and the side arm (i.e., the “shoulder” of the ligand framework). For example, dinickel(II) complexes of [L3]− and [L4]− were shown to feature a bridging hydroxide at d(Ni⋯Ni) = 3.50 Å in [L4Ni2(OH)]2+ versus an O2H3 bridge at d(Ni⋯Ni) = 4.48 Å in [L3Ni2(O2H3)]2+ [24]. Both HL3 and HL4 can thus be expected to provide high stability constants to their dicopper complexes due to the tacn side arms, but with very different Cu⋯Cu separations. At this stage of investigation the focus lies on the search for empirical structure–activity correlations, neglecting mechanistic details that most certainly are distinct for every individual system.
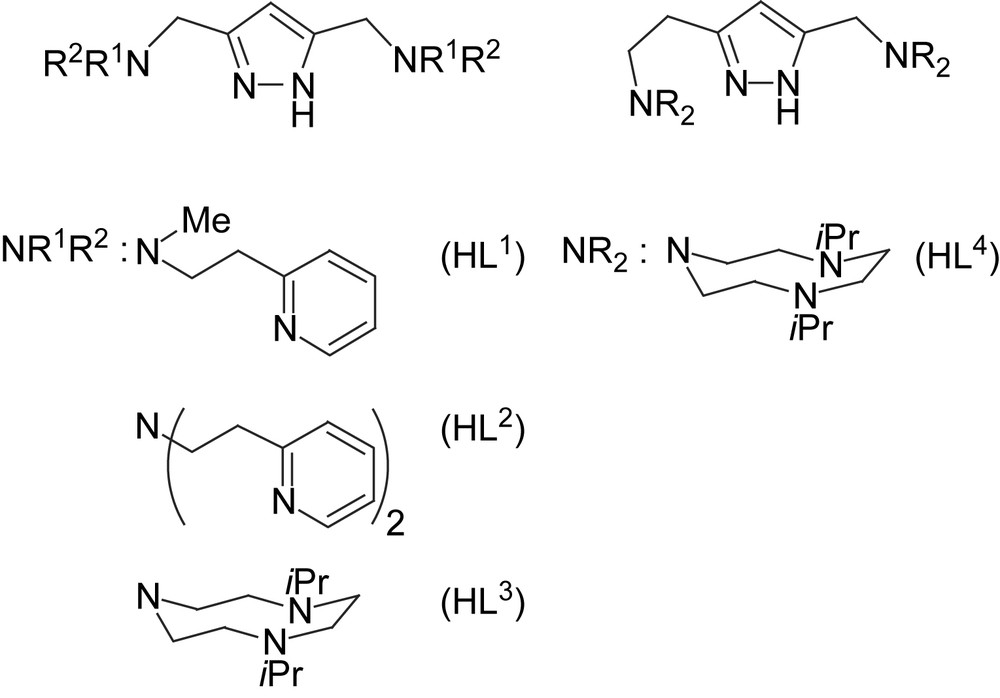
Pyrazole ligands used in this work.
2 Results and discussion
2.1 Synthesis and structural characterization
Reaction of the ligand HL1 with 1 equivalent of base (KOtBu) and 2 equivalents of Cu(NO3)2·3H2O affords complex [L1Cu2(MeOH)2(NO3)2]NO3 (5a). Its d–d-transition at 635 nm (ɛ = 290 mol l−1 cm−1) in methanol solution suggests the presence of tetragonal copper(II), which is confirmed by the single-crystal X-ray analysis. The molecular structure of the cation is depicted in Fig. 1, together with selected atom distances and bond angles. Complex 5a contains two copper ions in distorted square-pyramidal environment (τCu1 = 0.11, τCu2 = 0.38)1 [26], with the three N-atoms of the respective [L1]− compartment and a nitrate-O forming the basal plane. A methanol ligand occupies the apical position at long (Jahn–Teller) distances of >2.2 Å, and another O-atom of the semi-chelating nitrate forms an axial contact from the backside at >2.6 Å to complete an overall {4 + 1 + 1} coordination. Interestingly, the Cu-bound methanol molecules are involved in hydrogen bonding to the nitrate-O that ligate the adjacent Cu center, which results in double (Me)O–H⋯O(NO2) linkages between the two metal ions (dO⋯O = 2.75/2.76 Å). This arrangement forces the copper ions rather far apart (dCu⋯Cu = 4.36 Å) and displaces them quite drastically out of the pyrazolate plane, resulting in a severe torsion Cu–N–N–Cu of 35.1°. Pyrazolate-derived ligands with long side arms that give six-membered chelate rings such as in 1, 2, and 5a have been anticipated to suit well bimetallic complexes with shorter metal–metal separations of 3.5–3.9 Å, but longer separations appear to be easily accessible. Solvent insertion similar to the present case has previously been observed for dicopper(II) complexes of [L1]− with intramolecular (H)O–H⋯F bridges [25]. A dominant signal in the FAB mass spectrum of 5a at m/z = 613 characteristic for the ion [L1Cu2(NO3)2]+ suggests, however, that the methanol molecules are readily lost under reduced pressure.
In addition to 5a, a further complex [L1Cu2F(MeOH)2(BF4)]BF4 (5b) with the same pyrazolate ligand was employed in this work. The molecular structure of 5b has been determined previously [25] (Scheme 3). It also features a dicopper core based on [L1]− with a Cu⋯Cu distance of around 4.3 Å (4.290(1) Å), but a single (Me)OH⋯F bridge is now found within the bimetallic pocket. The fluoride ion has obviously been abstracted from one of the BF4− counteranions. Both metal ions exhibit an almost perfect square-pyramidal coordination geometry (τCu1 = 0.09, τCu2 = 0.01), where the apical positions are occupied at rather long distance by either a methanol molecule (dCu2–O2 = 2.271(2) Å) or a fluoride atom of a BF4− anion (dCu1–F2 = 2.602(2) Å). In methanol solution, 5b shows a visible absorption at 645 nm (ɛ = 200 mol l−1 cm−1) in accordance with square-pyramidal coordination.
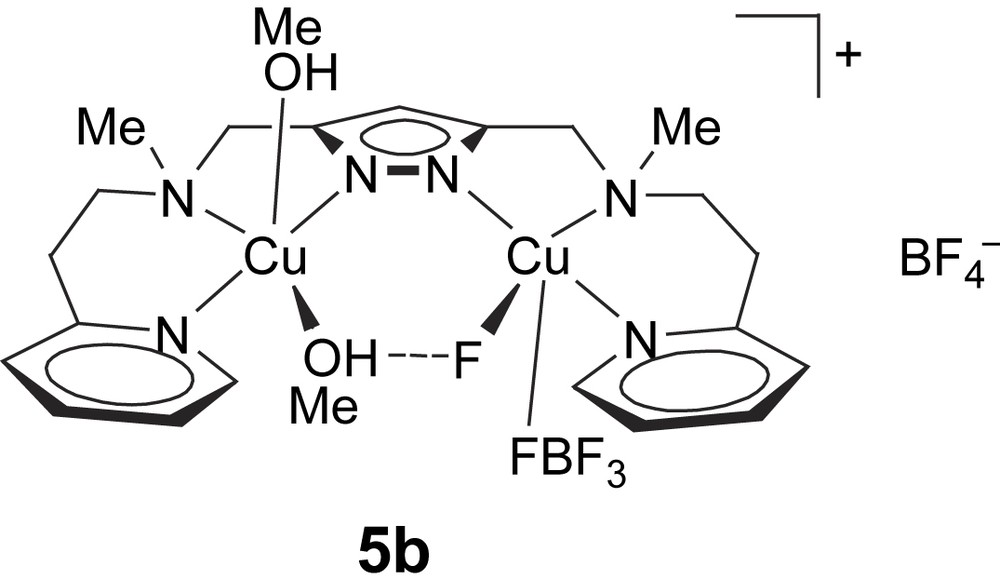
Complex [L1Cu2F(MeOH)2(BF4)]BF4 (5b).
Dicopper complexes of ligand [L2]− that contains additional donor side arms were obtained from HL2, 2 equivalents of base (KOtBu) and 2 equivalents of either Cu(ClO4)2·6H2O or Cu(CF3SO3)2, giving [L2Cu2(OH)](ClO4)2 (6a) or [L2Cu2(OH)](CF3SO3)2 (6b), respectively. As shown by X-ray crystallography both complexes, 6a and 6b, feature very similar cations in the solid state. The molecular structure of the cation of 6b is depicted in Fig. 2, along with selected atom distances and bond angles. Two independent but structurally similar molecules are found per unit cell.
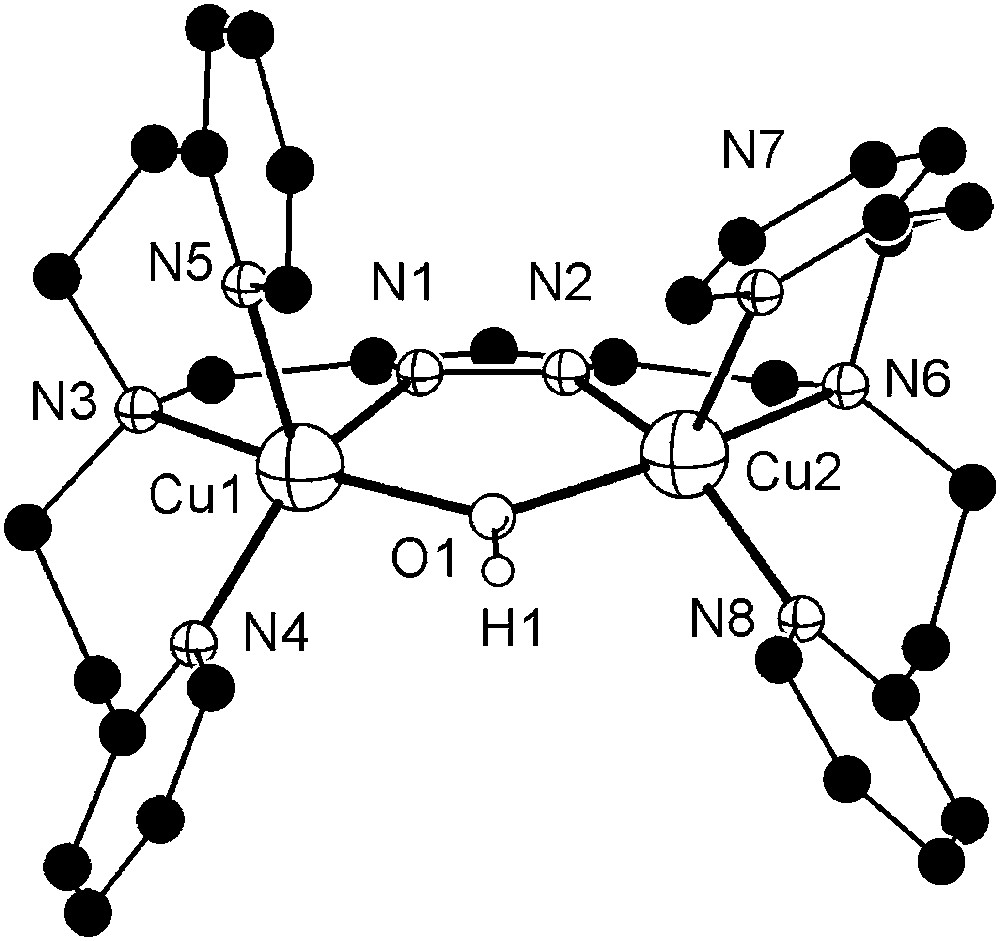
Molecular structure and numbering scheme of the cationic moiety of 6b. Most hydrogen atoms are omitted for clarity. Only one of the two crystallographically independent molecules is shown. Selected atom distances (Å) and bond angles: Cu1–N1 1.921(4), Cu1–N3 2.132(3), Cu1–N4 1.999(4), Cu1–N5 2.201(4), Cu1–O1 2.004(3), Cu2–N2 1.905(3), Cu2–N6 2.141(4), Cu2–N7 2.248(4), Cu2–N8 2.007(3), Cu2–O1 2.004(4), Cu1⋯Cu2 3.518(5); N1–Cu1–N3 79.4(2), N1–Cu1–N4 150.2(1), N1–Cu1–N5 101.8(2), N1–Cu1–O1 83.8(2), N3–Cu1–N4 94.9(1), N3–Cu1–N5 93.9(1), N3–Cu1–O1 161.8(1), N4–Cu1–N5 107.8(2), N4–Cu1–O1 96.2(1), N5–Cu1–O1 96.5(1), N2–Cu2–N6 78.2(1), N2–Cu2–N7 113.9(1), N2–Cu2–N8 152.2(1), N2–Cu2–O1 84.1(1), N6–Cu2–N7 92.5(1), N6–Cu2–N8 96.6(2), N6–Cu2–O1 161.0(1), N7–Cu2–N8 93.5(2), N7–Cu2–O1 101.1(1), N8–Cu2–O1 95.8(2). Masquer
Molecular structure and numbering scheme of the cationic moiety of 6b. Most hydrogen atoms are omitted for clarity. Only one of the two crystallographically independent molecules is shown. Selected atom distances (Å) and bond angles: Cu1–N1 1.921(4), Cu1–N3 2.132(3), Cu1–N4 ... Lire la suite
Both copper ions in 6b are hosted in their respective tetradentate binding pockets of [L2]− and are spanned by both the pyrazolate and a hydroxide ion. Their coordination geometry can be described as distorted square-pyramidal (τ = 0.13–0.22)1 [26], with one of the pyridyl-N in the apical positions at long Cu–N distances of 2.20–2.25 Å. One of the anions (ClO4− for 6a, CF3SO3− for 6b) forms an O–H⋯O hydrogen bond with the bridging hydroxide (not shown in Fig. 2). Because of the hydroxide bridge and favored by the long ligand side arms, the Cu⋯Cu distance in 6a and 6b is relatively short (3.44–3.52 Å) and is in the range assumed to be relevant for the catechol oxidase active site. UV/vis spectra (methanol) confirm the identity of the cations of 6a and 6b and the preservation of the square-pyramidal coordination environment in solution (λmax = 645 nm, ɛ = 260 mol l−1 cm−1). FAB mass spectra of both complexes reveal dominant signals at m/z = 671 characteristic for the ion [L2Cu2]+, suggesting that reduction to copper(I) with loss of the bridging hydroxide may readily occur under suitable conditions.
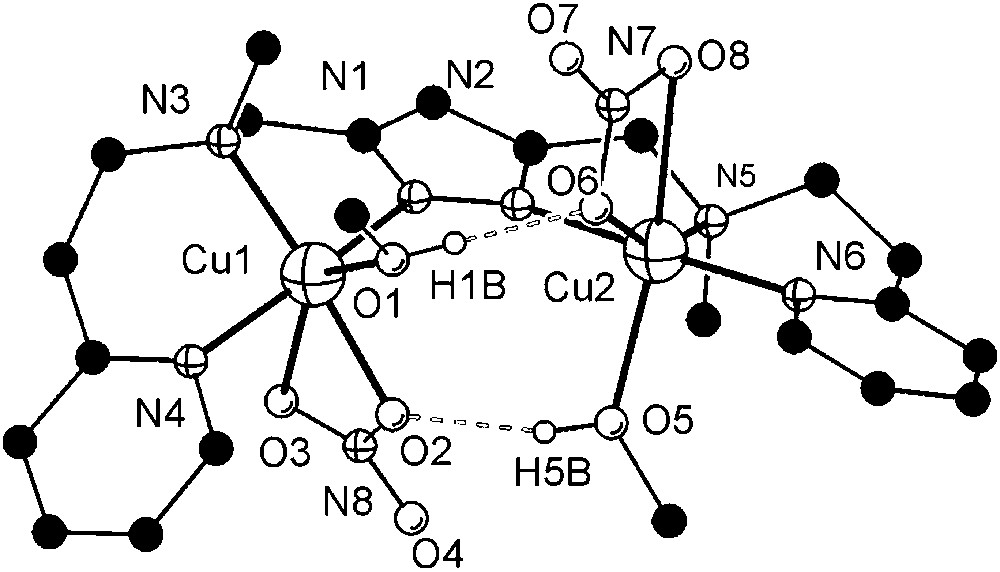
Dicopper(II) complexes of pyrazole/tacn hybrid ligands [L3]− and [L4]− that differ by the length of the spacer between the pyrazole and one of the tacn side arms were readily prepared from HL3 or HL4, 2 equivalents of base (KOtBu) and 2 equivalents of Cu(ClO4)2·6H2O. Unfortunately, only [L3Cu2(O2H3)](ClO4)2 (7) could yet be obtained in crystalline form. The molecular structure of its cation is depicted in Fig. 3, along with selected atom distances and bond angles.
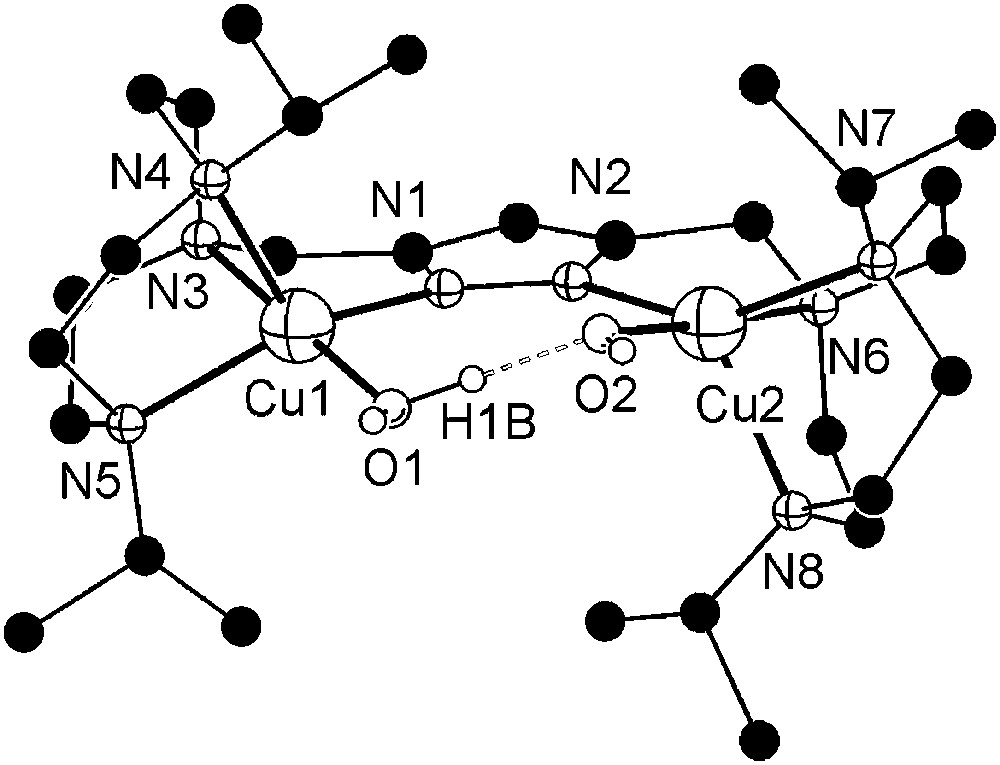
Molecular structure and numbering scheme of the cationic moiety of 7. Most hydrogen atoms are omitted for clarity. Selected atom distances (Å) and bond angles: Cu1–N1 1.998(4), Cu1–N3 2.056(4), Cu1–N4 2.206(4), Cu1–N5 2.125(4), Cu1–O1 1.905(4), Cu2–N2 2.024(4), Cu2–N6 2.048(4), Cu2–N7 2.174(4), Cu2–N8 2.200(4), Cu2–O2 1.896(4), Cu1⋯Cu2 4.412(1), O1⋯O2 2.424(6); N1–Cu1–N3 82.52(17), N1–Cu1–N4 121.13(16), N1–Cu1–N5 147.82(18), N1–Cu1–O1 95.43(17), N3–Cu1–N4 83.79(16), N3–Cu1–N5 83.15(16), N3–Cu1–O1 177.53(18), N4–Cu1–N5 85.60(16), N4–Cu1–O1 98.48(18), N5–Cu1–O1 97.97(17), N2–Cu2–N6 82.12(17), N2–Cu2–N7 146.73(16), N2–Cu2–N8 122.36(17), N2–Cu2–O2 95.42(16), N6–Cu2–N7 82.97(16), N6–Cu2–N8 84.23(17), N6–Cu2–O2 177.04(17), N7–Cu2–N8 85.33(15), N7–Cu2–O2 98.34(15), N8–Cu2–O2 98.50(17), O1–H1B⋯O2 168(7). Masquer
Molecular structure and numbering scheme of the cationic moiety of 7. Most hydrogen atoms are omitted for clarity. Selected atom distances (Å) and bond angles: Cu1–N1 1.998(4), Cu1–N3 2.056(4), Cu1–N4 2.206(4), Cu1–N5 2.125(4), Cu1–O1 1.905(4), Cu2–N2 2.024(4), Cu2–N6 2.048(4), Cu2–N7 ... Lire la suite
As anticipated from the constraints imposed by the ligand scaffold, 7 features a rather large Cu⋯Cu distance (4.41 Å) and coordination geometries of the copper ions intermediate between square-pyramidal and trigonal–bipyramidal (τCu1 = 0.49, τCu2 = 0.50)1 [26]. Distortions from square-pyramidal coordination are also apparent from the band positions of the weak ligand-field transitions at 700 and 1030 nm in solution. Due to the large metal–metal separation, an additional water molecule is incorporated besides the hydroxide bridge to give a (H)O–H⋯O(H) unit within the bimetallic pocket. The metal ions are located almost within the plane of the pyrazolate (torsion angle Cu1–N1–N2–Cu2 of 9.7°), while the O2H3 bridge forms an angle of 20.7° with that plane.
The complex [L4Cu2(OH)](ClO4)2 (8) could only be obtained as a green microcrystalline powder. Its molecular structure (Scheme 4) is assumed to be similar to that of the corresponding dinickel(II) complex, which has been characterized crystallographically [24]. Due to the longer spacer between the pyrazole and one of the tacn side arms in [L4]−, the dinickel complex [L4Ni2(OH)](ClO4)2 adopts a short Ni⋯Ni separation of 3.5 Å and hence accomodates a bridging hydroxide. A similar situation for 8 is supported by (i) FAB mass spectrometry showing dominant peaks at m/z = 773 (100% intensity) and 674 that are characteristic for ions [L2Cu2(OH)(ClO4)]+ and [L2Cu2(OH)]+, respectively, and (ii) by the elemental analysis that is in accordance with the proposed structure. The d–d transition at 626 nm in methanol solution suggests a largely square-pyramidal coordination environment for the copper ions in 8.
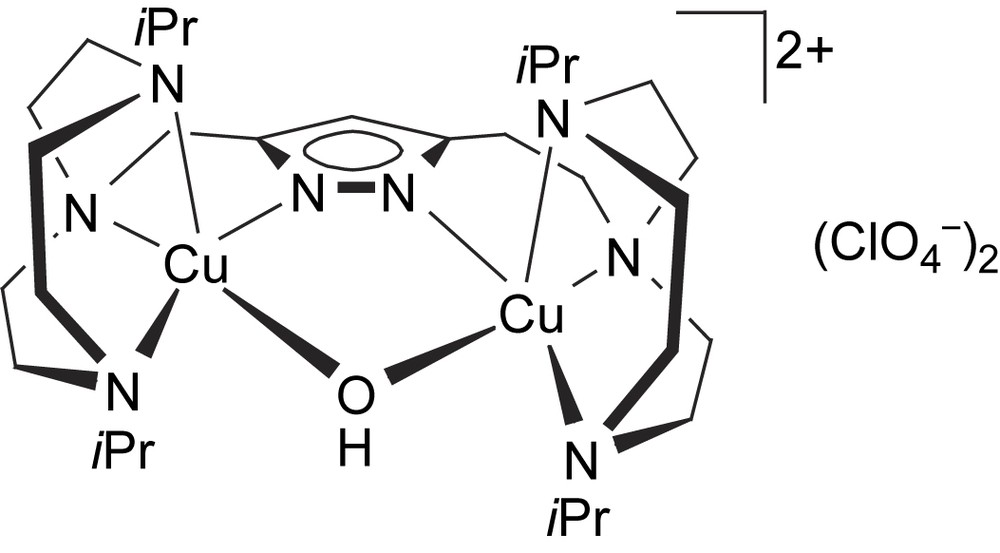
Proposed structure of [L4Cu2(OH)](ClO4)2 (8).
2.2 Cyclic voltammetry
Cyclic voltammetry measurements2 reveal a first irreversible reduction process in the range −0.5 to −0.9 V for all dicopper(II) complexes 1–6b with open-chain chelate arms, while reduction of complexes 7 and 8 with the pyrazole/tacn hybrid ligands takes place even below −1.0 V. The cyclic voltammogram of 5a is shown in Fig. 4 as an example. Anodic processes only occur at high potentials. Irreversibility of the reduction had to be expected because of the drastic differences in stereoelectronic requirements for copper(II) and copper(I). Generation of copper(I) is probably accompanied by rapid dissociation of the secondary bridging units, i.e., OH or (R)O–H⋯O(R), which are known to have a poor binding ability to copper(I) ions. However, irreversibility of the electrochemical redox processes does by no means preclude the suitability of the respective complexes as oxidation catalysts. The only crucial condition in this respect is that they can be reoxidized at appropriate potentials. According to previous work the redox potentials have to be in a suitable window for facile reduction of the catechol substrate on the one hand and the subsequent re-oxidation of the complex by molecular oxygen on the other hand [14,21]. It should be mentioned, however, that most previous investigations did not show a clear relation between the catalyst's redox potential and the catalytic activity [6].
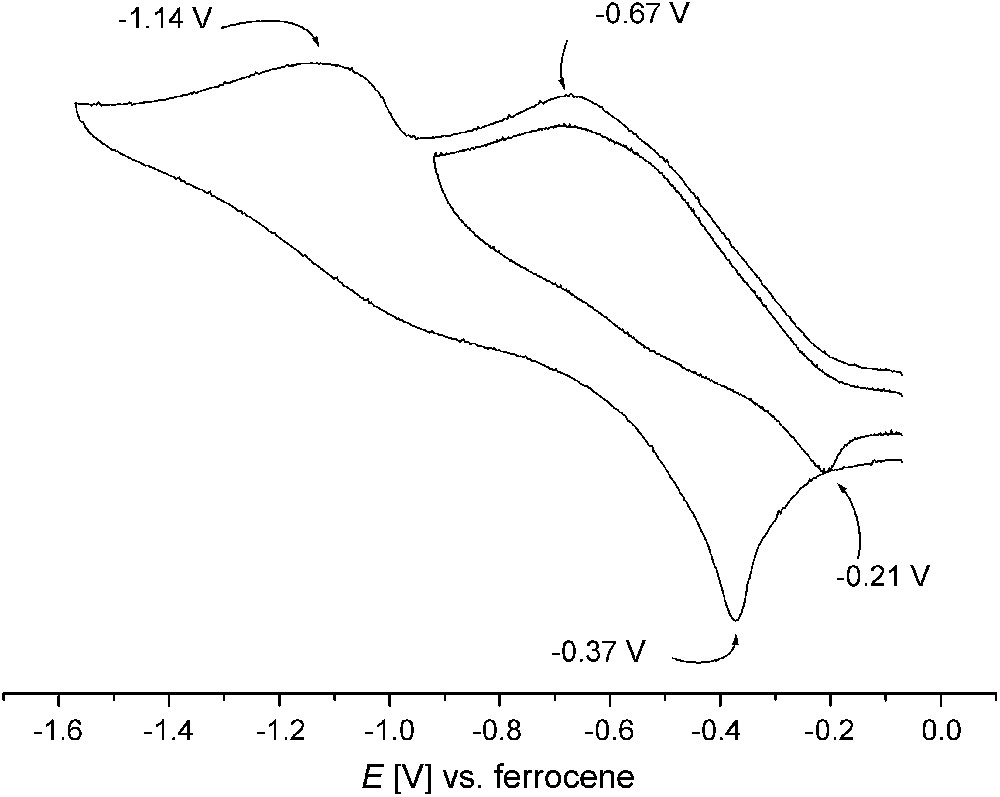
Cyclic voltammogram of 5a in acetonitrile (0.1 M NnBu4PF6, 100 mV s−1); peak potentials in volts versus the Cp2Fe+/Cp2Fe couple.
Even though the irreversibility of the electrochemical redox processes hampered their comparison for the various complexes, the present set of systems can be ranked qualitatively by their EpRed values obtained under similar conditions (Table 1). While comparable values of −0.75, −0.68 and −0.76 V were found for 1, 2 and 3, complex 4 exhibits greater stabilization of the copper(II) form as indicated by EpRed = −0.90 V. This is in line with the relatively low catecholase activity of this complex [12]. Among the new dicopper complexes reported here, complex 6b features the highest EpRed of −0.53 V, while the value for 5a is close to those of 1–3. The structural rigidity of the tacn ligand side arms in 7 and 8 is not suited to adapt to the very different stereoelectronic requirements of CuI and CuII, and hence these complexes have the lowest EpRed values.
Electrochemical properties of the complexes in acetonitrilea
| Complex | EpRed,1 [V] | EpRed,2 [V] |
| 1 | −0.75 | – |
| 2 | −0.68 | −1.08 |
| 3 | −0.76 | −1.03 |
| 4 | −0.90 | – |
| 5a | −0.67 | −1.14 |
| 6b | −0.53 | −1.11 |
| 7 | −1.07 | – |
| 8 | −1.38 | – |
a NnBu4PF6 (0.1 M), 200 mV s−1 for 1–4, 100 mV s−1 for 5a, 6b, 7, and 8; peak potentials in volts versus the Cp2Fe+/Cp2Fe couple.
2.3 Catecholase activity
Catecholase activities of complexes 5a, 5b, 6b, 7, and 8 were probed with the commonly used DTBC as a test substrate. The low redox potential of DTBC makes it easy to oxidize, and its bulky tert-butyl groups prevent unwanted side reactions such as ring opening or polymerization of the resulting quinone. The oxidation product 3,5-di-tert-butyl-o-quinone (DTBQ) displays a strong absorption at λmax = 400 nm whose appearance can be conveniently followed by UV/vis spectroscopy. Kinetics of the catecholase reactions mediated by the various dicopper complexes were analyzed by the initial rate method.
Exploratory experiments in methanol solution at 20 °C revealed only negligible activity of 8 and very low activity of 7 (kexp = 2.2 ± 0.2 h−1; v0 = kexp·[complex]). A slight increase in activity for 8 could be observed when the reaction was carried out in a mixture of methanol and aqueous TRIS buffer (29:1) at pH 8 and 40 °C.3 First-order dependence of the initial rate on catalyst concentration (kexp = 2.8 ± 0.1 h−1) as well as saturation kinetics at high concentrations of the substrate was found under those conditions. Interpretation of the experimental data on the basis of a Michaelis–Menten model, originally developed for enzyme kinetics, gave the following parameters: kcat = 3.9 ± 0.4 h−1, KM = (1.8 ± 0.1) × 10−3 mol l−1, and vmax = (8.1 ± 0.8) × 10−6 mol l−1 min−1 (determined from a Lineweaver–Burk plot). Due to their low activities, complexes 7 and 8 were not investigated in greater detail.
Complexes 5a, 5b, and 6 exhibit much higher activities in the same mixture of methanol and aqueous TRIS buffer (29:1) even at 20 °C. Since [L1]− with its few long chelate arms imparts relatively low stability to its dicopper complexes at higher pH, catecholase experiments were conducted at pH 7.3 for 5a,b. A linear dependence of the initial rate on catalyst concentration was found when the concentration was varied between 10−5 and 10−4 M (5a,b) or 10−6 and 10−5 M (6b) while [DTBC]0 was kept constant at a large initial excess of 2 × 10−3 M.4 Complexes 5a and 5b show an almost identical catalytic behavior with pseudo first-order rate constants kexp = 593 ± 4 h−1 (5a) and 629 ± 12 h−1 (5b) (Table 2), which suggests that the different moieties within the bimetallic pocket (double (Me)O–H⋯O(NO2) bridges in 5a versus (Me)O–H⋯F bridge in 5b) are rapidly displaced under the experimental conditions to give the same species in solution. This is corroborated by comparison of the UV/vis spectra of 5a,b in methanol and in methanol/TRIS buffer (29:1)3 (Fig. 5). In pure methanol, both λmax values and intensities of the d–d absorption bands for 5a and 5b are clearly different, while their spectra are basically identical in TRIS buffered solution. ESI mass spectrometric results for the different solutions are in accordance with the conclusions drawn from the UV/vis data: while a signal at m/z = 508 for [L5Cu2F]+ confirms the presence of the fluoride ligand for 5b in pure methanol, no such signal can be detected in TRIS buffered solution. In the latter medium, spectra for 5a and 5b are almost identical and show peaks at m/z = 489 for the fragment [L5Cu2]+ and at m/z = 613 characteristic for [L5Cu2(NO3)2]+ (note that TRIS buffer was used as the NO3− salt in these experiments).
Kinetic parameters of the oxidation of DTBC by the catalysts 5a, 5b, and 6ba
| kexp [h−1] | kcat [h−1] | KM [mol l−1] | vmax [mol l−1 min−1] | |
| 5ab | 593 ± 4 | 1125 ± 102 | (1.0 ± 0.1) × 10−3 | (3.8 ± 0.3) × 10−4 |
| 5bb | 629 ± 12 | 1432 ± 93 | (1.4 ± 0.1) × 10−3 | (4.8 ± 0.3) × 10−4 |
| 6bc | 5178 ± 62 | 6502 ± 239 | (7.5 ± 0.5) × 10−4 | (10.8 ± 0.3) × 10−4 |
a At 20 °C in a mixture of methanol and 0.27 M aqueous TRIS buffer (29:1), vmax was obtained from the Lineweaver–Burk plot.
b pH 7.3.
c pH 8.0.
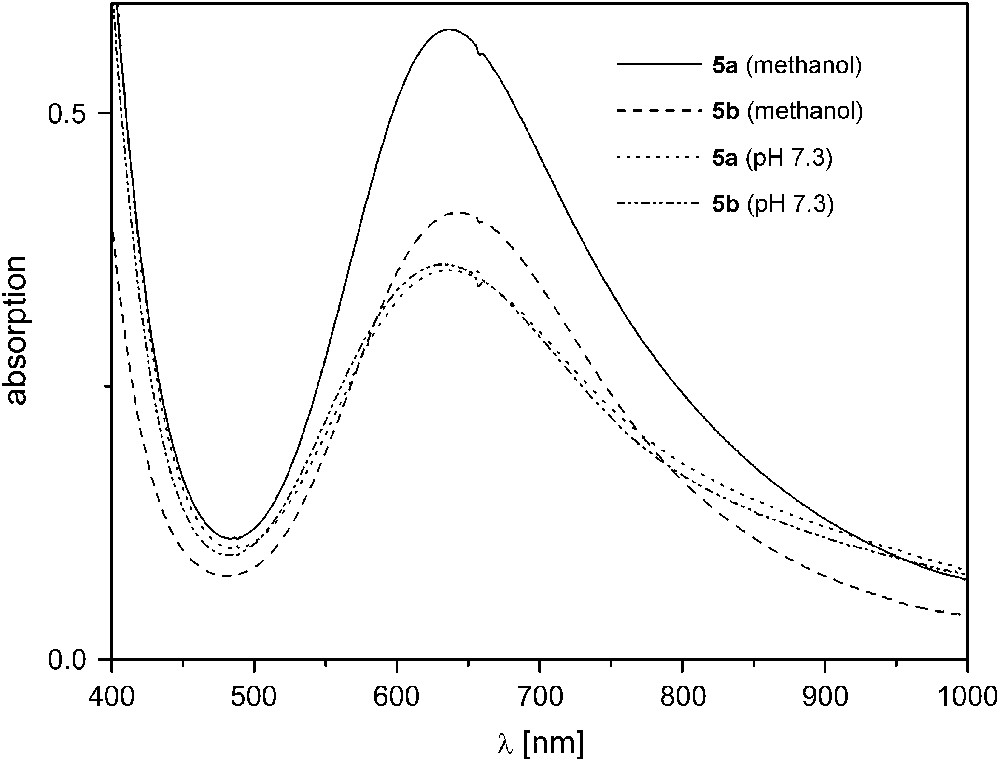
UV/vis spectra of 5a,b in pure methanol and in methanol/TRIS buffer (29:1) at pH 7.3.
A linear relationship between initial rates and catalyst concentrations was also found for 6b. The catalytic activity of 6b is the highest among all pyrazolate-based dicopper complexes examined so far (kexp = 5178 ± 62 h−1 at pH 8.0, Table 2). Saturation kinetics were observed for all complexes 5a,b and 6b when the substrate concentration [DTBC]0 was varied between 4 × 10−4 and 2.8 × 10−3 M while the catalyst concentration was kept constant at 2 × 10−5 (for 5a,b) or 10−5 M (for 6b). Lineweaver–Burk plots are shown in Fig. 6, and data derived from the Michaelis–Menten model are given in Table 2.
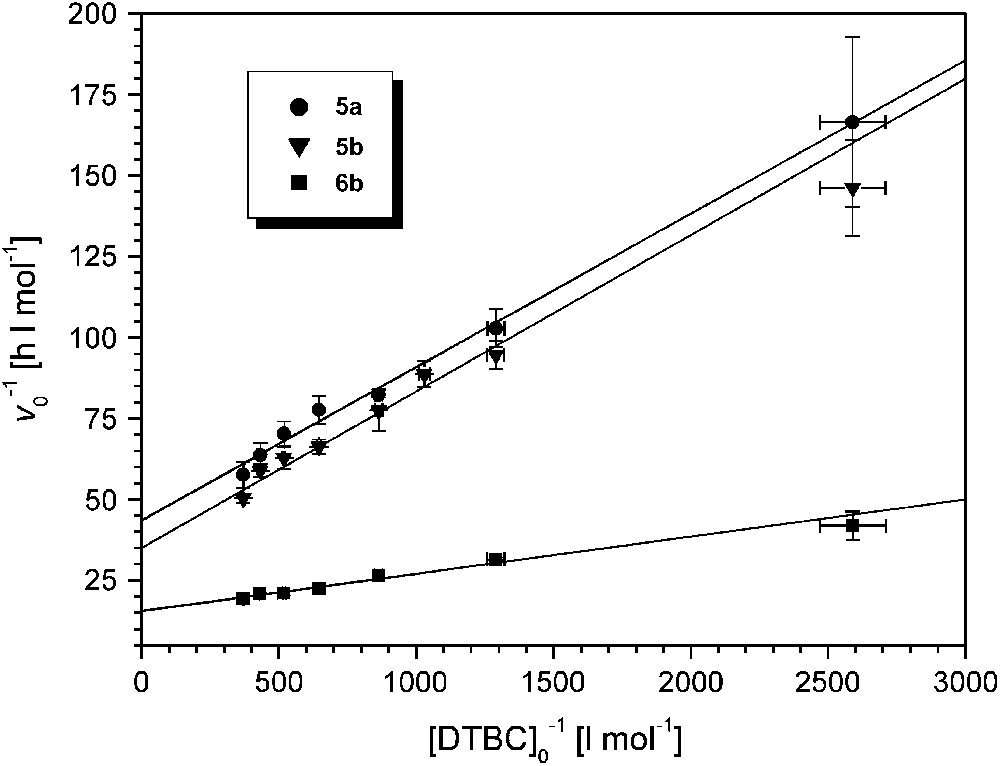
Lineweaver–Burk plots for the catalysts 5a, 5b (at pH 7.3) and 6b (at pH 8); [5a]0 = [5b]0 = 2 × 10−5 M, [6b]0 = 10−5 M; T = 20 °C.
2.4 Structure–activity correlations
The various catechol oxidase model systems 1–8 have as a common structural motif the pyrazolate bridge spanning the two metals, while differences are introduced via the chelating N-donor side arms that generate distinct binding sites for hosting the proximate copper ions. Given this structural relationship, comparison of the available data for the series of catalysts allows to establish some qualitative structure–activity correlations, even though kinetic data have been obtained under somewhat distinct conditions that proved most appropriate for the individual systems. In Fig. 7 the experimental pseudo first-order rate constants kexp are plotted versus the Cu⋯Cu separation of the bimetallic core as determined by X-ray crystallography. The ligand scaffolds certainly provide some structural flexibility, and the Cu⋯Cu distances observed for the solid state may differ to a certain extent in solution. However, the length and topology of the ligand side arms constrains the accessible range of metal–metal distances, and as a general trend it is clearly discernible that shorter Cu⋯Cu distances are advantageous for high catecholase activity.
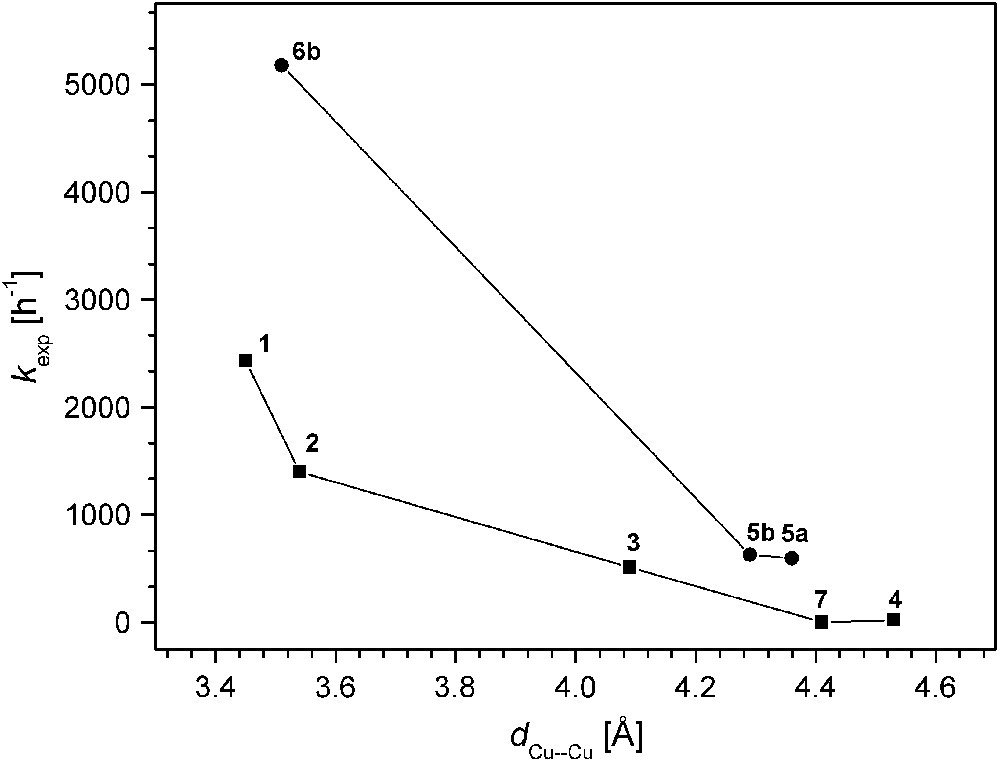
Catecholase activity versus Cu⋯Cu distance for the pyrazolate-based catalysts.
On the other hand, complex 8 that is believed to feature a very short Cu⋯Cu distance (as concluded from the structure of the corresponding dinickel analogue) exhibits extremely low activity, indicating that additional factors play a decisive role. Fig. 8 illustrates the relation between kexp and the redox properties (qualitatively expressed by the EpRed values). It is quite evident that all dicopper catalysts with EpRed below a threshold value of around −0.8 V are barely active. In the same tenor one may argue that the activity of 6b is highest because of its favorable combination of short Cu⋯Cu separation and comparatively high redox potential. It has to be emphasized, however, that the present structure–activity correlations are purely empirical and are not founded on any detailed mechanistic basis.
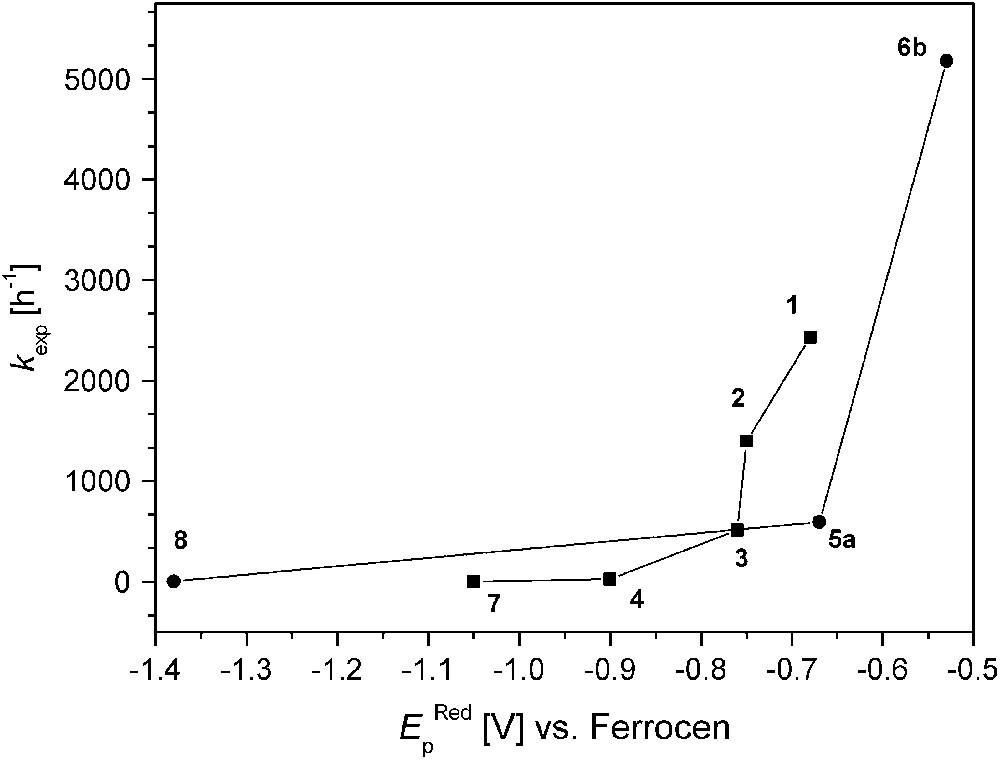
Catecholase activity and reduction peak potentials (EpRed values versus Cp2Fe+/Cp2Fe).
In view of the very high activity of 6b, the influence of pH was investigated in some more detail for this particular catalyst system. The AcOH/AcO−, MES, MOPS, and TRIS buffers were used to cover different pH ranges, and results are summarized in Fig. 9. It is apparent that the type of buffer has a dramatic effect on the catecholase activity. While the sluggish reactivity at low pH may be explained by protonation of the bridging hydroxide and/or inhibition by the acetate anion, the negligible reactivity in MEPS or MOPS buffered solution (pH 5.6–7.6) is unexpected and suggests some undesired interaction between the buffer and the catalyst. In contrast, use of either nitrate or chloride in the case of TRIS buffer has only a minor effect. Somewhat lower activities around the optimum pH 8.0 may again suggest competitive inhibition by the chloride, and in line with this assumption a Cl-bridged complex [L2Cu2Cl](CF3SO3)2 (9) could be isolated in independent experiments and structurally characterized (Fig. 10). In complex 9 the chloride ion blocks the binding site within the bimetallic pocket at a Cu⋯Cu separation of 3.83 Å.
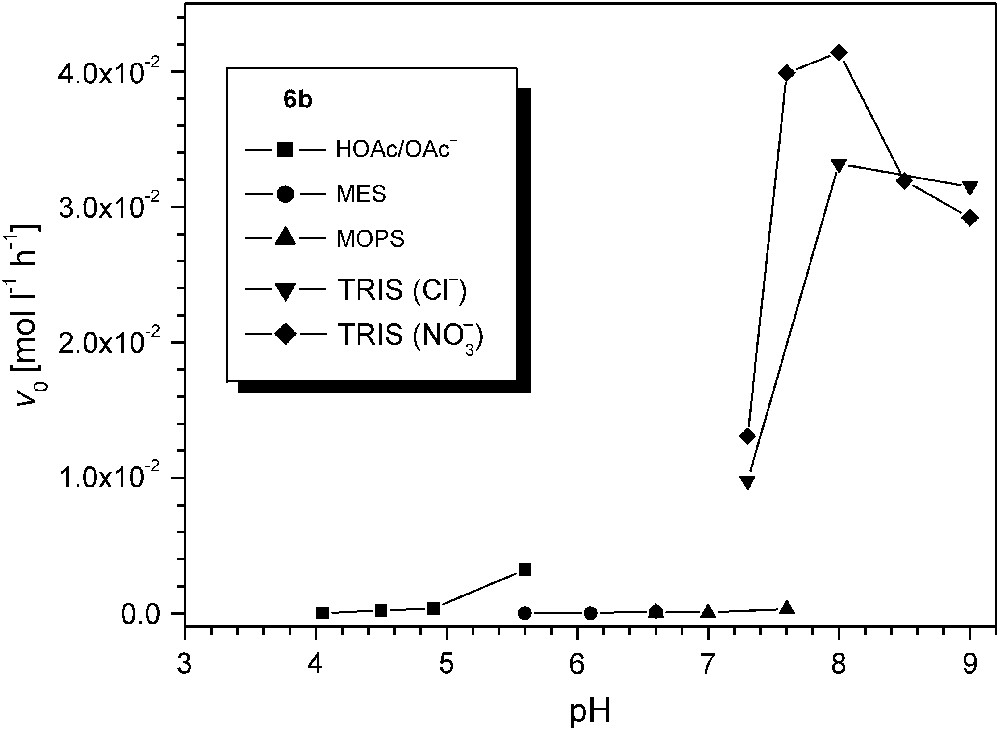
Effect of pH and buffer on the catecholase activity (initial rates v0) of 6b.
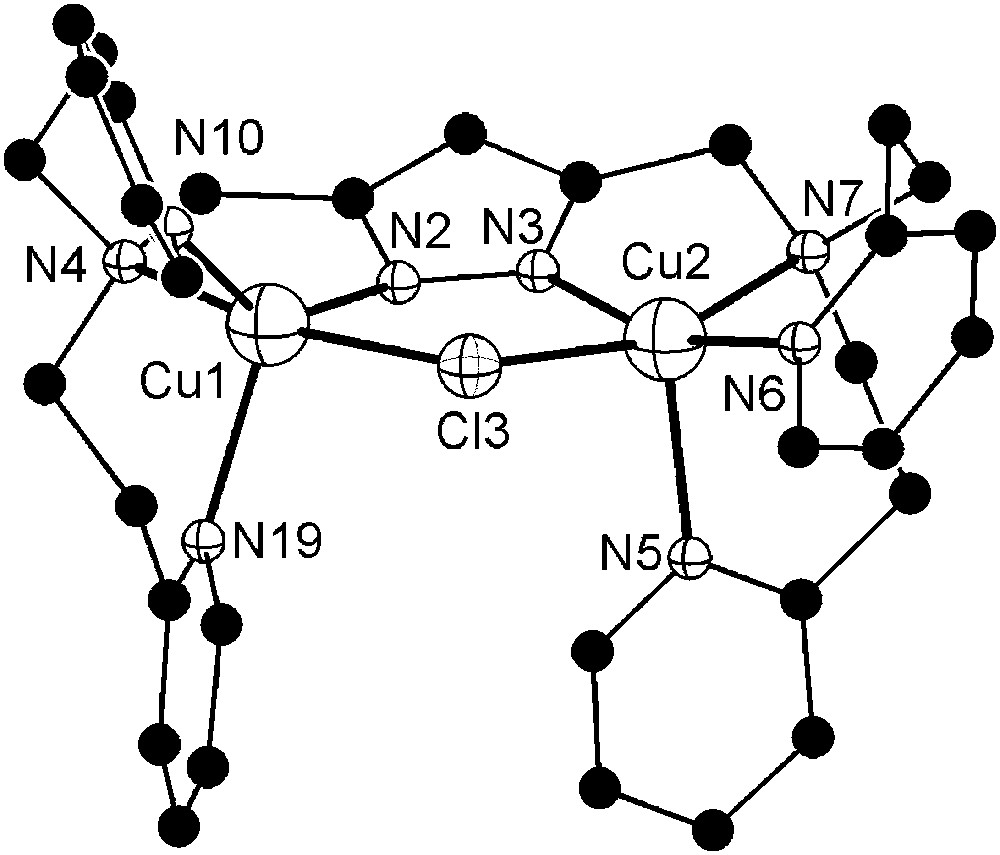
Molecular structure and numbering scheme of the cationic moiety of 9. Hydrogen atoms are omitted for clarity. Selected atom distances (Å) and bond angles: Cu1–N2 1.932(3), Cu1–N4 2.115(3), Cu1–N10 2.044(3), Cu1–N19 2.179(4), Cu1–Cl3 2.440(1), Cu2–N3 1.900(3), Cu2–N5 2.207(4), Cu2–N6 1.980(3), Cu2–N7 2.158(4), Cu2–Cl3 2.412(1), Cu1⋯Cu2 3.829(2); N2–Cu1–N4 81.5(1), N2–Cu1–N10 139.4(1), N2–Cu1–N19 111.6(1), N2–Cu1–Cl3 86.9(1), N4–Cu1–N10 96.0(1), N4–Cu1–N19 94.1(1), N4–Cu1–Cl3 167.3(1), N10–Cu1–N19 109.0(1), N10–Cu1–Cl3 89.5(1), N19–Cu1–Cl3 95.0(1), N3–Cu2–N5 102.0(1), N3–Cu2–N6 160.6(1), N3–Cu2–N7 80.0(2), N3–Cu2–Cl3 87.2(1), N5–Cu2–N6 97.2(1), N5–Cu2–N7 96.8(2), N5–Cu2–Cl3 99.0(1), N6–Cu2–N7 95.0(2), N6–Cu2–Cl3 92.6(1), N7–Cu2–Cl3 161.4(1). Masquer
Molecular structure and numbering scheme of the cationic moiety of 9. Hydrogen atoms are omitted for clarity. Selected atom distances (Å) and bond angles: Cu1–N2 1.932(3), Cu1–N4 2.115(3), Cu1–N10 2.044(3), Cu1–N19 2.179(4), Cu1–Cl3 2.440(1), Cu2–N3 1.900(3), Cu2–N5 2.207(4), Cu2–N6 1.980(3), ... Lire la suite
The catecholase activity of simple Cu2+ salts such as copper(II) nitrate, tetrafluoroborate, or triflate in TRIS buffer at pH 8.0 is much lower than the activity of 6b, which ascertains that TRIS does not extract the metal ions from the complex. Integrity of 6b under these conditions is confirmed by UV/vis spectroscopy. While at present the reason for the dramatic effect of the buffer remains unknown, it is obvious that great caution has to be exercised when comparing catalytic data from different studies employing different solvents and buffers.
3 Concluding remarks
A set of four new dicopper complexes with different pyrazolate-based compartmental ligands has been prepared and complements a series of related dicopper systems reported earlier. The thus available assortment of complexes featuring a common structural motif provides a suitable basis for establishing some qualitative structure–activity correlations. Distinctions between the various complexes are introduced by different chelate side arms of the ligand scaffolds, imparting different Cu⋯Cu distances and redox potentials. This leads to greatly differing activities in the catalytic oxidation of DTBC mediated by those complexes. All complexes investigated here (and most of the other pyrazolate-bridged complexes investigated earlier [12]) display a Michaelis–Menten type kinetic behavior, although detailed mechanistic information is not yet available for the individual catalysts. Caution is thus recommended when comparing and interpreting relative activities, but nevertheless some empirical relationships between molecular structure and reactivity are discernible from the present work. Here it is apparent that both Cu⋯Cu separation and redox potential play an important role: the shortest Cu⋯Cu distance accessible with the present set of systems (∼3.5 Å) is clearly advantageous for high activity (which decreases at larger distances), and activity is greatly diminished when the dicopper complex has a redox potential below a certain threshold value (qualitatively expressed here by an EpRed value of −0.8 V). Both parameters have to be considered simultaneously, and it is likely that additional factors play a role as well. Finally one should be very cautious when comparing activity data from different studies that use different solvents and buffers, since a dramatic effect of the buffer on activity is possible, as observed here for the most active complex 6b.
4 Experimental
4.1 General procedures and methods
HL1, HL2, HL3, and HL4 were prepared as described previously [24,25]. Solvents were dried according to established procedures and distilled prior to be used. All other chemicals were purchased from commercial sources and used as received. Yields of crystalline complexes given in the experimental procedures are not optimized. Microanalyses: Analytical Laboratory of the Institute of Inorganic Chemistry at Georg-August-University Göttingen. IR spectra: Digilab Excalibur, recorded as KBr pellets. Wavelengths in cm−1. UV/vis spectra: Varian Cary 1E or Cary-5000, measured in quartz cuvettes of 0.2 cm or 1 cm path length. Wavelengths λ in nm and extinction coefficients ɛ in l mol−1 cm−1. Mass spectra: Finnigan MAT 95 (FAB-MS). Cyclic voltammetry: Perkin–Elmer Model 263A instrument with glassy-carbon working electrode and platinum reference and counter electrodes, in 0.1 M NnBu4PF6/CH3CN. Ferrocene (the potential being 0.45 V versus SCE in DMF) was used as internal standard.
4.2 Syntheses of the complexes
4.2.1 [L1Cu2(MeOH)2(NO3)2]NO3 (5a)
To a solution of HL1 (295 mg, 0.81 mmol) in 150 ml methanol were added 1 equivalent of KOtBu (91 mg, 0.81 mmol) and 2 equivalents of Cu(NO3)2·3H2O (391 mg, 1.62 mmol) and the reaction mixture was stirred for 2 h. After removal of all volatile material under reduced pressure the residue was redissolved in a small amount of methanol, filtered, and the solution layered with Et2O to gradually afford blue crystals of the product 5a. Yield: 330 mg (0.45 mmol, 56%); IR (KBr):
4.2.2 [L2Cu2(OH)](ClO4)2 (6a)
To a solution of HL2 (120 mg, 0.23 mmol) in acetonitrile (150 ml) were added 1.9 equivalents of KOtBu (49 mg, 0.44 mmol) and 1.9 equivalents of Cu(ClO4)2·6H2O (163 mg, 0.44 mmol) and the dark green reaction mixture was stirred for 6 h. After removal of all volatile material under reduced pressure the residue was redissolved in a small amount of dichloromethane, filtered, and the solution layered with pentane and stored at 4 °C to gradually afford green crystals of 6a·2CH2Cl2. Yield: 67 mg (0.08 mmol, 33%); IR (KBr):
4.2.3 [L2Cu2(OH)](CF3SO3)2 (6b)
To a solution of HL2 (285 mg, 0.55 mmol) in methanol (150 ml) were added 1.9 equivalents of KOtBu (117 mg, 1.04 mmol) and 1.9 equivalents of Cu(CF3SO3)2 (377 mg, 1.04 mmol) and the reaction mixture was stirred for 2 h. After removal of all volatile material under reduced pressure the residue was redissolved in a small amount of acetone, filtered, and the solution layered with pentane to gradually afford green crystals of 6b·acetone. Yield: 83 mg (0.08 mmol, 15%); IR (KBr):
4.2.4 [L3Cu2(O2H3)](ClO4)2 (7)
To a solution of HL3 (177 mg, 0.34 mmol) in ethanol (70 ml) were added 2 equivalents of KOtBu (77 mg, 0.68 mmol) and 2 equivalents of Cu(ClO4)2·6H2O (253 mg; 0.68 mmol) and the reaction mixture was stirred for 12 h and then filtered. After removal of all volatile material under reduced pressure, the resulting residue was redissolved in a small amount of acetone and layered with Et2O to gradually afford green crystals of the product 7·acetone. Yield: 222 mg (0.24 mmol, 70%); IR (KBr):
4.2.5 [L4Cu2(OH)](ClO4)2 (8)
To a solution of HL4 (205 mg, 0.38 mmol) in methanol (150 ml) were added 2 equivalents of KOtBu (85 mg, 0.76 mmol) and 2 equivalents of Cu(ClO4)2·6H2O (282 mg, 0.76 mmol) and the green reaction mixture was stirred for 12 h. After removal of all volatile material under reduced pressure, the resulting residue was redissolved in a small amount of acetone, filtered, and layered with pentane. The product 8 gradually precipitated as a green powder which was isolated by filtration and dried under reduced pressure. Non-optimized yield: 110 mg (0.13 mmol, 34%); IR (KBr):
4.2.6 [L2Cu2Cl](CF3SO3)2 (9)
To a solution of HL2 (399 mg, 0.73 mmol) in dichloromethane (150 ml) were added 2.0 equivalents of KOtBu (164 mg, 1.46 mmol) and 2.0 equivalents of Cu(CF3SO3)2 (528 mg, 1.46 mmol) and the dark green reaction mixture was stirred for 6 h. After removal of all volatile material under reduced pressure the residue was redissolved in a small amount of dichloromethane, filtered, and the solution layered with diethyl ether to gradually afford green crystals of 9·CH2Cl2. Yield: 205 mg (0.19 mmol, 26%); IR (KBr):
4.3 Kinetic measurements
These were carried out at 20 °C in a thermostated quartz cell (d = 1 cm) with magnetic stirrer using a mixture of methanol and 0.27 M aqueous TRIS buffer (29:1) at pH 7.3 (for 5a,b) or pH 8.0 (for 6b). The increase of the absorption band of the product DTBQ at 400 nm (ɛ = 1810 l mol−1 cm−1) was followed by UV/vis spectroscopy. Each experiment was performed at least three times, and the obtained plots were analyzed by the initial rate method. In a first test series, solutions with varying complex concentration in the range 9.7 × 10−6–9.7 × 10−5 M (5a,b) or 9.7 × 10−7–9.7 × 10−6 M (6b) were prepared while the concentration of DTBC was kept constant (1.9 × 10−3 M). In a second test series, the complex concentrations were held constant at 1.9 × 10−5 M (5a,b) or 10−5 M (6b) while the catechol concentration was varied in the range 3.9 × 10−4–2.7 × 10−3 M. The rate law dependence on complex concentration was found to be first order while saturation kinetics were observed for the dependence on substrate concentration. A Michaelis–Menten analysis of the data was applied and vmax, KM and kcat determined from a Lineweaver–Burk plot. All errors were calculated conservatively. Blank experiments were carried out with simple copper(II) salts such as Cu(ClO4)2·6H2O, Cu(BF4)2·6H2O, or Cu(NO3)2·3H2O under identical conditions, and the observed catalytic activities of these salts were generally much lower than those of the dinuclear complexes with the respective anion.
4.4 Test for H2O2
First the DTBC was oxidized in the presence of the respective catalyst and air until an absorption A400 = 0.4 was reached. The reaction was then quenched with the same volume of 0.005 M H2SO4 and the product DTBQ as well as residual DTBC removed by extraction with CH2Cl2. To 2 ml of the remaining solution was added 1 ml of H2O in the reference cell. In the other cell 1 ml of a 0.3 M KI solution and catalytic amounts of lactoperoxidase for specific acceleration of the oxidation of I− to I3− were added. The development of the absorption band A353 (ɛ = 26 000 mol l−1 cm−1) of the resulting I3− was followed by UV/vis spectroscopy. In a blank experiment the same procedure was followed for a reaction mixture containing the catalyst, but no substrate. Additionally, each measurement was verified by H2O2 test strips from Merck. Both tests were negative for all examined catalysts.
4.5 X-ray crystallography
X-ray data were collected on a Stoe IPDS II diffractometer (5a), a four-circle diffractometer with Offset-Euler balance with a Bruker SMART 4K CCD-counter diffractometer (6a, 6b), a Nonius Kappa CCD diffractometer (7) using graphite monochromated Mo Kα radiation (λ = 0.71073 Å), and a Bruker SMART 6000 4K CCD diffractometer (9) using monochromated Cu Kα radiation (λ = 1.54178 Å) (Table 3). The structures were solved by direct methods and refined on F2 using all reflections with SHELX-97 [27]. Atomic coordinates and thermal parameters of the non-hydrogen atoms were refined in fully anisotropic models. Hydrogen atoms were either located in the difference Fourier map and refined isotropically or included using the riding model. SADABS was used to perform area-detector scaling and absorption corrections for 6a (Tmax/min = 1/0.7346) and 6b (Tmax/min = 1/0.7489) [28]. Crystals of 7 were racemically twinned (ratio of the two twin components 0.533(7):0.467(3)) and one of the CF3SO3− anions was disordered (occupancy factors: 0.912(2) and 0.088(2)). The carbon atom of the CH2Cl2 solvent molecule in 9 was found to be disordered about two positions (occupancy factor for both positions: 0.50(3)).
Crystal data and refinement details for 5a, 6a, 6b, 7, and 9
| 5a | 6a | 6b | 7 | 9 | |
| Empirical formula | [C23H35Cu2N8O8]+NO3− | [C33H38Cu2N8O]2+2ClO4−·2CH2Cl2 | [C33H38Cu2N8O]2+2CF3O3S−·C3H6O | [C32H66Cu2N8O2]2+2ClO4−·C3H6O | [C33H37ClCu2N8]2+2CF3O3S−·CH2Cl2 |
| Formula weight | 740.68 | 1058.55 | 1046.03 | 936.91 | 1091.32 |
| Crystal size [mm] | 0.30 × 0.20 × 0.20 | 0.43 × 0.39 × 0.11 | 0.41 × 0.32 × 0.21 | 0.15 × 0.10 × 0.05 | 0.60 × 0.45 × 0.34 |
| T [K] | 133 | 190 | 133 | 200 | 277 |
| Crystal system | Triclinic | Monoclinic | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | P-1 (no. 2) | P21/n (no. 14) | Pna21 (no. 33) | P21212 (no. 18) | P21/n (no. 14) |
| a [Å], α [°] | 8.382(2), 62.94(3) | 9.0111(8), 90 | 22.552(14), 90 | 17.540(4), 90 | 21.609(4), 90 |
| b [Å], β [°] | 13.924(3), 83.62(3) | 39.929(3), 106.223(2) | 13.21(2), 90 | 24.287(5), 90 | 13.260(3), 107.21(3) |
| c [Å], γ [°] | 14.606(3), 85.64(3) | 12.1896(10), 90 | 28.35(5), 90 | 9.955(2), 90 | 16.663(3), 90 |
| V [Å3] | 1508.1(5) | 4211.2(6) | 8449(21) | 4240.8(15) | 4560.9(16) |
| ρcalcd. [g cm−3] | 1.631 | 1.670 | 1.636 | 1.467 | 1.586 |
| Z | 2 | 4 | 8 | 4 | 4 |
| F(000) | 764 | 2160 | 4268 | 1976 | 2208 |
| μ [mm−1] | 1.483 | 1.454 | 1.193 | 1.192 | 4.302 |
| hkl range | ±9, ±16, −14 to 17 | −12–11, 0–53, 0–16 | −29–18, ±17, ±36 | ±22, −31–30, ±12 | −23–24, ±14, ±18 |
| θ range [°] | 2.45–24.63 | 2.81–28.70 | 1.79–27.59 | 1.43–27.00 | 2.14–59.97 |
| Measured refl. | 12 347 | 29 282 | 125 703 | 9705 | 28 340 |
| Unique refl. [Rint] | 5049 [0.0952] | 10 802 [0.0430] | 19 490 [0.0705] | 9246 [0] | 6582 [0.0231] |
| Observed refl. I > 2σ (I) | 3690 | 8292 | 16 630 | 7091 | 6273 |
| Refined parameters | 409 | 701 | 1164 | 519 | 579 |
| Goodness-of-fit | 0.983 | 1.067 | 1.071 | 1.120 | 1.060 |
| R1, wR2 (I > 2σ (I)) | 0.0597, 0.1409 | 0.0501, 0.1098 | 0.0459, 0.0931 | 0.0730, 0.1008 | 0.0606, 0.1773 |
| R1, wR2 (all data) | 0.0819, 0.1507 | 0.0704, 0.1193 | 0.0605, 0.0997 | 0.1070, 0.1094 | 0.0619, 0.1792 |
| resid. el. dens. [e·Å−3] | 1.446/−1.041 | 1.162/−1.008 | 0.690/−0.574 | 0.783/−0.443 | 1.272/−1.036 |
CCDC-617183 (5a), -617184 (6a), -617185 (6b), -617186 (7), and -619359 (9) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336 033; deposit@ccdc.cam.uk.
Acknowledgement
We thank José Antonio Cuesta-Seijo, Dr. Mathias Noltemeyer, Thomas Labahn, Denis Vidovic (Göttingen) and Hans Pritzkow (Heidelberg) for collecting the X-ray data. Financial support by the DFG (Me1313/7-1) is gratefully acknowledged.
1 The angular structural parameter τ is defined as τ = (β − α)/60, where α and β represent two basal angles with β > α. It is a measure of the degree of trigonality: a perfect TB-5 structure is associated with τ = 1, while τ = 0 is expected for an idealized SPY-5 geometry.
2 All values versus the Cp2Fe/Cp2Fe+ couple.
3 For these experiments, TRIS buffer was used as the NO3− salt.
4 The relationship between v0 and catalyst concentration is not strictly linear at higher catalyst concentrations, suggesting a more complex situation that may also involve oligomeric species.