

1 Introduction
Cyclopentadienyl ligands with side chains containing donor functionalities are extensively used in organometallic chemistry, giving unique chemical and physical properties to the complexes [1]. Because of their hemilability and assembling capacity, such ligands have found significant applications, especially in catalysis and the construction of molecular materials. It is noteworthy that among these functionalized cyclopentadienyl ligands, those substituted with a pyridyl group seem to have been coordinated to Fe ions exclusively, and the resulting ferrocene derivatives served as building blocks for oligonuclear organometallic assemblies [2,3]. In this paper we present the introduction in f-element chemistry of the 2-pyridyl tetramethylcyclopentadienyl ligand η5-C5Me4-2-C5H4N, hereafter denoted as Cp∗Py, with the synthesis and characterization of the uranium(IV) compounds (Cp∗Py)2UX2 (X = Cl, Me, BH4) and [(Cp∗Py)2U(BH4)][BPh4]; we also describe the crystal structure of the chloride complex (Cp∗Py)2UCl2 and the ethylene bridged ansa-metallocene derivative C2H4(η5-C5Me3-2-C5H4N)2UCl2 which was obtained in an unexpected and unreproducible manner from the reaction of (Cp∗Py)2UMe2 and CuCl2.
2 Results and discussion
Reaction of UCl4 with two equivalents of LiCp∗Py in THF gave the bis(cyclopentadienyl) compound (Cp∗Py)2UCl2 (1) which, after evaporation of the solvent and extraction with toluene, was isolated as an orange powder in 88% yield (Eq. (1)); orange crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of pentane into a THF solution.
| (1) |
The crystal structure of 1 is shown in Fig. 1 together with selected bond lengths and angles. The asymmetric unit contains half a molecule, the other half being generated by the binary axis. In contrast to what was observed with ferrocene type complexes containing the Cp∗Py ligand or its equivalent without methyl groups (CpPy), where the pyridyl moiety is either not coordinated or bound to a distinct metal centre in a dinuclear assembly [3], both the Cp∗ ring and the N atom are linked to the uranium(IV) ion in 1. This ligation mode of the Cp∗Py ligand is also found in the previously described compounds [(Cp∗Py)(Cp∗PyH)Fe]2[U8Cl24O4(Cp∗Py)2] [4a] and {(Cp∗Py)UCl(Et2O)}2(μ-O)2 [4b] which were, respectively, formed by reaction of UCl4 and (Cp∗Py)2Fe in CH2Cl2, and crystallization of 1 in a mixture of THF and Et2O, in the presence of adventitious traces of air. The average U–C distance of 2.78(11) Å is similar to those measured in the aforementioned compounds [2.72(9) and 2.78(8) Å] and in bis(pentamethylcyclopentadienyl) uranium(IV) complexes [5], while the U–N distance of 2.757(6) Å is significantly larger, by 0.2 Å, and is even out of the range of the U–N(Py) distances found in uranium(IV) compounds, which is 2.54–2.70 Å [6]. The impeded approach of the Py unit to the metal centre is likely due to the steric crowding of the coordination sphere; contacts as short as 1.97 and 2.07 Å are present between hydrogen atoms bound to C(1) and C(12′) or C(13′) (symmetry code ′ = −x, y, 0.5−z). This situation is also indicated by the displacements of atoms C(2) and C(5) out of the tetramethylcyclopentadienyl mean plane, 1.138(19) and 0.307(11) Å, and the dihedral angle between the five- and six-membered rings, 60.4(3)°, which are smaller than in the previous di- and octanuclear complexes (ca. 1.65 and 0.45 Å and 73°, respectively). The U–Cl distance of 2.6874(16) Å can be compared with the average value of 2.69(5) Å in (Cp∗)2UCl2(HNPPh2) (Cp∗ = η5-C5Me5) [7].
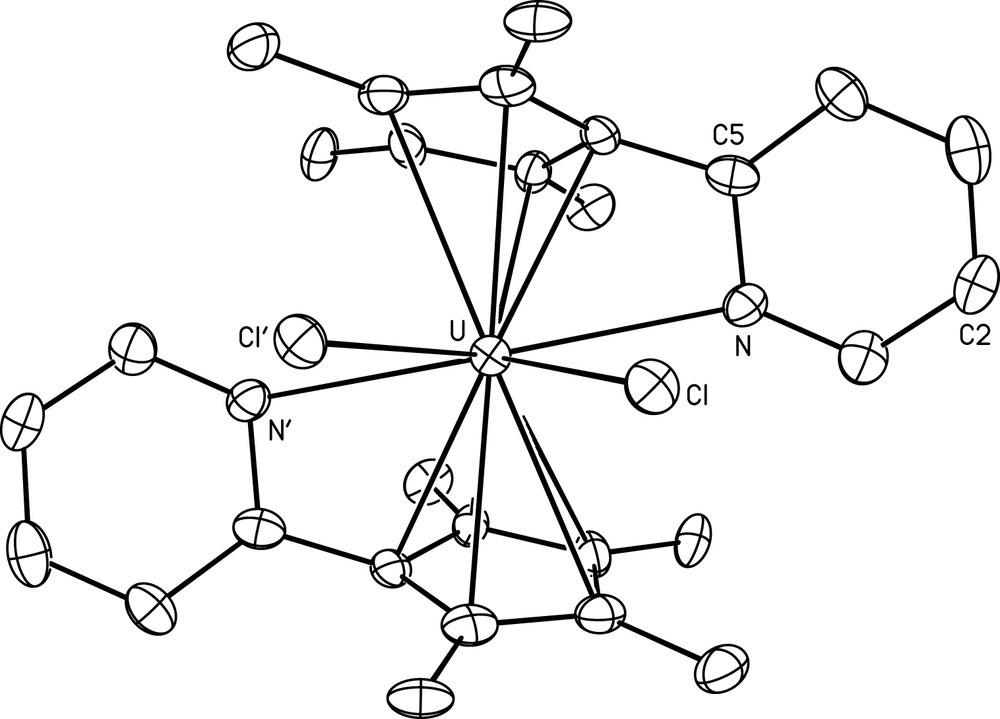
View of (Cp∗Py)2UCl2 (1) with displacement ellipsoids drawn at the 50% probability level. H atoms have been omitted. Symmetry code: ′ = −x, y, 0.5 − z. Selected bond lengths (Å) and angles (°): U–Cl 2.6874(16), U–N 2.757(6), 〈U–C〉 2.78(11); Cl–U–Cl 76.65(7), (Cp∗ centroid)–U–(Cp∗ centroid) 142.2.
Complex 1 was treated with a slight excess of LiMe or LiBH4 in THF for giving, after usual work-up, the pale brown methyl and dark red borohydride derivatives, (Cp∗Py)2UMe2 (2) and (Cp∗Py)2U(BH4)2 (3), in 77% and 23% yield, respectively, (Eqs. (2) and (3)). In the IR spectrum of 3, the strong singlet at 2466 cm−1 and the broad bands at 2213 and 2133 cm−1 are characteristic of a tridentate BH4 group [8].
| (2) |
| (3) |
The cationic compound [(Cp∗Py)2U(BH4)][BPh4] (4) was obtained as an orange powder in 72% yield by treatment of 3 with 1 equivalent of HNEt3BPh4 in THF (Eq. (4)). This protonolytic cleavage of the M–BH4 bond with an acidic ammonium salt was previously used for the synthesis of cationic complexes of zirconium [9], neodymium [10,11] and uranium [11,12].
| (4) |
Attempts to synthesize heterobimetallic complexes by addition of d transition metal salts to compounds 1–3 were unsuccessful; this failure is not so surprising in view of the strong chelate effect which prevents the hemilability of the Cp∗Py ligand. In one of these experiments with 2 and CuCl2 in THF, a black precipitate and a few dark red crystals were deposited from the solution, after 2 weeks at 20 °C. These latter compounds were found by X-ray diffraction analysis to be composed of the ansa-metallocene C2H4(η5-C5Me3-2-C5H4N)2UCl2 (5). The mechanism of formation of 5 is unclear but obviously involves C–H bond activation of a methyl group on each Cp∗Py ligand. Such activation of Cp∗ ligands have precedents in d transition metal chemistry, occurring under the influence of strong bases or thermolysis, and gave most generally tetramethylfulvene complexes and their derivatives [13]. Typical examples are the synthesis of Cp∗(C5Me4CH2)TiMe [13a] and Cp∗(C5Me4CH2)ZrPh [13b] from (Cp∗)2TiMe2 and (Cp∗)2ZrPh2, respectively; it has been shown that the η6 ligand in the zirconium compound has little fulvene π character and is best described as a σ,η5 ring-metalated group. The only report of intramolecular activation of a Cp∗ methyl C–H bond in an actinide complex concerns the formation of Cp∗(C5Me4CH2NAd)U(NHAd) (Ad = adamantyl) by heating the uranium(VI) bis(imido) compound (Cp∗)2U(NAd)2 in benzene [14]. Whereas coupling of Cp∗ ligands was observed in the synthesis of the dimeric complexes [(–CH2C5Me4)Co(Et2C2B4H3X)]2 (X = H, Cl, Br, I) by treatment of the monoCp∗ precursors Cp∗Co(Et2C2B4H3X) with BuLi in THF [15], complex 5 is, to the best of our knowledge, the first ansa-metallocene obtained by intramolecular ring-methyl activation of two cyclopentadienyl ligands. It is possible that the formation of 5 occurred via a bis(fulvene) intermediate; such reductive coupling of fulvenes by activated metals or low-valent metal species is well documented [16]. Despite many efforts, the synthesis of 5 could not be reproduced.
The crystal structure of 5 is shown in Fig. 2 together with selected bond lengths and angles. The molecular complex admits a pseudo-binary axis (approximate C2 point symmetry). The (Cp∗ centroid)–U–(Cp∗ centroid) angle in 5 is 113.1° as compared to 142.2° in 1. The opening of the coordination sphere by connecting the two cyclopentadienyl rings renders easier the approach of the pyridyl moiety to the metal centre, and the mean U–N distance of 2.584(5) Å is now unexceptional. The displacements of atoms C(2) and C(5) [C(16) and C(19)] from the proximal cyclopentadienyl mean plane, 1.353(16) and 0.332(9) Å [1.351(15) and 0.347(9) Å], and the dihedral angle between the five- and six-membered rings, 69.6(2)° [67.2(2)°], are larger than the corresponding values in 1, and also reflect the weaker steric constraints imposed to the Py unit. The average U–C and U–Cl distances of 2.80(12) and 2.684(3) Å, respectively, are similar to those found in 1; the Cl–U–Cl angles do not differ in 1 and 5, with values of 76.65(7)° and 76.12(4)°, respectively, while the N–U–N angle of 167.92(13)° is 18° larger in the ansa-metallocene.
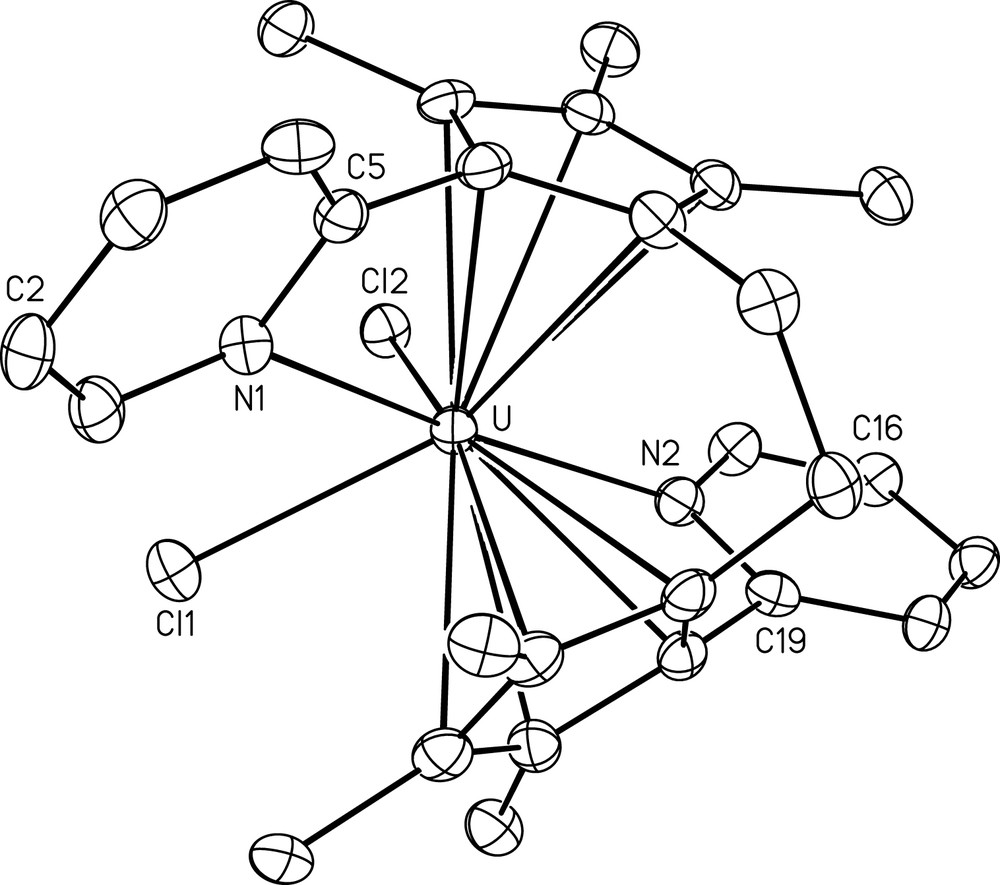
View of C2H4(η5-C5Me3-2-C5H4N)2UCl2 (5) with displacement ellipsoids drawn at the 40% probability level. H atoms have been omitted. Selected bond lengths (Å) and angles (°): U–Cl(1) 2.6816(13), U–Cl(2) 2.6872(12), U–N(1) 2.589(4), U–N(2) 2.579(4), 〈U–C〉 2.80(12); Cl(1)–U–Cl(2) 76.12(4), (Cp∗centroid)–U–(Cp∗ centroid) 113.1.
3 Experimental section
3.1 General
All reactions were carried out under argon with the rigorous exclusion of air and water (<5 ppm oxygen or water) using standard Schlenk vessel and vacuum line techniques or in a glove box. Solvents were dried by standard methods and distilled immediately before use. IR samples were prepared as Nujol mulls between KBr round cell windows and the spectra recorded on a Perkin–Elmer FT-IR 1725X spectrometer. The 1H NMR spectra were recorded on a Bruker DPX 200 instrument and referenced internally using the residual protio solvent resonances relative to tetramethylsilane (δ: 0); the spectra were recorded at 23 °C when not specified otherwise. Elemental analyses were performed by Analytische Laboratorien at Lindlar (Germany). The salt HNEt3BPh4 precipitated in water by mixing NaBPh4 and HNEt3Cl; LiCp∗Py [2a] and UCl4 [17] were prepared by published methods.
3.2 Synthesis of (Cp∗Py)2UCl2 (1)
A flask was charged with UCl4 (194 mg, 0.51 mmol) and LiCp∗Py (209 mg, 1.02 mmol) and THF (10 mL) was condensed in it. After stirring for 15 h at 20 °C, the solvent was evaporated off and the residue extracted with toluene (15 mL). The solution was evaporated to dryness, leaving an orange powder of 1 (318 mg, 88%). Anal. Found: C, 47.45; H, 4.42; N, 3.91. C28H32Cl2N2U calc.: C, 47.67; H, 4.57; N, 3.97%. 1H NMR (THF-d8) δ: 18.08 (s, 12H, Me), 15.25 (s, 2H, Py), 7.55 (s, 2H, Py), 2.50 (t, J = 6 Hz, 2H, Py), −0.33 (s, 2H, Py), −1.02 (s, 12H, Me).
3.3 Synthesis of (Cp∗Py)2UMe2 (2)
A flask was charged with UCl4 (201 mg, 0.53 mmol) and LiCp∗Py (216 mg, 1.05 mmol) and THF (10 mL) was condensed in it. The mixture was stirred for 15 h at 20 °C and the solvent was evaporated off. MeLi (29.4 mg, 1.3 mmol) was added and THF (10 mL) was condensed in. After 1 h at 20 °C, the solvent was evaporated off and the residue extracted with toluene (15 mL). The solution was evaporated to dryness, leaving a pale brown powder of 2 (318 mg, 88%). Anal. Found: C, 54.03; H, 5.63; N, 4.11. C30H38N2U calc.: C, 54.21; H, 5.76; N, 4.21%. 1H NMR (toluene-d8) δ: 26.56 (s, 2H, Py), 11.99 (t, J = 6 Hz, 2H, Py), 8.40 (t, J = 7 Hz, 2H, Py), 8.18 (s, 12H, Me), 4.11 (d, J = 7 Hz, 2H, Py), −12.99 (s, 12H, Me), −16.40 (s, 6H, Me).
3.4 Synthesis of (Cp∗Py)2U(BH4)2 (3)
A flask was charged with UCl4 (204 mg, 0.54 mmol) and LiCp∗Py (220 mg, 1.07 mmol) and THF (15 mL) was condensed in it. The mixture was stirred for 15 h at 20 °C and LiBH4 (30.0 mg, 1.4 mmol) was added. After 7 h at 20 °C, the solvent was evaporated off. A first extraction with toluene (15 mL) gave, after evaporation, a red powder of 3 (110 mg, 31%) contaminated with some impurities. A second extraction with toluene (15 mL) afforded the pure product (125 mg, 35%). Anal. Found: C, 50.46; H, 5.97; N, 4.37. C28H40B2N2U calc.: C, 50.63; H, 6.07; N, 4.22%. 1H NMR (toluene-d8) δ: 24.40 (s, 12H, Me), 7.86 (s, 12H, Me), 1.54 (t, J = 7 Hz, 2H, Py), −1.50 (d, J = 7 Hz, 2H, Py), −2.24 (t, J = 7 Hz, 2H, Py), −8.61 (d, J = 7 Hz, 2H, Py), −35.42 (q, J = 92 Hz, 8H, BH4).
3.5 Synthesis of [(Cp∗Py)2U(BH4)][BPh4] (4)
A flask was charged with 4 (40.0 mg, 0.060 mmol) and HNEt3BPh4 (24.0 mg, 0.057 mmol) and THF (10 mL) was condensed in it. After stirring for 2 h at 20 °C, the mixture was evaporated to dryness and the orange powder of 4 was washed with toluene (15 mL), filtered off and dried under vacuum (50 mg, 86%). Anal. Found: C, 64.31; H, 5.93; N, 2.99. C52H56B2N2U calc.: C, 64.48; H, 5.83; N, 2.89%. 1H NMR (THF-d8) δ: 48.82 (s, 2H, Me), 36.57 (s, 6H, Me), 13.30 (s, 2H, Py), 6.9–6.2 (m, 20H, BPh4), 3.01 (s, 2H, Py), −3.22 (s, 6H, Me), −8.68 (s 2H, Py), −23.33 (s, 6H, Me), −28.96 (s, 2H, Py), −34.93 (q, J = 97 Hz, 4H, BH4).
3.6 Crystals of the ansa-metallocene C2H4(η5-C5Me3-2-C5H4N)2UCl2 (5)
An NMR tube was charged with 2 (7.6 mg, 0.011 mmol) and CuCl2 (1.6 mg, 0.012 mmol) in THF-d8 (0.5 mL). After 2 weeks at 20 °C, a black precipitate was formed together with a few dark red crystals of 5.
3.7 Crystallography
Data were collected at 100(2) K on a Nonius Kappa-CCD area detector diffractometer [18] using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) and processed with HKL2000 [19]. The structures were solved by direct methods with SHELXS-97 and refined by full-matrix least-squares on F2 with SHELXL-97 [20]. Absorption effects were empirically corrected with the program DELABS in PLATON [21]. All non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were introduced at calculated positions and were treated as riding atoms with an isotropic displacement parameter equal to 1.2 (CH, CH2) or 1.5 (CH3) times that of the parent atom. The molecular plots were drawn with SHELXTL [22]. Crystal data and structure refinement details are given in Table 1.
Crystal data and structure refinement details for complexes 1 and 5
| 1 | 5 | |
| Chemical formula | C28H32Cl2N2U | C28H30Cl2N2U |
| M (g mol−1) | 705.49 | 703.47 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | P21/n |
| a (Å) | 18.0216(16) | 10.6046(5) |
| b (Å) | 8.3429(6) | 14.2218(6) |
| c (Å) | 17.9960(17) | 16.8635(6) |
| β (°) | 111.562(4) | 106.075(2) |
| V (Å3) | 2516.4(4) | 2443.85(18) |
| Z | 4 | 4 |
| ρcalcd (g cm−3) | 1.862 | 1.912 |
| μ (Mo Kα) (mm−1) | 6.682 | 6.880 |
| F(000) | 1360 | 1352 |
| Reflections collected | 8173 | 16480 |
| Independent reflections | 2329 | 4612 |
| Observed reflections [I > 2σ(I)] | 1957 | 3707 |
| Rint | 0.090 | 0.059 |
| Parameters | 150 | 304 |
| R1 | 0.036 | 0.031 |
| wR2 (all data) | 0.075 | 0.068 |
| S | 0.982 | 1.034 |
| Δρmin (e Å−3) | −0.95 | −1.22 |
| Δρmax (e Å−3) | 0.83 | 0.84 |
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (CCDC 612606 and 612607).