

1 Introduction
The growth of catalytic asymmetric synthesis has exploded in the past several decades [1] and is now arguably the most important synthetic method for the production of chiral compounds. This has made homogeneous hydrogenation of functionalized olefins one of the most studied enantioselective catalytic reactions [2a,b]; of equal practical interest is the hydrogenation of CO and CN bonds.
The most efficient catalysts for the hydrogenation of functionalized ketones are based on Rh- and Ru-diphosphine catalyst [3]. The method of choice for reducing unfunctionalized ketones, on the other hand, is Noyori's newly developed Ru-diphosphine/diamine catalyst system [4]. Although both Ru-diphospine/diamine complexes 1 [e.g., P-P = (S)-BINAP, N-N = (S,S)-1,2-diphenylethylenediamine] and the widely used Ru–arene/diamine complex RuCl(TsDPEN)(η6-arene) 2 [TsDPEN = N-(p-toluenesulfonyl)-(S,S)-1,2-diphenylethylene-diamine] (Fig. 1) contain a characteristic NH functionality, the catalytic activity of these two catalyst systems differs tremendously.
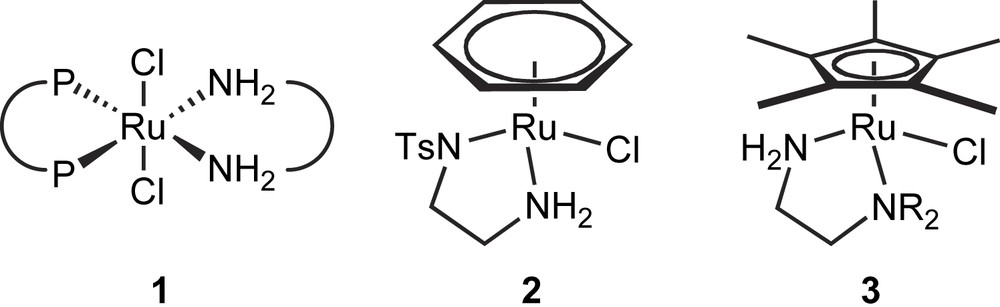
General hydrogenation and transfer hydrogenation complexes.
Complexes of the type 2 are typically used in transfer hydrogenation and yet exhibit almost no reaction as hydrogenation catalysts [5], while RuCp∗-1,2-diamine complexes of the type 3 do so easily.
Recently, it has been demonstrated that RuCp∗-1,2-diamine complexes of the general type 3, which are isoelectronic to the transfer hydrogenation catalyst 2 (Fig. 1), are active catalysts for the hydrogenation of ketones with H2 as the source of hydride [6,7]. These findings are some of the few known examples of homogeneous hydrogenation catalysts that are capable of activating molecular hydrogen without having at least one phosphine ligand around the metal center [8a–d]. These studies also showed that the most active catalysts were obtained with a diamine having one tertiary and one primary amino function. Based on these reports we envisaged that the structural motif of our newly developed thiazole [9] and oxazole [10] based ligands, could serve as chiral backbones for the synthesis of new chiral diamines suitable as ligands in the asymmetric phosphine free hydrogenation of various prochiral aryl ketones. These ligands have previously been used as N,P-coordinating ligands in the Ir-catalysed asymmetric hydrogenation of olefins with great success [9,10].
2 Results and discussion
2.1 Thiazole ligand synthesis
Two thiazole amine ligands 5 and 6, capable of forming six-membered ring chelates with transition metals were synthesized in a similar fashion, Scheme 1. Enantiomerically pure tosylates 1 and 2 were prepared as described in the literature [9]. Nucleophilic displacement by NaN3 in DMF at 80 °C yielded the corresponding azides 3 and 4, with less than 1% elimination product observed by 1H NMR. Azides 3 and 4 were then reduced to their corresponding amines 5 and 6 with H2 over Pd/C at 30 bar.
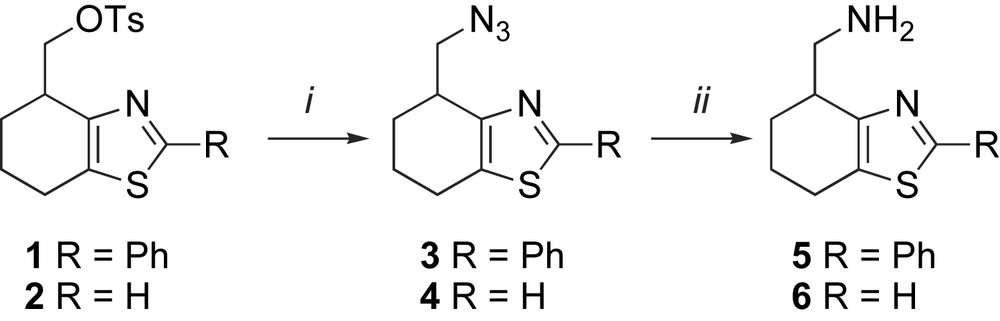
Synthesis of ligands 5 and 6. (i) NaN3, DMF, 80 °C: 3 73% and 4 63%; (ii) Pd/C, H2 (30 bar): 5 100% and 6 68%.
The same protocol was employed with racemic 1; compound 3 was then resolved via chiral HPLC [Chiralcel OD column, hexane/isopropanol 80:20, tR 9.51 min (R) and 15.1 min (S)] and subsequently hydrogenated giving both the optically pure enantiomers of 5.
Compound 11 [9] was synthesized in a four-step procedure starting from racemic 7, Scheme 2. The ester 7 was hydrolyzed to the corresponding carboxylic acid 8 and converted to amide 9 by formation of an activated ester by treatment with ammonia in situ [11] yielding amide 9 in high yield (97%).
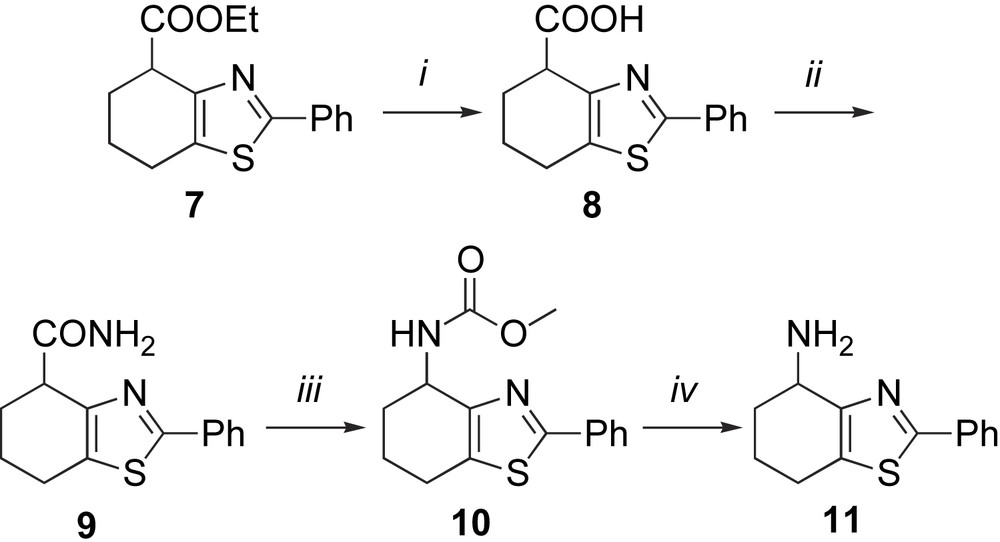
Synthesis of ligand 11. (i) NaOH, EtOH/H2O (6/5), reflux, 92%; (ii) (Boc)2O, pyridine, NH4HCO3, dioxane, RT, 97%; (iii) NBS, AcONa, MeOH, DMF, 60 °C, 16%; (iv) NaOH, EtOH, reflux, 68%.
Several attempts were made to convert amide 9 directly into amine 11 by Hoffmann rearrangements using general procedures such as Br2/NaOH/H2O [12] and Br2/NaOEt/EtOH [13]. Unfortunately, this gave very low conversion due to the low solubility of amide 9. A modified procedure was employed instead, using DMF as the solvent and NBS in presence of AcONa as Hoffmann rearrangement conditions to Br2 and NaOH [14], giving 10 in 16% isolated yield.
Hydrolysis of carbamate 10 in ethanol with an excess of NaOH gave the free amine rac-11, which was resolved by chiral HPLC [Chiralcel OD, hexane/isopropanol 80:20, tR 13.8 min (11a) and tR 19.5 min (11b)] (Scheme 2).
2.2 Oxazole ligand synthesis
Compound 14 was synthesized from previously reported enantiomerically pure 12 [10], Scheme 3. Alcohol 12 was converted into the corresponding azide 13, with inversion of configuration, in 70% yield and 98% ee by a Mitsunobu reaction using diphenyl phosphoryl azide as the azide source [15]. This reaction produced 6% of elimination byproduct that could not be removed by flash chromatography but was easily removed in the next step. Azide 13 was reduced to amine 14 with H2 over Pd/C at 30 bar, 83% isolated yield with 98% ee, Scheme 3.

Synthesis of ligand 14. (i) (PhO)2P(O)N3, DEAD, Ph3P, THF, 0 °C, 15 min, RT 1.5 h, 70%; (ii) Pd/C, isopropanol, H2 (30 bar), 83%.
2.3 Catalyst studies
In order to investigate the activity of these new ligands, we tested (S)-5, (R)-6, (−)-11a and (R)-14 in the hydrogenations of eight different ketones of the general type ArCOR. The results are summarized in Tables 1 and 2. All reactions were run at RT for specified time and at 30 bar H2.
Asymmetric Ru-catalysed hydrogenation of prochiral aryl ketones using ligands (S)-5 and (R)-6
| Entry | Ar | R | (S)-5a | (R)-6a | ||
| % Conv.b | % ee.c | % Conv.b | % ee.c | |||
| 1 | C6H5 | CH3 | 87 | 46 (S) | >99 | 14 (R) |
| 2 | p-CH3OC6H4 | CH3 | 57 | 39 (S) | 98 | rac |
| 3 | o-CH3OC6H4 | CH3 | 48 | 18 (R) | >99 | 16 (R) |
| 4 | p-CH3C6H4 | CH3 | 61 | 48 (S) | >99 | 15 (R) |
| 5 | o-CH3C6H4 | CH3 | 41 | rac | >99 | rac |
| 6 | p-ClC6H4 | CH3 | 98 | 31 (S) | 99 | 15 (R) |
| 7 | p-F3CC6H4 | CH3 | 25 | 14 (S) | 99 | 15 (R) |
| 8 | C6H5 | t-Bu | 3 | 12 (R) | >99 | 23 (R) |
a Reaction time is 2 h at 30 bar H2 (see Section 4).
b Determined by 1H NMR.
c Determined by chiral GC.
Asymmetric Ru-catalysed hydrogenation of prochiral aryl ketones using ligands (−)-11a and (R)-14
| Entry | Ar | R | (−)-11aa | (R)-14a | ||
| % Conv.b | % ee.c | % Conv.b | % ee.c | |||
| 1 | C6H5 | CH3 | >99 | 42 (S) | >99 | 43 (S) |
| 2 | p-CH3OC6H4 | CH3 | >99 | 57 (S) | >99 | 60 (S) |
| 3 | o-CH3OC6H4 | CH3 | >99 | 37 (S) | >99 | 47 (S) |
| 4 | p-CH3C6H4 | CH3 | >99 | 58 (S) | >99 | 60 (S) |
| 5 | o-CH3C6H4 | CH3 | >99 | 17 (S) | >99 | rac |
| 6 | p-ClC6H4 | CH3 | >99 | 54 (S) | 97 | 63 (S) |
| 7 | p-F3CC6H4 | CH3 | >99 | 43 (S) | 42 | 48 (S) |
| 8 | C6H5 | t-Bu | 56 | 78 (S) | 61 | 74 (S) |
a Reaction time is 0.5 h at 30 bar H2 (see Section 4).
b Determined by 1H NMR.
c Determined by chiral GC.
One can see that the presence of electron-donating groups, methoxy and methyl, in the para position of the ketone, results in reduced conversions compared to acetophenone (after 2 h). The reverse is true with the electron withdrawing chorine (Table 1, (S)-5 entries 1, 2, 4 and 6), however, the highly electron withdrawing p-trifluoromethyl group results in only 25% conversion of the corresponding ketone (Table 1, (S)-5 entry 7). Moving the electron-donating group from the para to the ortho position (Table 1, compare entries 2 and 4 with 3 and 5) resulted in a further decrease of conversion.
While, in the case of ligand 5, there appears to be a correlation between the electronic character of the substrate substituents and conversions; there is no such obvious trend with substituents and enantioselectivities.
The ligand 6 results in a much more active catalyst, with conversions greater than 98% in 2 h, but at the expense of selectivity, Table 1. In contrast to results obtained with many transfer hydrogenation catalysts [7], increasing the steric bulk of the R group leads to an increase in stereoselectivity (cf. ligand 6 entries 1 and 8, Table 1). Whereas for the catalyst derived from ligand 5 the selectivity for the same substrate is inverted.
Interestingly, changing to five-membered chelating ligands, 11 and 14, results in a more active and selective catalyst, Table 2. The ee's obtained with these catalysts follow the same pattern as we have previously reported [7].
3 Conclusions
In conclusion, we have investigated the scope of the previously reported oxazole and thiazole moieties in the asymmetric hydrogenation of ketones. These newly synthesized diamine ligands are highly active in the Ru-catalysed asymmetric hydrogenation of a number of aryl ketones. We have shown in this study that a five-membered chelate is preferable over a six-membered chelate.
4 Experimental
All reactions were run using dried glassware and magnetic stirring. THF was freshly distilled under N2 from a deep-blue solution of sodium-benzophenone ketyl prior to use. CH2Cl2 was freshly distilled under N2 from powdered CaH2 prior to use. Flash chromatography was performed using silica gel 60 Å (37–70 μm). Analytical TLC was carried out utilizing 0.25 mm precoated plates, silica gel 60 UV254 and spots were visualized by use of UV light and/or treatment with an ethanol solution of phosphomolybdic acid (5%) followed by heating. For NMR-spectroscopy, samples were dissolved in CDCl3 and run at room temperature. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on a 400-MHz spectrometer. Chemical shifts for protons are reported using the residual CHCl3 as internal reference (δ 7.26). Carbon signals are referenced to the shift from the 13C signal of CDCl3 (δ 77.0). Mass spectra were measured at 70 eV (EI). Melting points are reported as their uncorrected values.
The ee's of synthesized compounds were determined by chiral chromatography on either a Chiralcel OD-H or Chiralpak AS-H column (4.6 × 250 mm), using hexane/isopropanol as mobile phase. The ees of hydrogenated ketones were determined by chiral GC on CP-Chirasil-Dex column with 15 psi N2 as carrier gas and FID detection [7].
4.1 General procedure for the preparation of compounds 3 and 4
4.1.1 (R/S)-4-(Azidomethyl)-2-phenyl-4,5,6,7-tetrahydrobenzo[d]thiazole (3)
Compound rac-1 (4.80 g, 12.0 mmol, 1.0 eq) and NaN3 (1.17 g, 18.0 mmol, 1.5 eq) were suspended in DMF (20 mL) and heated slowly in oil bath to 80 °C over 1 h. Temperature was kept at 80 °C for 5 h until no starting material was detected by TLC (pentane/EtOAc 75:25). Water (30 mL) and Et2O (100 mL) were added. After separating the aqueous layer, the organic phase was washed with water (5 × 30 mL), dried (Na2SO4) and evaporated under vacuum to give 2.69 g of crude azide 3. After flash chromatography on silica (pentane/EtOAc 95:5) 2.38 g (73%) of pure azide 3 was obtained as a colourless oil. Separation on semi-preparative Chiralcel OD column [hexane/isopropanol 80:20, tR 9.51 min (R) and 15.1 min (S)] afforded two enantiomers as clear oils with >99% ee; Rf = 0.58 (pentane/EtOAc 90:10); [α]D23 +113.5 (S) enantiomer (c 1.2, CHCl3); IR (neat) νmax 2936, 2860, 2099, 1541, 1502, 1463, 1267, 979, 762 and 689 cm−1; 1H NMR δ: 1.73–1.88 (m, 2H, CH2), 1.92–2.13 (m, 2H, CH2), 2.79–2.84 (m, 2H, CH2CC), 3.15 (m, 1H, CHCH2N3), 3.60 (dd, J = 12.1, 8.3 Hz, 1H, CH2N3), 3.92 (dd, J = 12.1, 3.9 Hz, 1H, CH2N3), 7.34–7.45 (m, 3H, o/p-CH), 7.86–7.94 (m, 2H, m-CH); 13C NMR δ: 21.4, 23.7, 26.5, 37.5, 54.9, 126.2, 128.8, 129.6, 131.4, 133.9, 151.0, 164.9; MS (EI) (m/z) (rel. intensity) 271 (MH+, 100%), 270 (M, 14%), 215 (15%), 214 (69%), 213 (17%), 212 (10%) and 111 (13%).
4.1.2 (R)-4-(Azidomethyl)-4,5,6,7-tetrahydrobenzo[d]thiazole (4)
Prepared from enantiomerically pure (S)-2. Colourless oil, 63%; >99% ee; Rf = 0.56 (pentane/EtOAc 75:25); [α]D22 +8.80 (c 0.83, CHCl3); IR (neat) νmax 2941, 2864, 2102, 1543, 1450, 1418, 1274 and 909 cm−1; 1H NMR δ: 1.70–1.90 (m, 2H, CH2), 1.91–2.12 (m, 2H, CH2), 2.77–2.84 (m, 2H, CH2CC), 3.14 (m, 1H, CHCH2N3), 3.58 (dd, J = 12.1, 8.0 Hz, 1H, CH2N3), 3.82 (dd, J = 12.1, 3.9 Hz, 1H, CH2N3), 8.59 (s, 1H, CHN); 13C NMR δ: 21.4, 23.5, 26.5, 37.3, 54.8, 131.0, 149.8, 150.6; MS (EI) (m/z) (rel. intensity) 195 (MH+, 100%) and 138 (10%).
4.2 General procedure for the preparation of compounds 5 and 6
4.2.1 (S)-(2-Phenyl-4,5,6,7-tetrahydrobenzo[d]thiazol-4-yl)methanamine (5)
To a solution of (S)-3 (0.16 g, 0.60 mmol) in EtOAc (3 mL) was added Pd/C (10%) (10 mg, 0.6%).
The reaction vessel was placed in high-pressure equipment and charged with H2 (30 bar) and stirred overnight (reduction is complete after several hours). After filtering through Celite the solids were washed with Et2O. The solution was evaporated under vacuum to give 0.15 g (quantitative) of amine 5 as a yellowish oil; Rf = 0.51 (methanol/Et3N 80:20); [α]D22.7 +66.9 (c 1.0, CHCl3); IR (neat) νmax 3365, 3293, 3062, 2933, 2861, 2214, 1598, 1581, 1538, 1462, 1336, 1313, 977, 910, 763, 732 and 690 cm−1; 1H NMR δ: 1.55–1.86 (m, 4H, CH2, NH2), 1.89–2.04 (m, 2H, CH2), 2.71–2.81 (m, 2H, CH2CC), 2.86 (m, 1H, CHCC), 2.93 (dd, J = 12.4, 5.8 Hz, 1H, CH2NH2), 3.06 (dd, J = 12.3, 6.0 Hz, 1H, CH2NH2), 7.30–7.43 (m, 3H, o/p-CH), 7.83–7.91 (m, 2H, m-CH); 13C NMR δ: 21.7, 23.7, 26.7, 40.5, 46.8, 126.0, 128.6, 129.3, 130.1, 133.9, 153.5, 164.3; MS (EI) (m/z) (rel. intensity) 245 (MH+, 51%), 244 (M, 10%), 216 (19%), 215 (100%), 214 (66%) and 121 (17%).
4.2.2 (R)-(4,5,6,7-Tetrahydrobenzo[d]thiazol-4-yl)methanamine (6)
Yellowish oil, 68%; Rf = 0.48 (methanol/Et3N 80:20); [α]D24.2 +34.5 (c 1.3, CHCl3); IR (neat) νmax 3391, 2935, 1634 and 1415 cm−1; 1H NMR δ: 1.52 (br s, 2H, NH2), 1.61–1.85 (m, 2H, CH2CHCC), 1.91–2.04 (m, 2H, CH2CH2CC), 2.70–2.82 (m, 2H, CH2CC), 2.88 (m, 1H, CHCC), 2.95 (dd, J = 12.3, 6.1 Hz, 1H, CH2NH2), 3.06 (dd, J = 12.3, 5.5 Hz, 1H, CH2NH2), 8.57 (t, J = 0.7, 1H Hz, CHN); 13C NMR δ: 21.7, 23.6, 26.7, 40.4, 46.5, 129.8, 149.5, 153.0; MS (EI) (m/z) (rel. intensity) 169 (MH+, 9%), 140 (13%), 139 (88%), 138 (100%), 111 (15%) and 77 (12%).
4.3 Procedures for the preparation of compounds 8–14
4.3.1 2-Phenyl-4,5,6,7-tetrahydrobenzo[d]thiazole-4-carboxylic acid (8)
To a solution of thiazole ester 7 (12.68 g, 46.1 mmol, 1.0 eq) in 96% ethanol (60 mL) was added 2 M NaOH (50 mL, 0.10 mol NaOH, 2.2 eq) and the reaction mixture was refluxed for 1 h. After evaporation of ethanol, water (200 mL) and CH2Cl2 (200 mL) were added. The aqueous phase was acidified with conc. HCl to pH approx. 5. After separating the organic layer, the aqueous layer was extracted several times with CH2Cl2 (4 × 100 mL), restoring the pH to 5 after each extraction. The combined organic extracts were dried (Na2SO4) and evaporated under vacuum to give 10.99 g (92%) of carboxylic acid 8 as a white solid. mp 156.9–159.8 °C; Rf = 0.28 (pentane/AcOH 80:20); IR (KBr) νmax 3050, 2945, 2863, 1948, 1735, 1700, 1550, 1500, 1462, 1394, 1261, 1222, 1177, 981, 860 and 761 cm−1; 1H NMR δ: 1.84 (m, 1H, CH2), 1.97–2.20 (m, 2H, CH2), 2.35 (m, 1H, CH2), 2.72–2.84 (m, 2H, CH2), 3.85 (m, 1H, CH), 7.38–7.48 (m, 3H, o/p-CH), 7.79–7.89 (m, 2H, m-CH), 12.2 (br s, 1H, COOH); 13C NMR δ: 22.1, 23.5, 25.1, 42.0, 126.3, 129.1, 130.6, 131.8, 132.4, 146.4, 166.2, 172.3; MS (EI) (m/z) (rel. intensity) 261 (MH+, 25%), 260 (M, 100%), 215 (51%) and 214 (42%).
4.3.2 2-Phenyl-4,5,6,7-tetrahydrobenzo[d]thiazole-4-carboxamide (9)
To a solution of compound 8 (10.31 g, 39.8 mmol, 1.0 eq) in dioxane (100 mL) was added pyridine (2 mL, 24.8 mmol, 0.62 eq), (Boc)2O (12.06 g, 55.3 mmol, 1.4 eq) and NH4HCO3 (3.98 g, 50.3 mmol, 1.3 eq). Solution was stirred overnight (after several hours the amide starts to precipitate). Most of the dioxane was evaporated under vacuum and water (20 mL) was added. The solids were filtered on a Buchner funnel, washed with water (40 mL) and dried in exicator to give 9.92 g (96.5%) of amide 9 as a fine white powder. mp 177.4–181.9 °C; Rf = 0.15 (pentane/AcOH 80:20); IR (KBr) νmax 3387, 3164, 2946, 1683, 1616, 1545, 1376, 1172, 973, 881, 775, 692 and 570 cm−1; 1H NMR δ: 1.84–2.03 (m, 3H, 2 × CH2CH2CHCO and CH2CHCO), 2.45 (m, 1H, CH2CHCO), 2.70–2.88 (m, 2H, CH2CC), 3.77 (m, 1H, CHCO), 5.75 (br s, 1H, NH2), 7.21–7.48 (m, 4H, o/p-CH and NH2), 7.78–7.92 (m, 2H, m-CH); 13C NMR δ: 21.4, 23.7, 24.6, 42.1, 126.2, 128.9, 129.9, 131.9, 133.4, 148.2, 165.4, 174.0; MS (EI) (m/z) (rel. intensity) 259 (MH+, 25%), 258 (M, 100%), 216 (15%), 215 (82%), 214 (88%), 213 (49%), 212 (13%), 180 (16%), 121 (20%), 111 (18%), 104 (15%) and 77 (14%).
4.3.3 Methyl 2-phenyl-4,5,6,7-tetrahydrobenzo[d]thiazol-4-ylcarbamate (10)
Compound 9 (1.00 g, 3.87 mmol, 1.0 eq) and AcONa (0.95 g, 12 mmol, 3.0 eq) were suspended in DMF (15 mL). Methanol (3.1 mL, 76 mmol, 20 eq) was added and suspension was immersed into ice bath whereby a solution of NBS (0.76 g, 4.3 mmol, 1.1 eq) in DMF (3 mL) was added dropwise. The suspension was heated slowly in oil bath to 60 °C over 2 h. After cooling, the solution was neutralised by 5% NaHCO3 (10 mL) and extracted by Et2O (50 mL). The organic phase was washed with water (3 × 10 mL), dried (Na2SO4) and evaporated under vacuum to give 0.785 g of crude material. Recrystallization from CH2Cl2:pentane gave 0.17 g (16%) pure thiazole carbamate 10 as fine white powder. mp 150.7–157.0 °C; Rf = 0.26 (pentane/EtOAc 75:25); IR (KBr) νmax 3282, 2949, 2248, 1706, 1545, 1464, 1337, 1253, 1192, 1071, 977, 909, 766, 733 and 692 cm−1; 1H NMR δ: 1.69–1.87 (m, 2H, CH2CH2CHNH), 1.93 (m, 1H, CH2), 2.08 (m, 1H, CH2), 2.47–2.72 (m, 2H, CH2), 3.71 (s, 3H, CH3), 4.81 (m, 1H, CHNH), 5.84 (br s, 1H, NH), 7.30–7.44 (m, 3H, o/p-CH), 7.74–7.88 (m, 2H, m-CH); 13C NMR δ: 19.8, 23.4, 29.8, 47.9, 51.9, 126.3, 128.7, 129.7, 133.0, 133.5, 149.9, 156.6, 165.3; MS (EI) (m/z) (rel. intensity) 290 (20%), 289 (MH+, 100%), 288 (M, 33%), 230 (16%), 229 (84%), 214 (24%), 213 (62%), 212 (20%) and 180 (14%).
4.3.4 (R/S)-2-Phenyl-4,5,6,7-tetrahydrobenzo[d]thiazol-4-amine (11)
Compound 10 (63 mg, 0.22 mmol, 1.0 eq) and NaOH (94 mg, 2.4 mmol, 11 eq) were dissolved in 96% ethanol (5 mL) and refluxed for 2 h. After evaporation of the solvent in vacuum, the residue was dissolved in water/Et2O (5:5 mL) and acidified with conc. HCl until pH < 1. After vigorous stirring and removal of the organic phase, the aqueous phase was basified with 2 M NaOH solution until pH > 12 and extracted by CH2Cl2 (3 × 5 mL). The combined organic extracts were dried (Na2SO4) and evaporated under vacuum to give 34 mg (68%) amine 11 as a colourless oil that crystallised on standing. mp 146.8–149.6 °C (racemic). Amine was separated on a semi-preparative Chiralcel OD column (hexane/isopropanol 80:20) to give two enantiomers 11a (tR 13.8) and 11b (tR 19.5), >99% ee; Rf = 0.58 (methanol/Et3N 95:5); [α]D23.6 −78.1 (c 0.75, CHCl3) 11a; [α]D23.7 +67.4 (c 0.75, CHCl3) 11b; IR (KBr) νmax 3327, 3056, 2943, 1690, 1465, 1203, 1171, 1127, 976, 828, 798, 760 and 689 cm−1; 1H NMR δ: 1.64–1.86 (m, 2H, CH2CH2CC), 1.96 (m, 1H, CH2CHNH2), 2.14 (m, 1H, CH2CHNH2), 2.72 (ddd, J = 12.2, 5.7, 1.5 Hz, 2H, CH2CC), 3.90 (br s, 2H, NH2), 4.17 (m, 1H, CHNH2), 7.27–7.47 (m, 3H, o/p-CH), 7.74–7.93 (m, 2H, m-CH); 13C NMR δ: 20.7, 23.6, 31.2, 47.8, 126.3, 128.8; MS (EI) (m/z) (rel. intensity) 232 (MH+, 16%), 231 (M, 100%), 230 (17%), 229 (37%), 215 (11%), 214 (36%), 213 (91%), 203 (15%), 202 (85%), 201 (24%), 175 (27%), 121 (18%).
4.3.5 (R)-6,6-Dimethyl-2-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-4-azide (13)
Compound 12 (1.00 g, 4.11 mmol, 1.0 eq) and triphenyl phosphine (1.29 g, 4.92 mmol, 1.2 eq) were dissolved in dry THF (15 mL) and cooled to 0 °C. Diphenyl phosphoryl azide (1.06 mL, 4.92 mmol, 1.2 eq) and 85% DEAD (0.93 mL, 5.0 mmol, 1.2 eq) were added and the solution was stirred for 10 min at 0 °C and then for 1.5 h at RT. The solvent was evaporated under vacuum and the residue was chromatographed on a silica column (Et2O/pentane 10:90) to give 0.77 g (70%) of crude azide as a colourless oil, containing 6% of elimination product (1H NMR) that could not be separated. Product was used as is in the next step. Rf = 0.47 (pentane/Et2O 90:10); HPLC: Chiralcel OD-H column (4.6 × 250 mm), hexane/isopropanol 80:20, 0.5 mL/min, tR 9.31 min minor (S) and 13.2 min major (R), 98% ee; [α]D23.3 −30.2 (c 1.42, CHCl3); IR (neat) νmax 2958, 2929, 2871, 2098, 1646, 1553, 1488, 1449, 1253, 1058, 1025, 921, 775, 710 and 693 cm−1; 1H NMR δ: 1.03 (s, 3H, Me), 1.17 (s, 3H, Me), 1.68 (dd, J = 13.5, 7.9 Hz, 1H, CH2CHN3), 1.95 (ddd, J = 13.4, 5.9 Hz, 1.1, 1H, CH2CHN3), 2.46 (ddd, J = 16.6, 1.6, 1.1 Hz, 1H, CH2CC), 2.58 (dd, J = 16.6, 2.1 Hz, 1H, CH2CC), 4.75 (ddt, J = 7.8, 6.0, 1.7 Hz, 1H, CHN3), 7.36–7.46 (m, 3H, m/p-CH), 7.97–8.06 (m, 2H, o-CH); 13C NMR δ: 26.8, 29.8, 32.6, 35.2, 42.2, 53.6, 126.1, 127.4, 128.5, 130.0, 132.5, 148.5, 161.1; MS (EI) (m/z) (rel. intensity) 269 (MH+, 35%), 268 (M, 12%), 227 (21%) and 226 (100%).
4.3.6 (R)-6,6-Dimethyl-2-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-4-amine (14)
Compound 13 (0.43 g, 1.6 mmol) was dissolved in isopropanol (4 mL) followed by addition of Pd/C (10%) (0.10 g, 2.3%); the vial was placed in the high-pressure equipment and charged with H2 (50 bar) and stirred for 14 h at RT. The solution was filtered through Celite and evaporated. Et2O (20 mL) and water (20 mL) were added and the solution was acidified by conc. HCl until pH < 1; after vigorous stirring and removal of the organic phase the aqueous phase was washed with another portion of Et2O (10 mL). The aqueous phase was basified with conc. NaOH (aq) until pH > 12, extracted with CHCl3 (2 × 20 mL), the combined organic phase was dried (Na2SO4) and the solvent was removed under vacuum to give 0.33 g (83%) of amine 14 as a yellowish oil. Rf = 0.61 (methanol/Et3N 95:5); HPLC: Chiralpak AS-H column (4.6 × 250 mm), hexane/isopropanol 95:5, 0.5 mL/min, tR 17.4 min minor (S) and 21.69 min major (R), 98% ee; IR (neat) νmax 3362, 2956, 1645, 1550, 1486, 1449, 1388, 1367, 1285, 1062, 1025, 775, 727, 700 and 693 cm−1; 1H NMR δ: 1.00 (s, 3H, CH3), 1.15 (s, 3H, CH3), 1.36 (dd, J = 13.0, 9.7 Hz, 1H, CH2CNH2), 1.77 (brs, 2H, NH2), 1.91 (ddd, J = 13.0, 5.7 Hz, 1.6, 1H, CH2CNH2), 2.44 (dt, J = 16.3, 1.6 Hz, 1H, CH2C = C), 2.55 (dd, J = 16.3, 2.6 Hz, 1H, CH2CC), 3.90 (dddd, J = 9.9, 5.7, 2.7, 1.6 Hz, 1H, CHNH2), 7.33–7.47 (m, 3H, m/p-CH), 7.93–8.03 (m, 2H, o-CH); 13C NMR δ: 26.1, 31.5, 32.9, 35.9, 44.4, 47.3, 126.2, 128.2, 128.9, 130.0, 138.2, 146.7, 161.0; MS (EI) (m/z) (rel. intensity) 243 (MH+, 13%), 242 (M, 15%), 226 (13%), 225 (13%), 189 (71%), 187 (16%), 186 (100%), 171 (25%), 158 (14%), 104 (13%) and 77 (15%).
4.4 General procedure for hydrogenations
A Schlenk tube was charged with appropriate diamino ligand (0.0190 mmol), Ru-complex [Cp∗RuCl]4 (0.00475 mmol) and 3 mL isopropanol, under N2 atmosphere. The solution was stirred at RT for 30 min; 0.110 M solution of tBuOK in isopropanol (0.170 mL, 0.0190 mmol) and substrate (1.90 mmol) was added. The reaction mixture was loaded via a syringe into N2 filled test tube containing a stirring bar. Syringe was washed with isopropanol (1 mL) and the solvent was combined with reaction mixture. Test tube was placed in the high-pressure equipment and purged several times with H2 and a pressure of 30 bar of H2 was applied for specified time. The pressure was released and solution was first evaporated under reduced pressure, diluted by Et2O (1 mL) filtered through a short plug of silica, and the plug was washed with Et2O (3 mL). The combined solvent was evaporated under vacuum and residue was analysed by 1H NMR (CDCl3) for the conversion and chiral GC (Et2O) for the ee.