

1 Introduction
The application of catalytic reactions in industrial processes, in drug detoxification, in environment bioanalysis and in biodegradation of pollutants makes biocatalysts very attractive compounds to be studied. Among biocatalysts, metal containing enzymes play important roles in numerous chemical and biological transformations. Therefore, increasing attention is being devoted to the possibility of tailoring synthetic catalysts that mimic natural metalloenzymes [1–9].
In metalloenzymes, catalytic activity and stability of the metal cofactors are regulated by specific interactions with amino acid residues in the protein cavities [10,11]. First, the protein matrix shapes the primary coordination shell of the metal ion dictating the composition, number, and geometry of the ligands. In addition, residues in the second shell, the immediate surroundings of the primary coordination sphere, influence a variety of structural and chemical facets, including hydrogen-bonding interactions to the ligands, pKa values of ligand groups, and the control of the metal center oxidation state and redox potential. Finally, neighbouring side chains also exert steric and chemical controls over the ability of the metal ion(s) to bind or discriminate the substrate and to accommodate conformational changes [10,11].
Thus, it is clearly evident that the structures of the metal-binding sites in proteins reflect a delicate interplay between opposing requirements for tight binding of the cofactor, which drives toward static, coordinately saturated geometries, versus function, which requires the cofactor to be coordinately unsaturated, sometimes with unusual geometries, and properly positioned for binding substrates and undergoing dynamic changes in configuration during catalysis.
Therefore, numerous efforts are continuously aimed to a better understanding of the factors that modulate metalloprotein activity, with the ultimate goal of developing metalloenzymes with novel structures and functions [1–9]. The study of natural metalloenzymes and related mutants, generated by site-directed mutagenesis, as well as the study of smaller complexes containing designed organic ligands, has clarified several fundamental properties of natural proteins [12–17]. For example, mutation studies have shown that modest changes brought to an enzyme's structure can have large effects on its catalytic properties. Chemical modification of a single amino acid residue can change the fundamental activity of the enzyme, and a single site mutation can alter its enantioselectivity [12,13].
Even though impressive progress has been made with both strategies, the study of large proteins may be hampered by their extreme complexity, and it is often difficult to design small molecule complexes that can bind multiple substrates and metal ions in water. Therefore, due to the inherent complexity of enzyme structure, tailoring synthetic biocatalysts still is a daunting challenge, since it requires the development of sophisticated molecular architectures that distil the quintessential elements believed to be responsible for the activities.
This problem has been recently approached with the stratagem of synthesizing scaled-down polypeptides that self-assemble with metal ions and cofactors to form metalloprotein mimetics.
These structures are simple enough to avoid much of the ambiguity of interpretation associated with large proteins, and yet should simultaneously be of sufficient size and chemical diversity to allow the construction of functional sites. Thus, these mimetics – which stand at the crossroads of traditional small molecule models and large proteins – should allow one first to understand the properties of natural proteins, and second to construct economical catalysts and sensing devices.
The development of peptide-based models takes advantage of recent progress in both design and synthesis of peptides and proteins. Relatively simple rules have been established for the development of stable and well-defined protein and metalloprotein models, using either minimalist design strategies or de novo design approach [5–7,18,19]. We have approached the challenge of reproducing metalloprotein active sites using both strategies.
We centered our attention on iron-containing proteins, and we developed models for heme proteins (mimochromes) [20–26], iron–sulfur proteins [27,28], and diiron–oxo proteins (DFs) [29].
All these models have a common characteristic: they are fully or partially C2-symmetric systems, intended to reproduce the quasi-symmetrical structure of a metalloprotein. The use of C2 symmetry is particularly advantageous: it simplifies the design, synthesis and structural characterization of the models, even though it leaves open the problem of possible diastereomeric forms.
A miniaturization process was applied in the development of the first two classes of artificial metalloproteins [20–28]. By a detailed analysis of the crystal structures of natural heme proteins and iron–sulfur proteins, we successfully selected the minimum set of constituents necessary for an accurate reconstruction of the active sites' structures. In particular, we selected the type and number of constituents to be assembled, to include all the elements of the primary and secondary coordination shell to the metal. The results obtained on these two class of metalloprotein models, indicated that these simple, structurally defined systems provide a good opportunity for investigating how changes in the polarity, solvent accessibility, and electrostatic potential surrounding the active site affect its electrochemical midpoint potential and chemical reactivity. These molecules are very promising systems in the field of metalloenzymes model compounds: they have a high degree of functional similarity with respect to their natural counterparts, and they are excellent scaffolds to engineer new peptide–metal complexes, with different metal cofactors.
In the field of diiron–oxo proteins, we used a de novo design approach for the development of artificial models. In particular, we developed the DF (Due Ferri) family of artificial proteins, as models of diiron and dimanganese metalloproteins, with the aim of reproducing the functional specificity of the natural systems and with the ultimate goal of developing new metalloenzymes with functions unprecedented in nature [29–42]. The DF proteins were developed through an iterative process of design and rigorous characterization, which allowed us to diagnose the problems associated with the initial models, and to improve the models in subsequent designs. Thus, the problem of designing the DF family was approached through several steps. The first involved the design of stable, uniquely folded proteins that contain the metal binding site; we engineered DF1, which was an antiparallel dimer of helix–loop–helix motif, able to bind the dimetal cofactor near the center of the structure [30–32]. To improve the functional properties of the initial model, DF1 was subjected to several changes in the sequence, as well in the loop structure (DF1 and DF2 subsets) [33–40]. The design further evolved with the construction of versions in which the four helices are distinct chains that come together by non-covalent self assembly (DFtet) [41–43]. Each class has different advantages. For example, the symmetric nature of the dimeric derivatives simplifies interpretation of data, whereas analogues of the four-chain DFtet constructs can be mixed and matched to allow easy generation of combinatorial diversity.
In this paper, we will summarize all the results we obtained to date from our work on the DF family of proteins, by using both experimental and theoretical studies.
2 DF1: a minimal model for diiron proteins
Diiron sites are found in a functionally diverse class of proteins involved in oxygen binding and activation [44–48]. Diiron proteins catalyze hydroxylation, desaturation, and epoxidation reactions on a variety of alkyl and aryl substrates [44–51], and a diiron site in ribonucleotide reductase, which participates in DNA biosynthesis, is responsible for the formation of organic radicals [52]. The hemerythrins are responsible for the transport of O2, and ferritins are important for iron oxidation (ferroxidase activity), storage and transport [53–57]. There are many structural and mechanistic commonalities amongst this class of diiron proteins.
Although the folds of these proteins are frequently complex, a simple four-helix bundle (Fig. 1) houses their active sites [58–62]. The helices have a slight left-handed tilt as in classical four-helix bundles [63] and antiparallel four-stranded coiled coils [64]. Thus, their geometries may be discussed in terms of the heptad nomenclature of coiled coils [64–66].
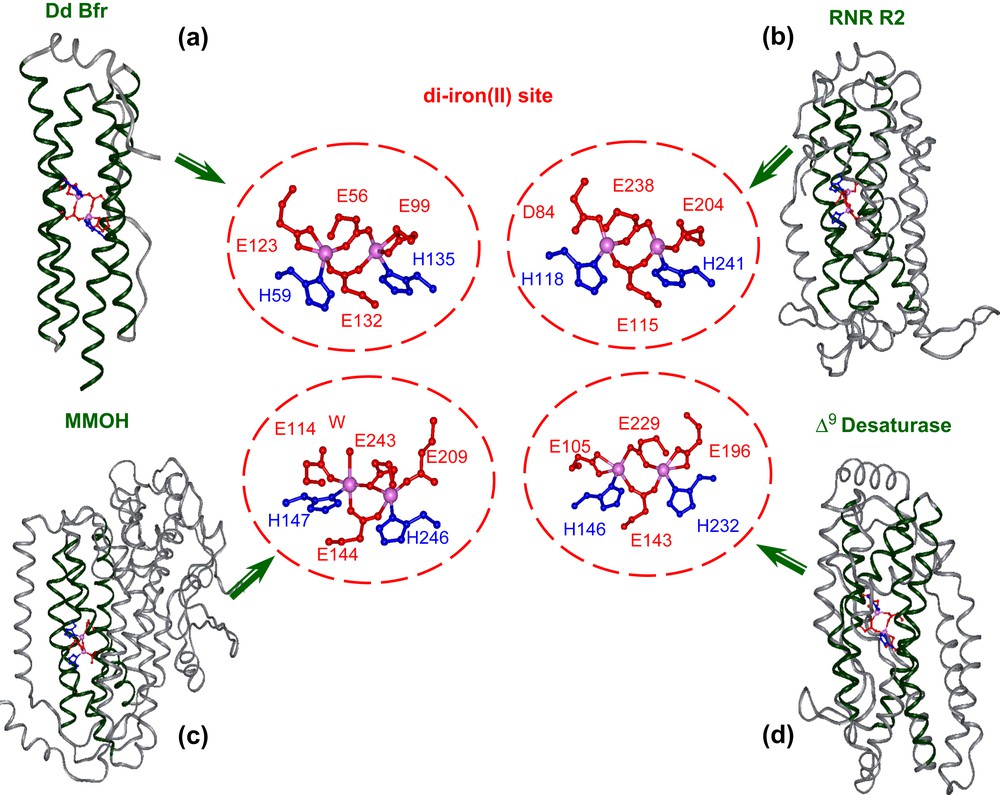
Four-helix bundle motif housing the diiron site and details of the metal binding site in: (a) bacterioferritin (PDB code 1NF4), (b) ribonucleotide reductase (PDB code: 1XIK), (c) hydroxylase component of methane monoxygenase (PDB code: 1FYZ), (d) Δ9-ACP desaturase (PDB code: 1AFR), as derived from X-ray crystallography.
A detailed analysis of the active site of several natural diiron proteins (see Fig. 1) reveals a highly symmetric arrangement of the liganding side chains [58–62]. Almost invariably, each helix contains a single coordinating Glu residue in an “a” position of the heptad motif, which projects towards the center of the bundle. Two carboxylates bridge both metal ions, while the other two interact with a single metal ion in a monodentate or bidentate chelating interaction. Two His residues at “d” position, three residues from the two bridging Glu side chains coordinate the iron as monodentate ligands, thus forming the Glu–Xxx–Xxx–His binding motif common to this class of proteins. Small side chains generally occur at the analogous “d” positions of the other two helices. Importantly, these side chains control the access of oxygen and substrates to the diiron center.
Other residues also help to shape the active sites of diiron proteins. The top and bottom of the active site are defined by the side chains projecting from “d” and “a” of neighboring heptads. The sides of the active site are defined by residues at positions “g” and “c” on two faces of the four-helix bundle, and “b” and “e” on the other two faces (Fig. 2). The residues at the b/e faces of the helical bundle tend to be tightly packed and appear to serve a structural role. In contrast, the c/g faces appear to be important for function; one g/c face presents side chains that help to define the entry and binding of substrates. The opposite c/g face may help to tune the properties of the coordination site by hydrogen bonding to the coordinating His residues, thus forming second-shell interactions.
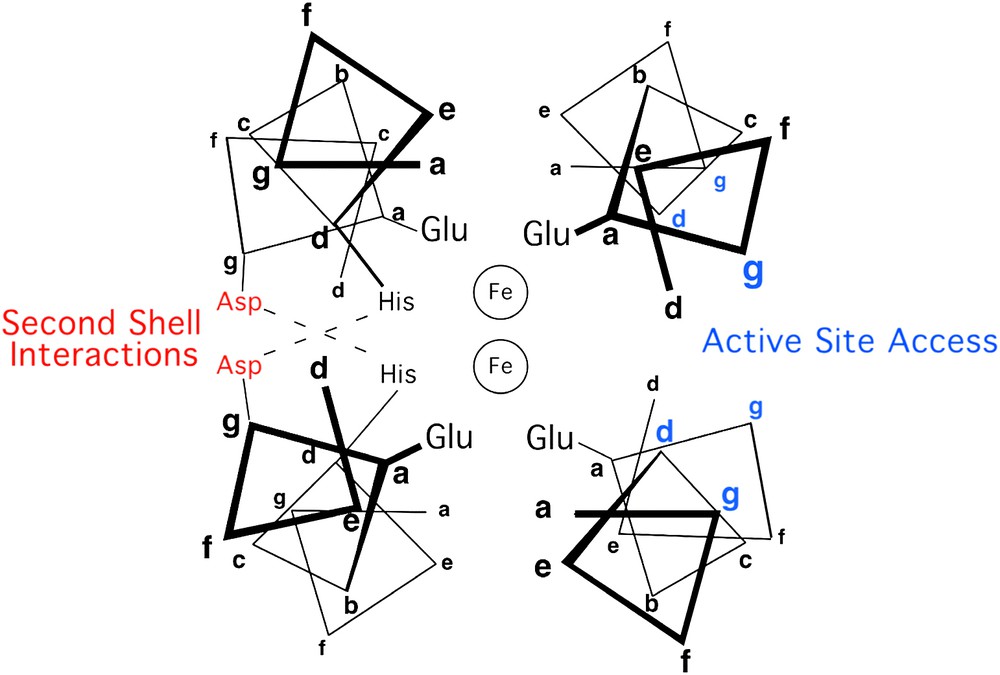
Helix-wheel representation of the antiparallel four-stranded coiled coil in proximity of the metal-binding site, in diiron protein. Residue positions are labeled according to the heptad repeat generally applied to coiled coils.
On the basis of this analysis, we designed the first model, DF1, which is made up of two 48-residue helix–loop–helix (α2) motifs, able to specifically self-assemble into an antiparallel four-helix bundle [30–32]. Fig. 3 reports the amino acid sequence of the α2 motif together with the intended helical secondary structure. To provide a Glu4His2 liganding environment for the diiron center, each α2 subunit contains a Glua–Xxxb–Xxxc–Hisd sequence in helix 2, which donates a His side chain (His39/His39′) as well as a bridging Glu carboxylate (Glu36/Glu36′) to the site. A second Glu carboxylate ligand on helix 1 (Glu10/Glu10′) provides a fourth protein ligand per metal ion. Liganding side chains were placed in the appropriate rotamers to allow interaction with a diiron center. The final positions of the helices were dictated by three different requirements: (1) the geometry of the liganding site was restrained to bind diiron with two bridging Glu carboxylates, two non-bridging Glu side chains, and the Nδ of two His side chains; (2) the helical packing angles and distances were constrained to match those typically observed in the active sites of diiron proteins; (3) precise 2-fold symmetry between the two pairs of helices was enforced.
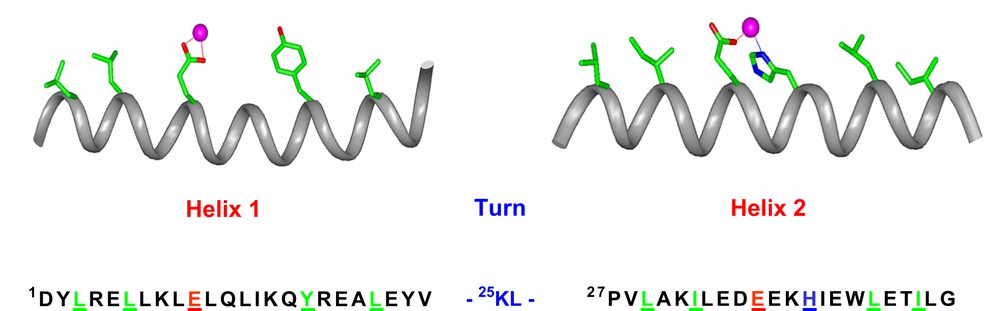
The sequence of DF1. The DF1 sequence is indicated, together with the intended secondary structure. Residues that are expected to be essential for folding and/or function are shown in stick representation on the helices and are underlined in the sequence.
Each metal ion is five-coordinate, and a sixth vacant site lies on adjoining faces of the two metal ions, providing a potential site for binding of exogenous ligands. Satisfaction of side chain packing requirements was ensured by positioning hydrophobic side chains to fill the core as dictated by the steric environment of the backbone structure. In addition, Glu and Lys residues were chosen for helix-favoring, solvent-accessible sites, to provide water solubility and to drive the assembly into the desired antiparallel topology. Finally, an idealized γ–αL–β inter-helical loop was included between the two pairs of helices.
The DF1 sequence was also carefully engineered to include second-shell interactions, which are crucial for defining structural and functional properties of metal-binding sites. Thus, in DF1 a Tyr residue at position 17 donates a second-shell hydrogen bond to the non-bridging E10′ of the other monomer in each of the α2 subunits (the same interaction exists between the symmetrically related pair, Y17′ and E10). Similarly, an Asp residue at position 35′ forms a hydrogen bond with the imidazole Nɛ of the His ligand in the neighboring helix of the dimer. This Asp is further involved in a salt bridge interaction with a Lys at position 38. This hydrogen-bond network consisting of Lys/Asp/His in DF1 is similar to that observed in the active sites of natural proteins, such as methane monooxygenase, where the lysine residue is replaced by an arginine, to form an Arg/Asp/His cluster (see Fig. 4).
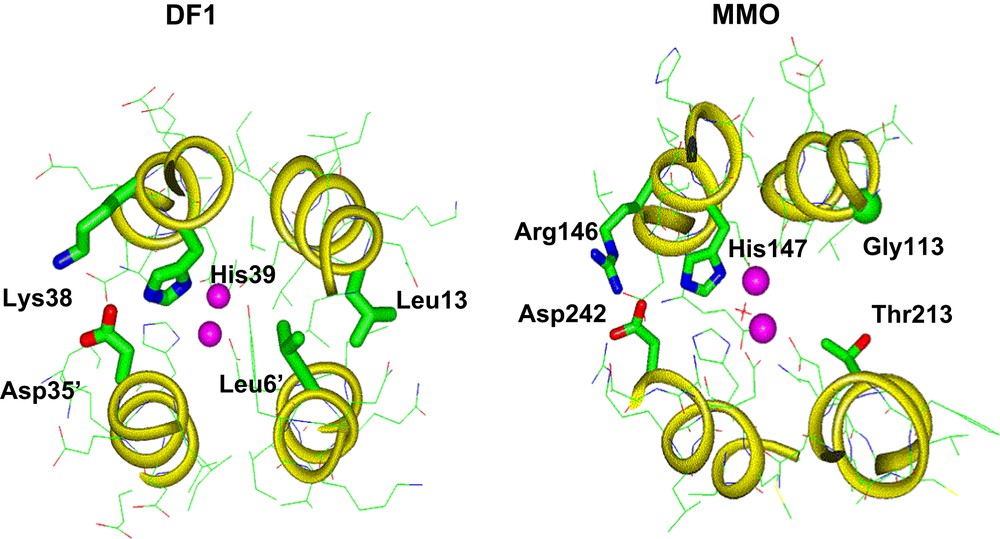
Hydrogen-bonded network consisting of Lys/Asp/His in DF1 (left; PDB code 1EC5) versus Arg/Asp/His, as observed in MMO (right; PDB code 1MTY).
DF1 was prepared in good yield by standard solid-phase methodology. The design effectively resulted in the construction of a protein that adopts a folded, native-like helical conformation in aqueous solution, able to bind divalent metal ions such as Zn(II), Co(II), and Fe(II) [31]. The successful assembly of the protein in the desired fold was proven by several techniques. The CD spectrum for both the apo- and metal-reconstituted protein is consistent with its proposed helical structure. Protein dimerization was ascertained by analytical ultracentrifugation analysis, which indicated that the protein sedimented as a single homogeneous species with an apparent molecular weight of 11,600 Da, consistent with the value expected for a dimer (11,760 Da).
Fig. 5 shows the visible spectrum of the di-Co(II)–DF1 complex, and Table 1 reports spectroscopic data of several Co(II)-substituted DF derivatives. Data for the di-Co(II) derivative of bacterioferritin are also reported, for comparison. Inspection of Table 1 shows that all Co(II)-substituted DF derivatives exhibit absorption bands around 520 nm, 550 nm, and 600 nm, with associated molar extinction coefficients per Co(II) ion of approximately 150 M−1 cm−1 for the most intense 550-nm band. These values are in good agreement with those reported in the literature for the Co(II) derivative of bacterioferritin that contains an identical 4-Glu, 2-His ligation environment [67]. It is well known that Co(II) complexes show distinctive UV–vis spectra, which are sensitive to the metal coordination geometry and to the nature of the ligands [68]. The molar extinction coefficients of the Co(II) d–d transitions increase as the coordination number decreases. Typical values range from approximately 10–20 M−1 cm−1 to 100–150 M−1 cm−1 to 400–600 M−1 cm−1 for hexa-, penta- and tetra-coordinate complexes, respectively [68]. Thus, for all the di-Co(II)–DF complexes, the positions and intensities of the d–d transitions in the visible region are consistent with a pentacoordinate geometry at the cobalt site.
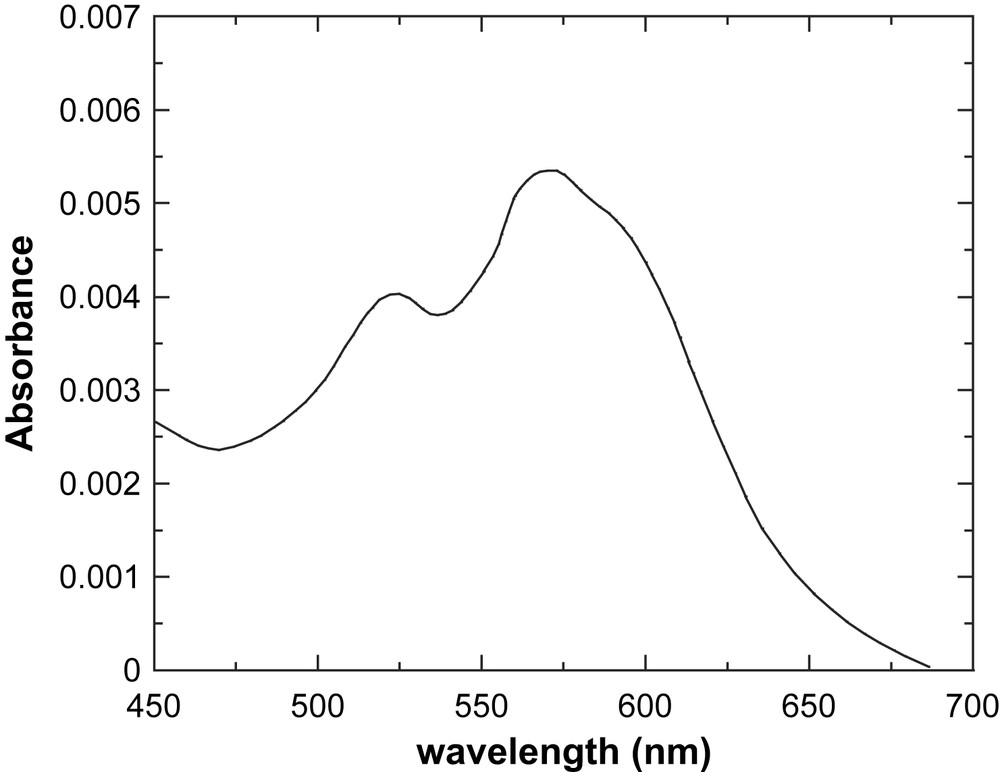
Visible spectrum of DF1 cobalt complex (1.4 × 10−5 M, MOPS buffer pH = 7.0; 1.0-cm path-length cell).
Spectroscopic data of Co(II)-substituted DF derivatives, compared to Co(II)-bacterioferritin
| Protein | λ (nm) | ɛ (M−1 cm−1) | λ (nm) | ɛ (M−1 cm−1) | λ (nm) | ɛ (M−1 cm−1) |
| DF1a | 524 | 143 | 574 | 190 | 594 | 165 |
| DF2b | 520 | 140 | 550 | 155 | 600 | 90 |
| DFtetc | 520 | 125 | 550 | 140 | 597 | 80 |
| DFscd | 529 | 124 | 550 | 136 | 600 | 86 |
| Bacterioferritine | 520 | 126 | 555 | 155 | 600 | 107 |
Detailed information of the overall DF1 structure were obtained from the analysis of the crystal structure of the di-Zn(II) form of the protein (Fig. 6), as well as the NMR structure of the apo-form [31,32]. The overall structure is very similar to the intended designed model, consisting of an antiparallel pair of helical hairpins with the desired topology. The structure of the active site is very close to that found in the diferrous and dimanganous forms of naturally occurring proteins [60,69]. Each metal ion is 5-coordinate, with two Glu carboxylates (Glu36 and Glu36′), each interacting with both zinc ions in a 1,3 syn–syn bidentate bridging interaction, and two Glu side chains (Glu10 and Glu10′), each binding individual ions in a chelating manner (Fig. 6b). Two His residues complete the ligation environment about the dimetal site, by Nδ atom coordination to individual ions. The Zn(II)–Zn(II) distance is 3.9 Å, close to the distance observed between metal ions in the dimanganous and diferrous forms of natural diiron proteins. The pentacoordinate ligand arrangement with two 1,3 bridging carboxylates and two chelating carboxylates is also nearly identical to that observed in diferrous Δ9-ACP desaturase [58] and di-Mn(II)–bacterioferritin [69].
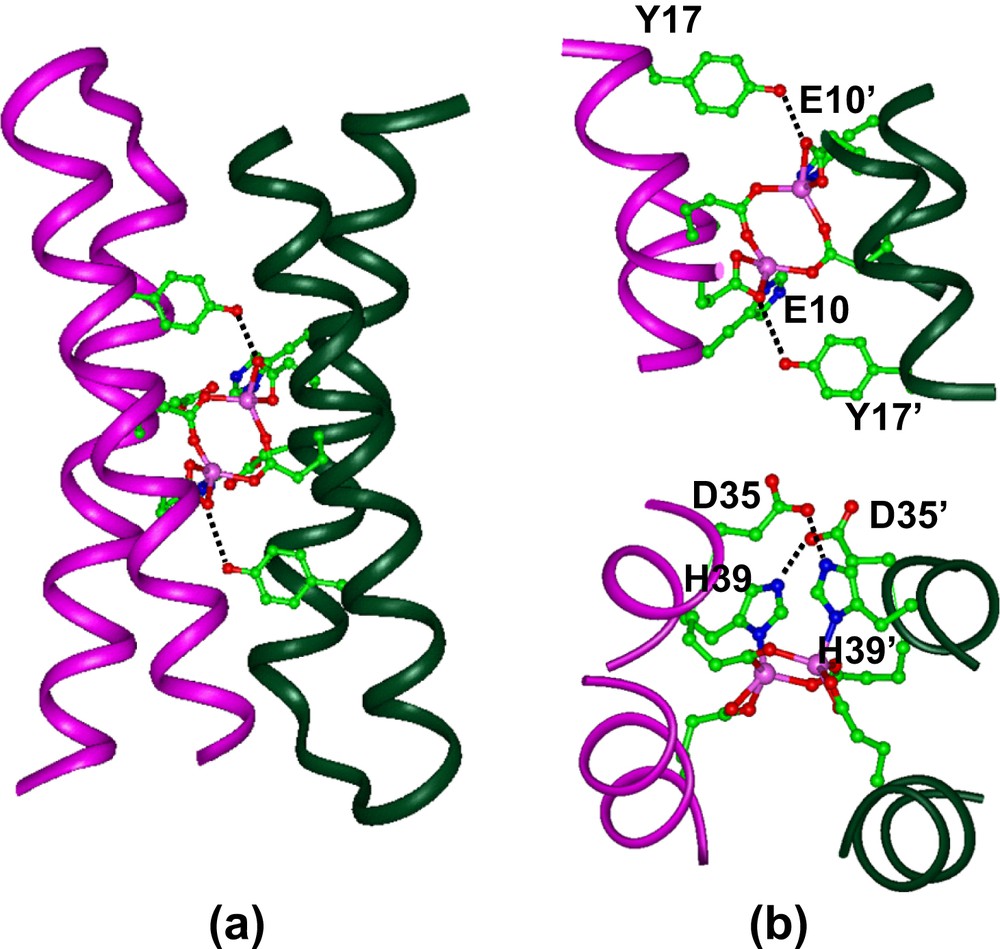
(a) Crystal structure of di-Zn(II)–DF1. (b) Structure of the binding site, showing the second-shell hydrogen bonds between Tyr17 and Glu10′ (top), and between Asp35 and His39′ (bottom).
The ligands surround the metal ions, except for a vacant pair of sites along one face of the dimetal center. The vacant sites are oriented trans to the His ligands, and are well oriented for interaction with exogenous ligands. Further, the intended second-shell interactions were all actually observed in the experimental structure (Fig. 6b).
DF1 was also characterized in its di-Mn(II) form, by X-ray analysis [35]. The three-dimensional structure is very similar to the di-Zn(II) derivative, both in the overall protein structure, and in the dimetal site coordination geometry.
All the results obtained on the prototype DF1 clearly showed that our approach for the development of artificial diiron proteins was successful. Further, the folding of DF1 was dictated neither by the binding of the metal cofactor, nor by the nature of the metal ion. In fact, solution structure determination by NMR analysis of the apo-form indicated that the four-helix bundle fold and the metal binding site are largely preorganized in the apo-protein [32]. The solution structure of apo-DF1 agrees well with the model structure, and it is nearly identical to the crystal structure of di-Zn(II) and di-Mn(II) forms [31,35].
Unfortunately, although DF1 behaves as a native-like protein irrespective of its ligation state, it had several limitations. To constitute the metalloprotein, DF1 has to be denatured and refolded in the presence of metal ions. Further, the diiron complex was not able to bind exogenous ligands. Therefore, the next step was redesigning DF1, with the aim of improving the properties of the prototype and constructing catalytically active analogs.
3 Engineering an active site cavity into DF1
The design of DF1 was particularly challenging, because it required the burial of six polar ionizable groups in the hydrophobic core of a four-helix bundle. To drive the four-helix bundle formation, the interior of DF1 was efficiently packed with a large number of hydrophobic side chains, resulting in a protein with maximal stability. The requirements for a high degree of conformational stability are often contrary to those for binding and enzymatic functions, which in turn require a certain level of conformational freedom.
DF1 was not able to support any activity, probably because access to its dimetal center is blocked by a pair of symmetrically related hydrophobic Leu residues at position 13 of the sequence, L13 and L13′. In order to introduce an active site cavity into the protein, DF1 was re-designed: modeling suggested that replacement of Leu side chains at position 13 and 13′ with smaller Ala or Gly residues might open up the active site, providing a cavity large enough to allow small molecules to access the metal center (see Fig. 7). Indeed, natural diiron and dimanganese proteins, with substrate-access channels, tend to have small side chains at these positions. Therefore two DF1 analogs, L13A–DF1 and L13G–DF1, were synthesized [32,34,35,37,38].
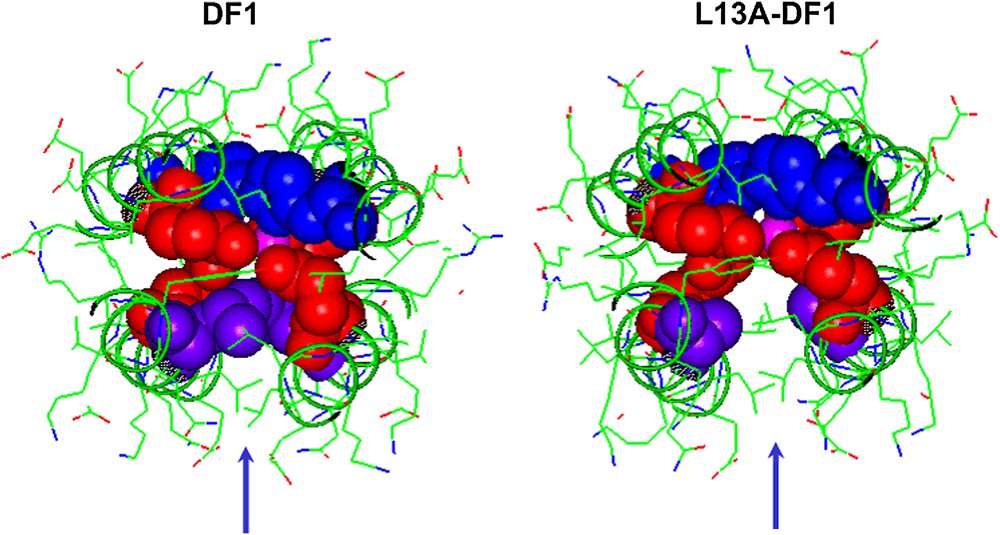
Engineering an active site cavity into DF1. Top view of the active site cavity in DF1 (left) and L13A–DF1 (right). The liganding Glu (red spheres) and His (blue spheres) side chains are reported. The replacement of Leu13/13′ (violet spheres) with Ala creates an access channel to the dimetal site. (For interpretation of the references to colour in figure legends, the reader is referred to the web version of this article.)
The analysis of the folding and binding properties of these new proteins allowed us to outline the stability/function tradeoffs associated with DF models. In order to determine the thermodynamic cost of carving an active site access channel within a protein, the thermodynamics of folding of the three mutants were analyzed [32]. The thermodynamic stabilities of DF1, L13A–DF1, and L13G–DF1 were determined by chemical denaturation with Gdn·HCl, by monitoring the loss of the helical CD signal at 222 nm. The Gdn denaturation curves for DF1, L13A-DF1 and L13G-DF1 markedly depend on the total protein concentration, as expected for a monomer–dimer equilibrium. Globally fitting of the thermodynamic parameters to a two-state equilibrium, between folded dimers and unfolded monomers, gave the free energies of dimerization of the three proteins. DF1 has exceptional stability, with a dissociation constant of approximately 0.001 femtomolar (fM); the corresponding values for L13A-DF1 and L13G-DF1 were 100 fM and 0.6 nM. The difference in stability associated with mutation of Leu 13 to Ala [ΔΔG (0)] was found to be 5.6 kcal/mol dimer (2.8 kcal/mol monomer). This value is within the range expected for mutating a single buried Leu to an Ala in a native protein [71,72]. The mutation of position 13 from Ala to Gly resulted in a further destabilization of 5.2 kcal/mol (2.6 kcal/mol monomer), reflecting contributions from both a decrease in the helix propensity of Ala relative to Gly [≈1 kcal/mol monomer [66,73] and a decreased hydrophobic driving force [≈1.3 kcal/mol monomer [72]].
In order to confirm the presence of the designed cavity, the crystal structures of the two mutants were solved. The L13A–DF1 mutant was crystallized as di-Mn(II) complex in the presence of excess of Mn(II) ions [38]. The asymmetric unit contains three dimers, whose structures are nearly identical to that of di-Zn(II)– and di-Mn(II)–DF1. Mn(II) ions are also involved in inter-subunit contacts. Dimers within a given unit cell are linked by five “external” crystallographically independent manganese ions. The electron density of the metal binding site and the surrounding site is well-defined (Fig. 8). As intended in the design, the dimetal binding site lies at the bottom of a deep pocket water-filled. A trilobate peak of electron density was found in a depression formed between the coordinating glutamate residues. This density has been interpreted as a dimethyl sulfoxide molecule, derived from the crystallization buffer, bridging the two Mn ions. An exogenous bridging organic ligand was observed in the crystal structures of several natural diiron and dimanganese proteins. In addition to DMSO, a number of ordered water molecules fill the pocket leading into the di-Mn(II) site. These water molecules fill the aqueous channel formed by the leucine replacements in DF1. Consistent with the design, all the second-shell interactions were retained in the crystal structure of the di-Mn(II)–L13A–DF1 complex.
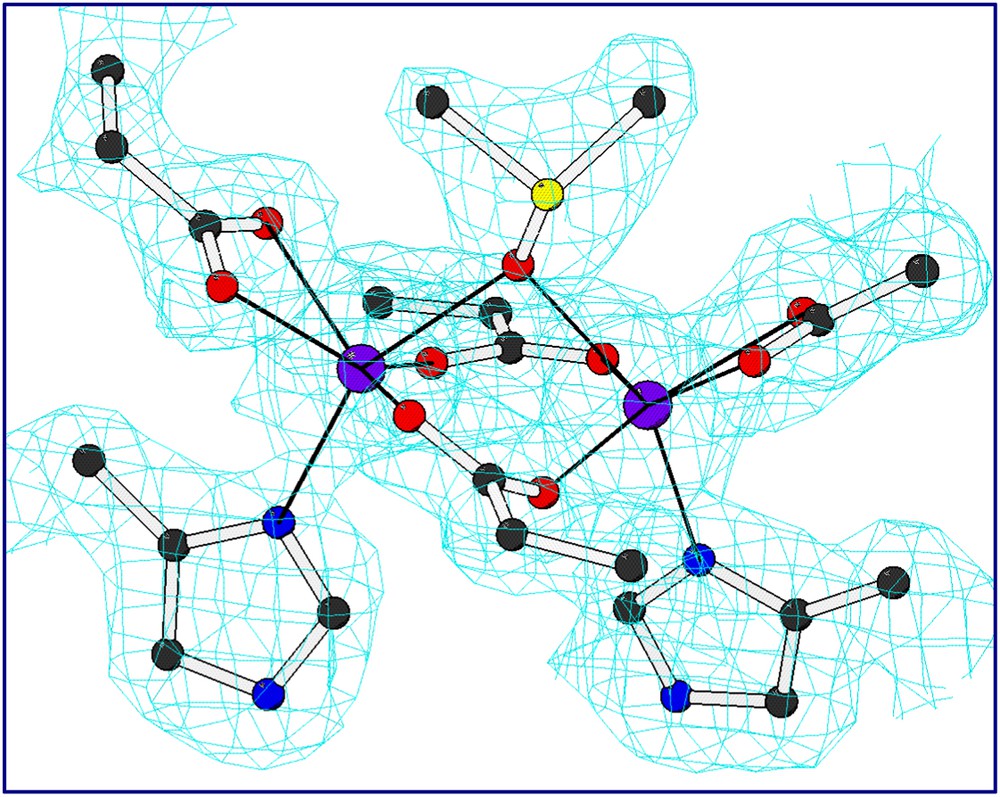
Electron density map of the dinuclear metal binding site of di-Mn–L13A–DF1 (2Fo–Fc map (contour levels are 1σ) for the liganding residues including the exogenous DMSO ligand.
The replacement of the two leucine residues in DF1 with the smallest amino acid, glycine, indeed further increases the channel dimensions, as observed by the analysis of the crystal structure of the di-Mn(II)–L13G–DF1 [37]. The structure displays four crystallographically independent dimers in the asymmetric unit (Fig. 9). Despite the structural similarity with di-Mn(II)–L13A–DF1, a significant difference was found in the electron-density map around the dimetal site. Each of the four crystallographically independent dimers has a larger number of water molecules without DMSO molecules in the protein core with respect to the Ala13 DF1 variant. Fig. 10 depicts the active site cavity in L13A–DF1 and L13G–DF1: the water-filled access channel has been expanded in the expected manner, upon substitution of Ala 13/13′ with Gly.
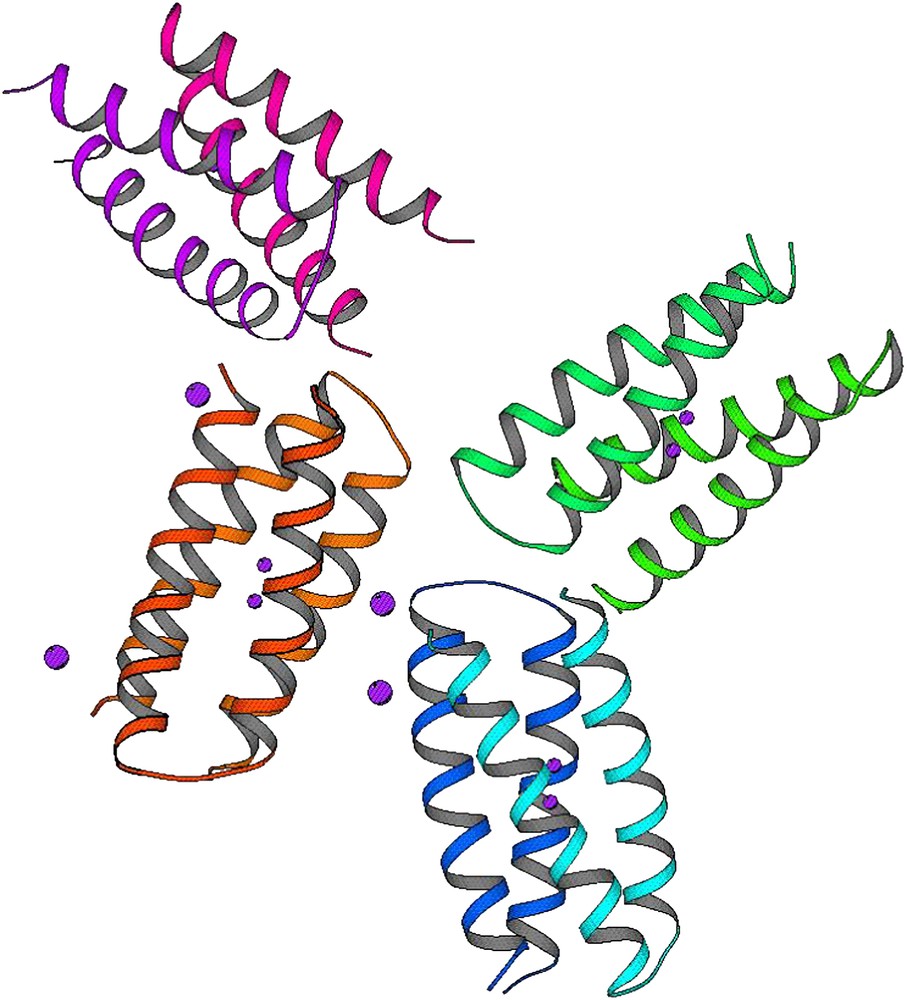
di-Mn–L13G–DF1 crystal structure: the four crystallographically independent dimers of the asymmetric unit. Violet spheres indicated the Mn(II) ions.
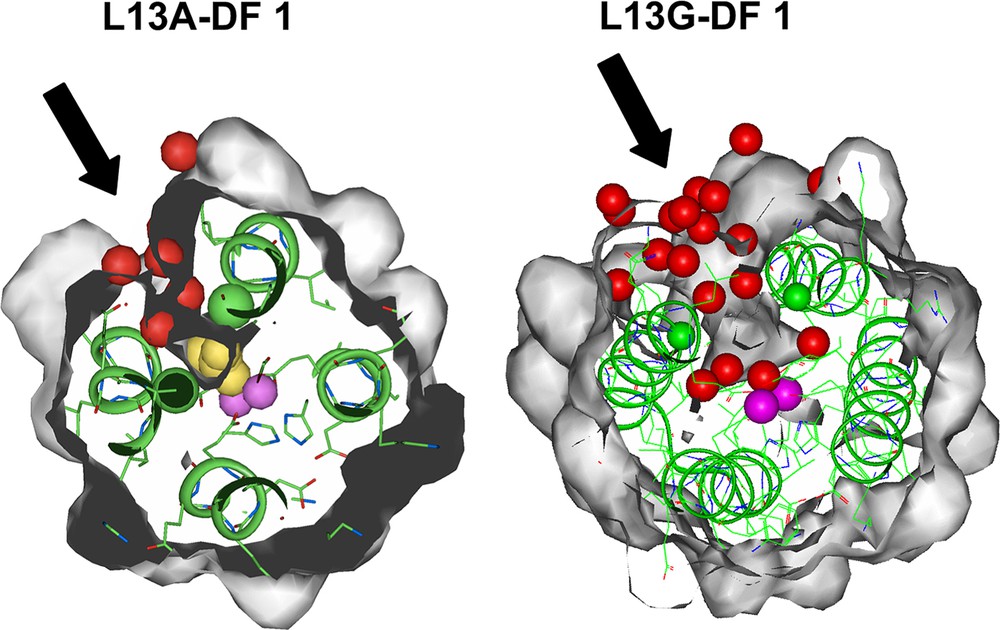
Top view of the active site cavity in L13A–DF1 (left) and L13G–DF1 (right). The access channel, indicated by black arrows, expands upon Ala to Gly mutation. The yellow spheres (in L13A–DF1) indicate the dimethyl sulfoxide molecule, the red spheres indicate the water molecules, and the violet spheres indicate the metal ions.
The more striking feature found in the di-Mn(II)–L13G–DF1 structure is the presence of two different dimanganese coordination environments (Fig. 11). In three of the four crystallographically independent dimers (AB, CD and EF), a water molecule bridges the two metal ions, while in the other dimer (GH) two terminal water molecules are coordinated to the two manganese ions trans to the histidine ligands. The inter-metal distance is shorter in three of the dimers (3.6 Å) and longer in the fourth (4.2 Å). These different modes of binding led to changes in the quaternary structure (Fig. 12). By superimposing the C-terminal helices (2 and 2′) of the four crystallographic dimers, it can be seen that they are virtually invariant among the structures. In contrast, the N-terminal helices (1 and 1′) occupy different positions in the GH dimer relative to the others. In particular, the two copies of helix 1 undergo a shift in opposite directions (approximately 0.7 Å) along the Z axis, away from the metal-binding site. Indeed, this shift increases the length of the metal–metal distance. This sliding-helix mechanism may be extended to natural metalloproteins, in order to accommodate changes in their coordination environment. However, such motions have not been observed in the parent natural metalloproteins, probably because it might be more difficult to be observed in a complex, large molecule.
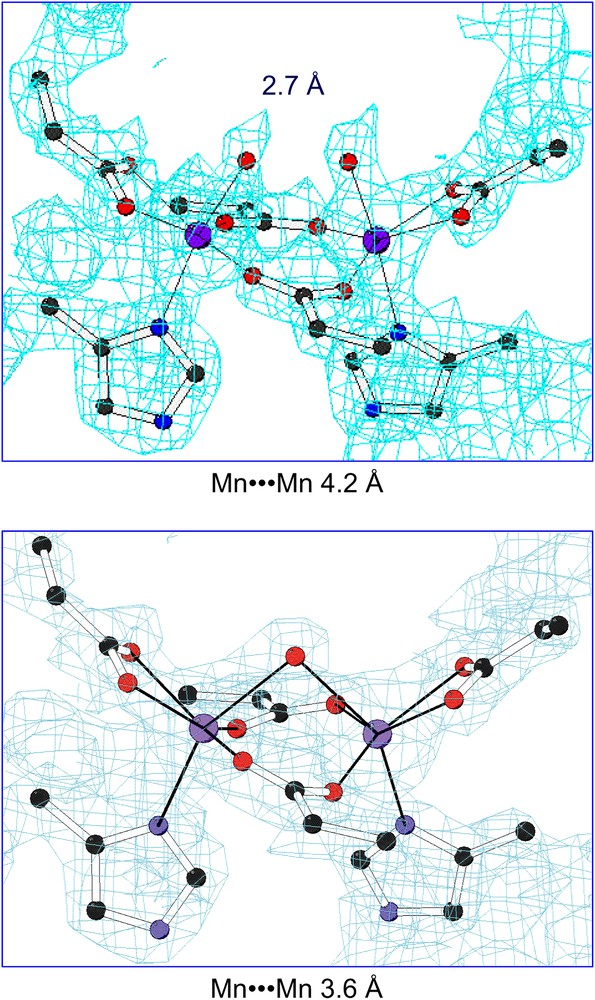
2Fo–Fc electron density maps (contour levels are 1.5σ) of the dinuclear metal-binding site of di-Mn–L13G–DF1 in the AB dimer (top) and in the GH dimer (bottom).
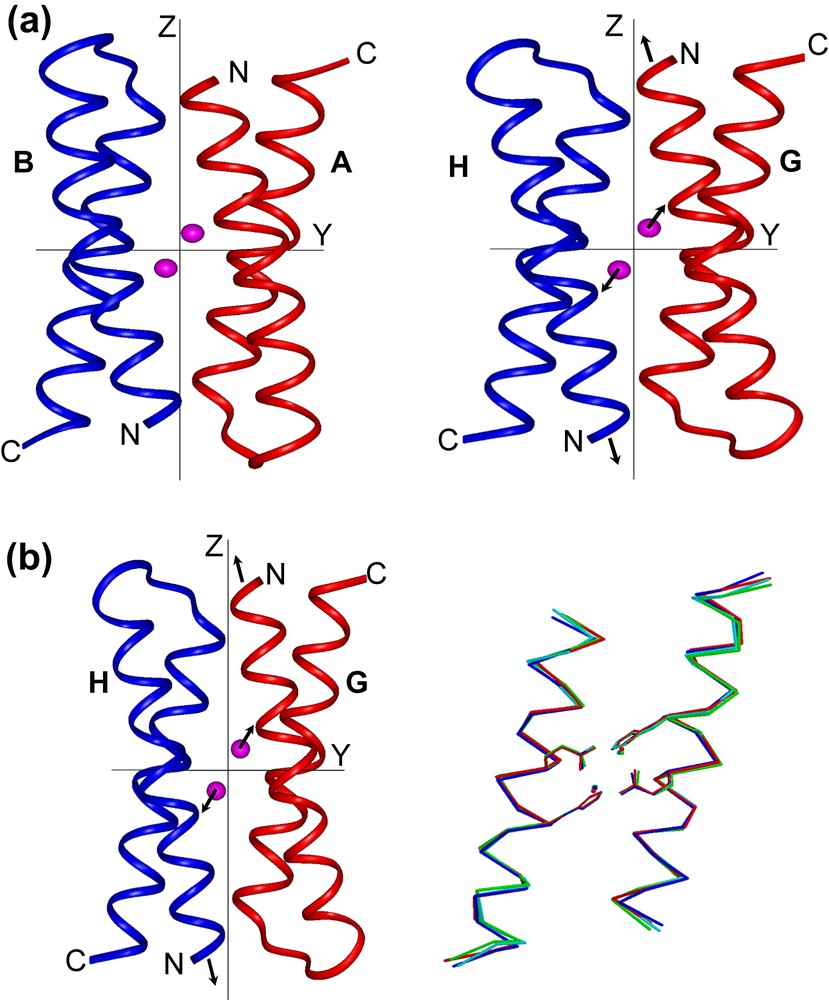
(a) Crystallographically independent dimers of di-Mn–L13G–DF1, AB (left) and GH (right), viewed along the x axis of a common orthogonal reference system. The arrows in the GH dimer indicate the shifts of N-terminal helices and metal ions with respect to the other dimers. (b) Superimposition of helices 1 and 1′ (left) and helices 2 and 2′ (right) of the four crystallographically independent dimers of di-MnII–L13G–DF1. Note the movement of helices 1 and 1′ of the GH pair (red) relative to the remaining helices (AB, blue; CD, green; EF, cyan).
In summary, the three-dimensional structures of the di-Zn(II)–DF1, di-Mn(II)–L13A–DF1 and di-Zn(II)- L13G–DF1 are very similar, indicating that the introduction of the access channel and changing the metal ion from Zn(II) to Mn(II) did not greatly affect the structure of the protein. Further, the analysis of the thermodynamic consequences of introducing a substrate-access channel into the structure of DF1 shows that the destabilization is extreme; the Leu-to-Gly mutation destabilizes the dimer by 10 kcal/mol. Thus, although this functional mutation extracts a large thermodynamic price, the extreme stability of DF1 provides adequate stability to compensate for the mutation.
The limited solubility of DF1 analogs in aqueous solution has facilitated the formation of X-ray diffraction-quality crystals, thus allowing a deep investigation of their structures in the solid state; on the other hand, it has impeded further solution characterization in the absence of organic solvents. We therefore tried to improve the properties of DF1 molecules by designing two recombinant versions of L13A–DF1, DF2 and DF2t [33,34,36,39,40], that have several changes in their sequence, as well in the loop structures, with respect to DF1 (see Fig. 13a for a comparison of their amino acid sequences). In order to enhance the water solubility and to reduce the tendency to aggregate in solution, several hydrophobic residues on the surface were converted to polar residues in DF2. Further, a careful examination of the DF1 structure showed that the inter-helical turn adopts a strained conformation, which might account for these problems. Thus, the loop was re-designed in DF2.

Sequence alignment of DF family proteins respect to the prototype DF1. The liganding residues are highlighted in green, second-shell ligands in pink, and residues involved in the active site access in yellow. (a) DF2 subset and (b) DFtet subset: the residues in the A-subunits that were varied are underlined.
We first performed an analysis of the structures and sequence preferences of inter-helical hairpins, in a large database of crystallographically characterized proteins. A very limited number of inter-helical turns were found to occur with high frequency. Furthermore, these turns occur only within well-defined structural contexts. Three classes of turns were found to occur with high frequency: the Rose and Shellmann αL–β, which differs for the H-bonding pattern, and the longer αR–β–αR–β. In DF2, the sequence was changed to stabilize the αL–β inter-helical turn; in DF2t the longer αR–β–αR–β loop was introduced between the helices.
DF2 and DF2t were expressed in Escherichia coli. The solution properties of DF2 and DF2t were thoroughly studied by sedimentation equilibrium ultracentrifugation, CD-monitored guanidine denaturation, and NMR solution characterization [34,40]. Thermodynamic measurements showed that DF2t was slightly more stable than DF2 in the apo-state. Both proteins are strongly stabilized by divalent cations, which they bind with a stoichiometry of two ions per four-helix bundle. Both had similar affinities for metal ions, although DF2t bound Mn(II) an order of magnitude greater than DF2 bound this ion.
The structural characterization in solution and in the crystal state of both proteins [36] confirmed that they formed the desired dimeric helix–loop–helix structure (Fig. 14). Small differences between the crystal and the solution structure are restricted to the loop regions, with the NMR structure showing two loop conformational families, in both the proteins. Analysis of the loop regions revealed that the 3-residue loop, β–αL–β loop, engineered in DF2t, was more defined than the 2-residue αL-β loop, present in DF1 and DF2 (Fig. 15). This finding may be responsible for the higher stability and the better expression level of DF2t with respect to DF2.
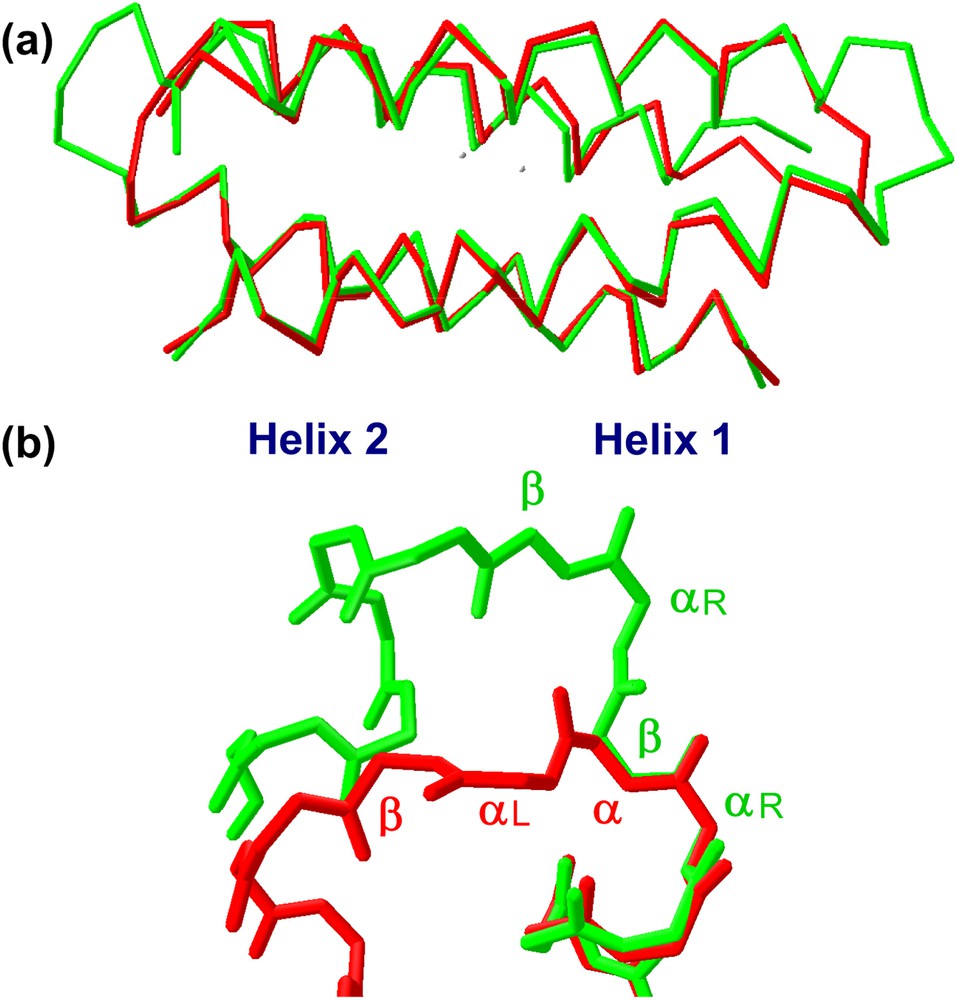
(a) Comparison of DF2 (red) and DF2t (green) crystal structures. (b) Backbone structure of the turns in DF2 (red) and DF2t (green).
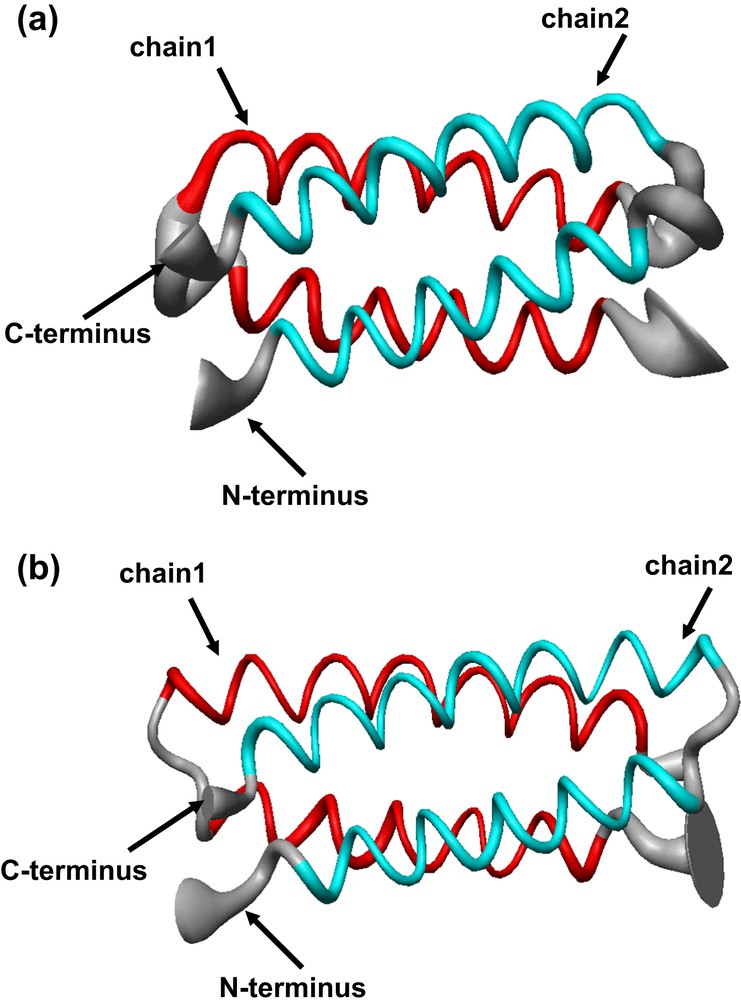
Solution structure of (a) di-Zn(II)–DF2 and (b) di-Zn(II)–DF2t, represented as a tube of radius proportional to the rmsd of each residues. The thinner segment of the ribbon in the DF2t loop region evidences a comparatively lower positional variability of the backbone atoms with respect to that observed in DF2 (thicker region).
With the aim of correlating structural with functional properties, we examined the ligand binding and spectroscopic properties of all the DF1 and DF2 analogs. In contrast to the prototype DF1, all the other members of DF1 and DF2 subsets, containing either Ala or Gly at the access site position, can be reconstituted with metal ions without unfolding the apo-protein, thus suggesting that the metal binding site is readily accessible to small molecules. All the DF1 analogues showed an absorption spectrum of the diferric form typical of oxo-bridged diferric systems, and all the proteins are able to bind azide (Fig. 16). Further, the UV–vis spectrum shows small changes upon addition of acetate, benzoate or 4-hydroxybenzoate anions, suggesting that the carboxylate groups of these ligands interact with the metal center, similarly to natural diiron–oxo proteins [38].
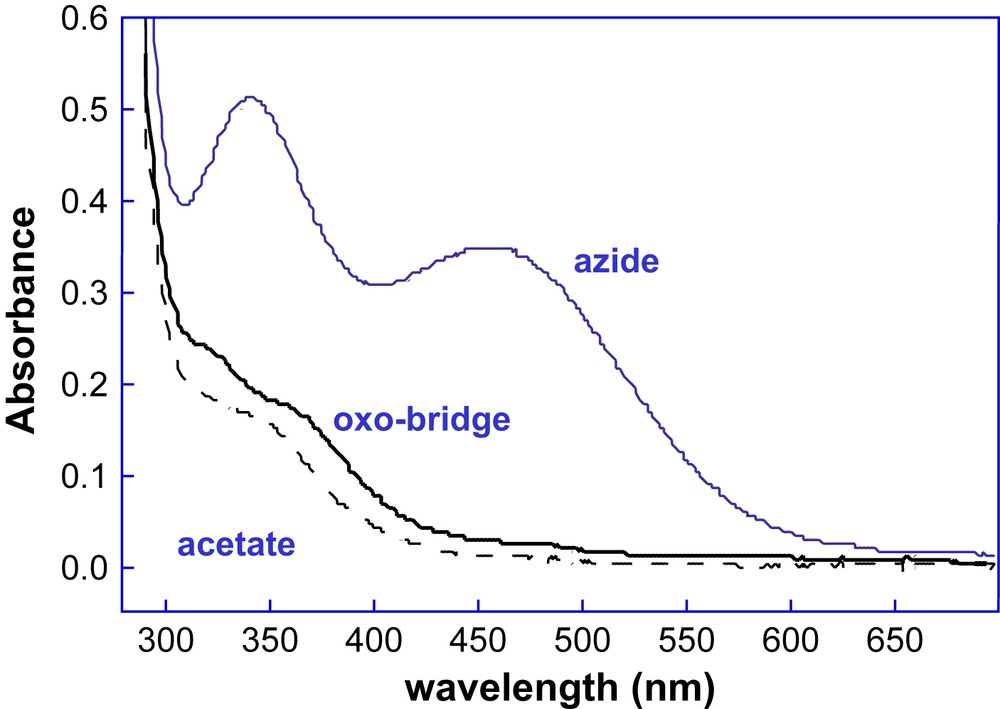
Spectrum of di-Fe(III)–DF2 (solid black lines) and spectral changes that accompany the addition of the small molecules sodium azide (blue line) and sodium acetate (black dashed line).
This last finding was confirmed by the crystal structure resolution of the diferric complex of DF2t [39]. The diiron site is characterized by a cluster consisting of two μ-1,3 bridging Glu carboxylates, two chelating Glu carboxylates and two Nδ bound His ligands (Fig. 17). Further, the diiron(III) center is able to accommodate two additional non-protein ligands. In fact, two exogenous ligands were apparent in the electron density map: an oxo ligand was modeled at the μ-bridging position, cis to both Fe–NδHis bonds, and an acetate ion, from the crystallization buffer, was modeled in the other electron density peak. An extensive series of second-shell hydrogen bonds restrain the protein Glu ligands to nearly the same positions as those observed in divalent metal-bound DF proteins, despite the accommodation of exogenous μ-oxo and acetate ligands. Thus, the diferric complex of DF2t does not show the same degree of flexibility of the carboxylate ligands, typically found in the natural diiron proteins and thought to be important for catalysis. In fact, changes in the coordination mode of active site ligands in natural diiron proteins are well precedented, as they have been structurally characterized in methane monooxygenase and in the R2 subunit of ribonucleotide reductase [74,59]. In all cases, the ligating modes of the active site carboxylate ligands have been shown to be flexible (carboxylate shifts) [70,75]. This flexibility is not surprising, as carboxylate ligands are able to adopt several coordination modes.
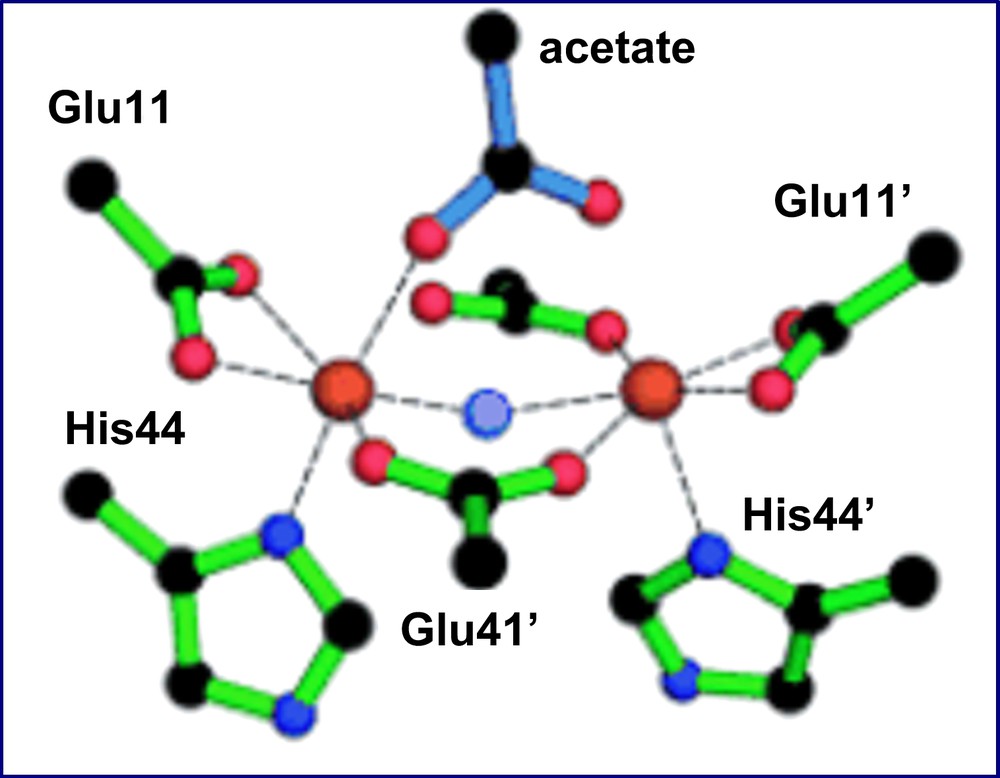
The diiron(III) cluster ligands and σA-weighted Fo–Fc omit map peaks modeled by acetate and μ-oxoligands (4.0σ) as observed in the di-Fe(III)–DF2t crystal structure.
In light of the catalytic relevance of the carboxylate shift phenomenon [76], a theoretical analysis was carried out on the di-Zn(II)–DF1 active site, in order to investigate the structural and dynamical behavior of the metal cofactor. First principles (Car-Parrinello) [77] and hybrid QM/MM molecular dynamic simulation [78] indicated that carboxylate shift from chelated to monodentate occurs during the simulations, at least on one of the two bridging glutamates. The calculations also showed that the second-shell interactions contribute significantly to the structural stability of the active site, and that the bulk solvent water molecules play a critical role in fine-tuning the dynamic of the system.
4 Searching for functionality: DFtet an heterotetrameric system
The results we had obtained so far on the analysis of all DF analogs clearly indicated that three main aspects must be considered in order to obtain stable and functional models: (i) the correct design of inter-helical turn; (ii) the residues responsible for the primary and second-shell ligands; (iii) the residues that define the active site access channel.
To search for a catalytically active system based on the DF structure, it is helpful to evaluate how systematic changes in the structure affect substrate-binding and catalytic properties.
DF1 is an extremely stable scaffold to tolerate significant changes in the amino acid sequence, without affecting its overall fold. The extreme thermodynamic stability of DF1 makes it a particularly apt framework for substitutions, which are important for function and for probing specific mechanistic questions, more difficult to analyze on a complex, large protein.
Although some DF molecules in the diferric forms are able to bind exogenous ligands, such as phenol and acetate, catalytic activity still requires changes in the second-shell ligands, in particular in defining the access to the diiron site. For example, modeling suggests that for the accessibility to the metal site, not only residues at position 13 play a critical role, as already described, but also residues at position 9 should be crucial. However, as the number of mutation increases, a very large number of combinations are generated. Thus an exhaustive analysis would require the preparation and purification of significant quantities of hundreds to thousands of variants, to be screened for binding of small molecules, and reactivity toward a variety of substrates.
To advance this goal, we tried to bridge the gap between “rational design” and combinatorial approaches through the development of four-helix bundle based on heterotetrameric systems [41]. A heterotetrameric system consisting of four disconnected helices, which could be separately synthesized, purified, and then combinatorially assembled, is uniquely suited for the production of an array of any desired helical bundles from a significantly smaller number of peptides. For an ABCD heterotetramer (in which each of the monomers was different), 104 combinations could be prepared by combining only 10 variants of each of the individual chains. Previously, strategies have been developed for the design of two-stranded coiled coils that specifically self-assemble to form AB heterodimers, with minimal formation of homodimeric species [79]; similarly, ABC heterotrimers [80–82] and A2B2 heterotetrameric [83] coiled coils have been designed.
We have applied this strategy to the development of DF1 analogs, by designing either an A2B2 heterotetrameric complex [41] and a three-component AA′B2 heterotetrameric complex [42]. We named these analogs DFtet. The design of heterotetrameric systems presents significant challenges for protein design. Specifying a unique topology is a more complex problem for a heterotetrameric system, as in DFtet, than it was in a homodimeric system, such as DF1 or DF2. In particular, it was important to destabilize both homooligomeric folds as well as undesired heterotetrameric topologies. To achieve this goal, a new computational design algorithm was developed. This design methodology included the concept of “negative design” [84–88,30] to prevent alternate topologies from occurring and as well as a “minimalist” approach [88,89] to minimize extraneous structural variables.
The algorithm was first applied for the design of A2B2 heterotetramers (Fig. 13b), where A and B are 33-residue peptides. The first step in the design process involved the specification of the four-helix bundle backbone, which was generated using the parameters obtained from the analysis of DF1. The helices of DFtet were extended relative to those of DF1 (33 residues in DFtet versus 24 residues in DF1) to increase the stability of the system. Helix A contains only the Glu ligands, and helix B contains the EXXH metal-binding motif. By inspection of the sequence alignment reported in Fig. 13b, it can be seen that the residues within the region of the binding site are the same as the corresponding residues of DF1. Similarly to L13A–DF1, an Ala residue was inserted at position 19 (position 13 in DF1) to create an open cavity. Residue 18 in DFtet–B (corresponding to residue 38 of DF1) was mutated from Lys to Arg, in order to strictly resemble the Asp–Glu–Xxx–Arg–His motif of natural diiron proteins such as MMO [90] and Δ9-ACP desaturase [60] (see Fig. 4).
The nature of the residues at the b/e and g/c interfacial positions (see Fig. 2 for the heptad nomenclature) was chosen to specifically stabilize only one of the possible topologies for an antiparallel A2B2 heterotetramer. These positions were allowed to correspond to Lys and Glu amino acids, and the energy of the desired folds and of the alternatively folded structures was considered in the calculation. The final sequences for A and B peptides correspond to those which showed the minimum number of unfavorable contacts in the desired topology (Fig. 18).
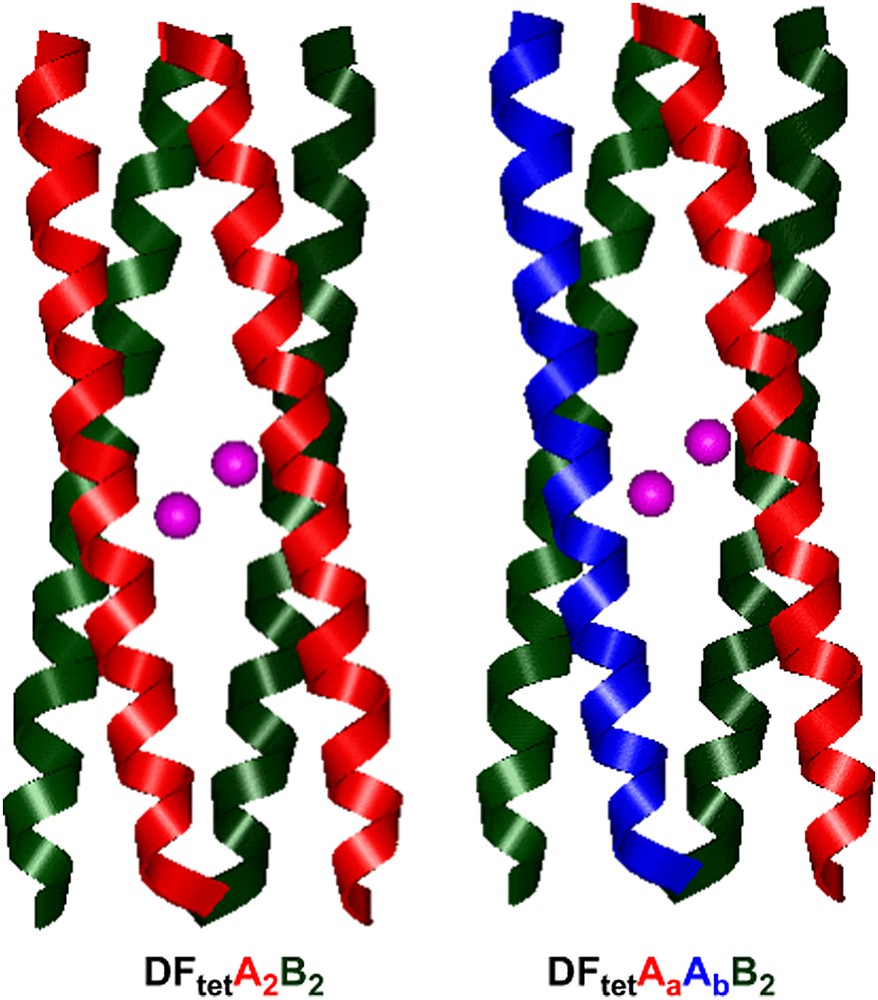
Models of DFtet.
The design of DFtet was successful: DFtet–A and DFtet–B are unfolded at neutral pH; however, when mixed in a 1:1 ratio, they assemble into a helical bundle with the expected stoichiometry [41]. Furthermore, the peptide is tetrameric and stable over a wide temperature range, being thermally unfolded only at low concentrations and at an elevated pH. DFtet also binds metal ions in the proper stoichiometry, and the spectroscopic properties of the Co(II) and Fe(III) derivatives strongly suggest that the ligand environment is precisely as intended. The match of the Co(II) spectrum between DFtet and DF1/DF2 analogs (see Table 1), and the identical kinetic scheme for Fe(II) oxidation provide good evidence that the metal-binding sites of these proteins are very similar. Thus, the intended topology appears to have been achieved.
The potentiality of DFtet for the preparation and screening of a library of DF variants has been demonstrated by the results we achieved with a less symmetrical version of the original heterotetramer DFtet. DFtet was re-designed for constructing a new system based on a three-component AA′B2 complex [42]. In order to prepare this complex, two electrostatically complementary versions of the A peptide were prepared, which should specifically assemble with two equivalents of B to form the desired AA′B2 heterotetramer. Along one face of the helix of the original A, negatively and positively charged groups had been placed near the N and C termini, respectively, to stabilize an antiparallel arrangement of the helices. The sequences of these two helices were varied such that one contains a strip of Glu along the entire length of the interface, and the other contains Lys and Arg at equivalent positions (Fig. 13b). The peptide with the acidic replacements was named helix Aa and the other helix Ab. Additional substitutions were also made in the second-shell interactions, to probe specific mechanistic questions. The non-bridging Glu residues in DF1 form second-shell interactions with two Tyr side chains (see Fig. 6), whereas in natural proteins only one of the structurally equivalent Glu residues is generally constrained by a second-shell interaction.
The assembly of the three peptide chains, Aa, Ab and B in the expected 1:1:2 stoichiometry to give the desired AaAbB2 heterotetramer was successful, as proven both by CD and size-exclusion chromatography studies [42]. The substitutions made to the AaAbB2 four-helix bundle, with respect to the prototype DF1, were interesting, as demonstrated by the results we obtained on the diiron complex of this new protein. On addition of ferrous ions and oxygen, the protein forms a product, entirely different from those formed in the original DF1 protein and DFtet complex.
Fig. 19 shows the UV–vis spectra of AaAbB2 measured at various time after the addition of Fe(II) in the presence of oxygen. The spectra are characterized by the presence of a strong band centered near 625 nm with an extinction coefficient of approximately 1000 M−1 cm−1. Although additional spectroscopic studies are necessary to confirm the identity of this species, its UV–vis spectrum is similar to that found in the cis-μ-1,2 peroxo-bridged diferric intermediates observed in a variety of diiron proteins, such as ferritin (λmax = 650 nm, ɛ = 1000 M−1 cm−1) [91,92] but at somewhat shorter wavelength than the corresponding species in the enzymes Δ9-ACP desaturase (λmax = 700 nm, ɛ = 1200 M−1 cm−1) [93], methane monooxygenase (λmax = 725 nm, ɛ = 1800 M−1 cm−1) --for a review, see Ref. [75]--and a mutant of the R2 subunit of ribonucleotide reductase (λmax = 700 nm, ɛ = 1800 M−1 cm−1) [94].
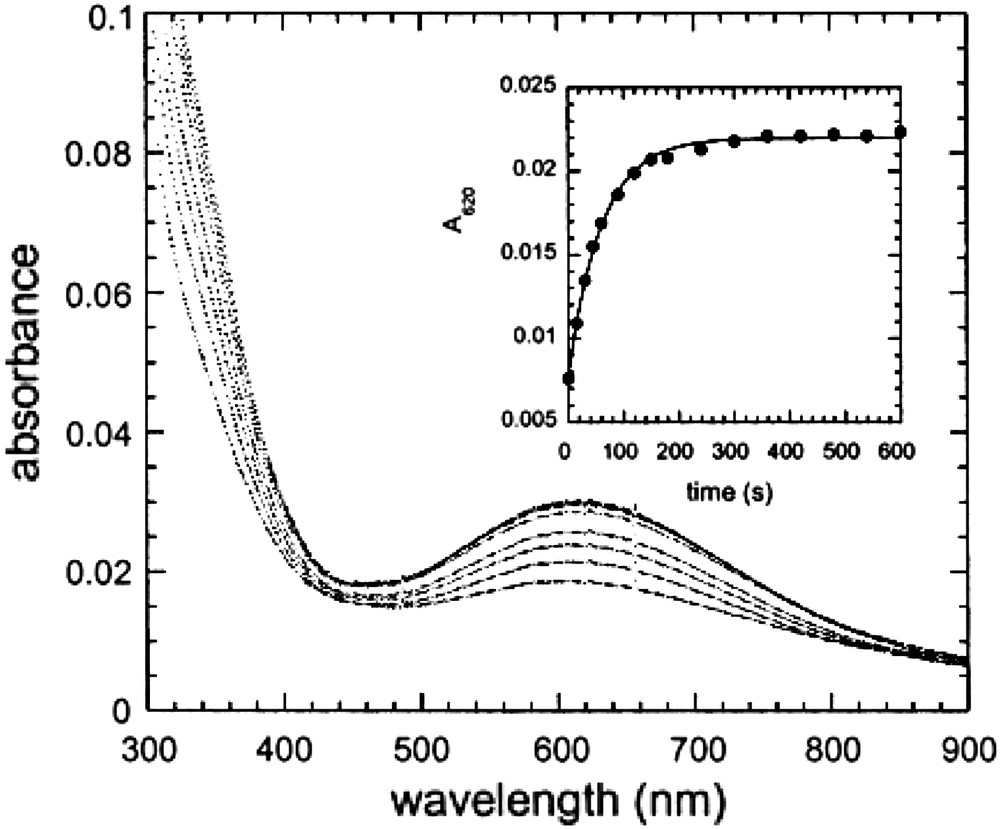
Spectra of AaAbB2 (25 μM in tetramer) measured at various times after the addition of ferrous ammonium sulfate, final concentration 50 μM, in air saturated buffer (150 mM Mes, pH 6.5/150 mM NaCl). The spectra were measured at 15, 30, 45, 60, 90, 120, 150, and 180 s after mixing. Inset: the kinetics of peroxide formation followed by the increase in absorbance at 620 nm.
The visible band could be also assigned to a tyrosinate–Fe(III) ligand-to-metal charge-transfer band [95]. Further studies are currently in progress.
Further design refinements to DFtet allowed us to develop an O2-dependent phenol oxidase [43]. The interior of the symmetrical DFtet A2B2 was sculpted for creating a pocket capable of binding a substrate, the 4-aminophenol. Modeling suggested that when this molecule is inserted in the active site of the original DFtet A2B2, and modeled such that its phenolic oxygen bridges the metal ions, it makes unfavorable contacts with Leu 15 and Ala 19 (corresponding to positions 9 and 13, respectively, of DF1) (see Fig. 20a). The steric bulk of these residues was therefore reduced in variants in which Ala 19 is changed to Gly and Leu 15 is changed to either Ala or Gly in both of the A chains (see Fig. 20b).
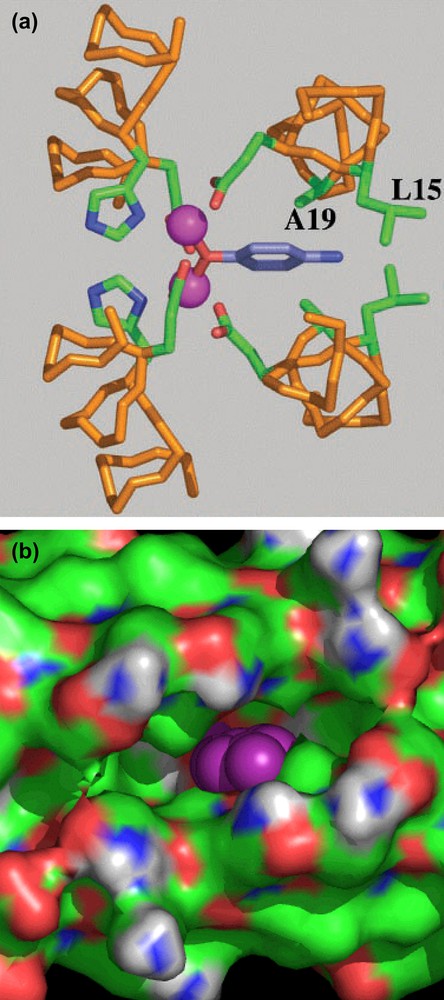
(a) Computer model of 4-aminophenol bound to the active site of DFtetA2B2. (b) Solvent-accessible surface associated with G4–DFtet. The aminophenol ring into the pocket is represented in purple.
A number of symmetrical variants, G4–DFtet (in which Leu 15 and Ala 19 of both A chains were substituted with Gly), L2G2–DFtet (in which Leu 15 was retained and Ala 19 was changed to Gly), A2G2–DFtet and G2A2–DFtet (which have Gly or Ala at the indicated positions) were screened for activity. In particular, all the variants were screened for their ability to react with Fe(II) and O2. Some variants produced a relatively stable intermediate, resembling the same diferric peroxo species formed in the DFtet AaAbB2. The variant with the fewest steric restrictions, namely G4–DFtet, showed increasingly rapid formation of the oxo-species with no detectable intermediates. The same variant showed the greatest binding ability toward phenol, which binds to the diferric site with high affinity.
Finally, we measured the two-electron oxidation of 4-aminophenol to the corresponding quinone monoimine, catalyzed by the proteins in atmospheric O2. Again, G4–DFtet showed a ≈ 1000-fold rate enhancement, relative to the background reaction when the initial rate of the reaction in the presence and absence of the protein was compared. The G4–DFtet catalyzed this reaction for at least 100 turnovers. Changing either of the Gly residues at positions 19 or 15 to Ala gave a protein whose rate was decreased between 2.5- and 5-fold. All the results on DFtet variant clearly showed that the catalytic efficiency is sensitive to changes in the size of a methyl group in the protein, illustrating the specificity of the design.
All these data demonstrate the promise of the combinatorial approach for identifying potential candidates for defined applications. Having tested the potential of the system, the next step is to prepare a library of variants, with the active site cavity designed for a specific functionality.
5 Conclusions
Principles and methods for protein design have matured, and protein design has deeply contributed to our understanding of the principles of protein folding and stabilization. It has now become possible to design structurally defined protein models. Our studies in the design of diiron proteins suggest that it should now be possible to approach the de novo design of highly functionalized metalloproteins with well-defined three-dimensional structures.
We applied different and complementary strategies for the construction of diiron proteins' model systems. To create the prototype scaffold we followed the symmetry concept, trying to reproduce in a symmetric model the quasi-symmetrical structure of a metalloprotein. The use of C2 symmetry has been shown to be particularly advantageous, because it simplifies the design, reduces the size of the molecules to be synthesized, and simplifies their structural characterization.
The C2 symmetric DF1 protein, based on the dimerization of a helix–loop–helix motif, has set the stage for the development of the other proteins. The studies on DF1 analogs and on the more soluble versions DF2 and DF2t, revealed the crucial features required for defining the properties of an artificial diiron protein. What we have learned by the lesson on DF molecules based on the helix–loop–helix motif can be summarized as follows.
The designed four-helix bundle motif allows the proper burial of the first-coordination-sphere ligands. All of our designed DF proteins bind divalent metal ions in the proper stoichiometry of two per protein. Further, all the spectroscopic and structural data suggest that all these proteins share the same pentacoordinate ligand environment, with each metal ion having three carboxylate ligands and one His ligand.
Of crucial importance in modulating the functional properties of the proteins are the residues at positions “g” and “d” that define the shape and accessibility of the active site cavity (see Fig. 2). Residues with different steric hindrance at the above-mentioned positions influence both stability and activity. The analysis of the thermodynamics of folding showed that stability decreases as the bulk of the residues at positions “d” decreases from Leu to Ala to Gly. On the opposite, as the active site cavity increases the chemical properties of the proteins increasingly resembles those of the natural enzymes. In fact, L13G-DF1 and L13A-DF1 variants, as well as the DF2 subset, bind exogenous ligands and display ferroxidase activity, in contrast to DF1, which was extremely stable and not able to support any function. Further evidences of the correlation between the catalytic activity and the size of the active site cavity came from the results obtained on the DFtet subset, in which the influence of the residues at the “g” positions was also evaluated. The results obtained on the variant with the fewest steric restrictions, namely G4–DFtet, reveal that changes, as small as the size of a single methyl group in the active site residues, have a significant effect on the protein functional properties. In fact, G4–DFtet shows exceptional substrate specificity: the substrate 4-methoxyanaline, in which the hydroxyl group of 4AP is converted to a methoxy, was not a substrate for the protein. Also, 4-aminoaniline, in which the phenolic hydroxyl is replaced by an amino group, was oxidized at a rate only 2-fold greater than the background reaction.
All the results we obtained so far were included in the design of the last protein, DF3, which belongs to the DF1 subset. This new symmetric helix–loop–helix dimer encompasses all the main features thought to be crucial for function: (i) Gly residue at both positions 9 and 13; (ii) new inter-helical loop with respect to DF1. The new protein shows improved solubility and active site accessibility, while retaining the unique native-like structure, as assessed by NMR structural characterization (manuscript in preparation).
In conclusion, throughout this paper we tried to illustrate the importance of using a very stable and highly optimized protein model for functional design, which can withstand multiple changes into the sequence without affecting the overall fold. DFs encompass these properties: their thermodynamic stability makes them apt framework for substitutions, which are important for function and for probing specific mechanistic questions, more difficult to analyze on a complex, large protein.
Acknowledgments
The authors wish to thank the European Commission for a Marie Curie postdoctoral fellowship to RTMT.