

1 Introduction
Molecular chirality is pervasive in the chemistry of life and plays a key role in biology as well as in technology. A variety of functions are dependent on the stereochemistry of the molecules and most of them are permitted only when a positive matching between the chiral elements of a substrate and of a reagent (or a receptor) occurs (chiral recognition).
Chirality may exert its influence in many fields of our daily life. We are well aware that the physical properties of stereoregular (tactic) polymers, either man-made or natural, are quite different from those of stereorandom (atactic) macromolecules. We know as well that stereo-ordered assemblies of chiral molecules are required for specific optical and electronic properties to be displayed by a molecular device.
Life itself is dependent on chirality because the interactions of living systems take place almost always with enantiopure compounds. Biosynthetic and metabolic routes which regulate the chemistry of life are made possible because chiral entities of highly organized asymmetry such as enzymes, receptors and binding sites of living organisms recognize and differentiate the substrates by their own chirality.
The pharmacological activity of two enantiomers of a chiral drug is different because biological messengers and cell receptors are chiral and the molecules of a pharma drug must match their asymmetry. Most frequently, only one enantiomer of a chiral molecule (the eutomer) displays the desired effect, whereas the other (the distomer) is either inactive or antagonist or is endowed with a different pharmacological profile.
A survey of about twenty years ago covering 1850 pharmacologically active compounds used in drug manufacturing pointed out that the totally synthetic products amounted to 1327 (of which 528 chiral), while the remaining 523 (of which 509 chiral) were natural or semi-synthetic derivatives. At the time of the survey, almost all the chiral natural derivatives (509; 98.5%) were sold as single enantiomers. The opposite was true in the case of the totally synthetic chiral drugs, where only 61 out of 528 (12%) were sold as single isomer. The overall number of single-enantiomer drugs out of a total of 1045 chiral molecules amounted to 570 (54% of chiral drugs; 31% of the totality of drugs).
Since then, the single-enantiomer business has become a rapidly growing segment in the drug market and at present it accounts for over one third of the total drug sales. It has topped € 100 billion for the first time in 1998, increasing a further 16% up to € 115 billion in 1999 [1]. Since 1999 this segment has grown at an average annual rate of 8% and it is expected to attain € 200 billion by 2006.
Continuing growth of this sector is rooted in the post-genomic research, which is providing new chiral targets for synthetic drugs, and in the regulatory pressures, which will increasingly require chiral molecules to be produced and marketed as single enantiomers.
Further stimulus will come from the interest of pharma industries in redeveloping a single-enantiomer form of the pharmas that they have been previously selling as a racemate. This ‘chiral switch’ provides the industries with a tool for responding to the increasingly demanding constraints of regulatory institutions and, at the same time, for extending the patent life of a protected product.
This trend will incite fine chemicals suppliers to expand their catalogue of chiral intermediates and to design new asymmetric routes to chiral products. These efforts are expected to stimulate further research in the area of asymmetric synthesis, with emphasis on metal- and bio-catalyzed processes and on chiral separations.
2 Possible routes for the preparation of chiral non-racemic compounds
Discovery and development of truly efficient procedures for the preparation of enantiopure products have been for a long time a great challenge for synthetic chemists (and sometimes still now it is). The methodologies viable for the production of chiral non-racemic, possibly enantiopure, compounds can be classified as indicated below:
- – isolating a chiral product from natural sources;
- – physical methods;
- – chemical and biotechnological methods.
2.1 Isolating a chiral product from natural sources
Processing vegetal crops affords essential oils which are mixtures more or less complicated that contain several chiral and achiral compounds. Separation of a single chiral component can be accomplished in several cases by applying established purification protocols.
2.2 Physical methods
- (a) Crystallization of a racemic mixture can occasionally proceed with separation of a conglomerate of enantiomorphous crystals of the two enantiomers. Pasteur's separation of racemic sodium ammonium tartrate by mechanical sorting of the enantiomorphous crystals is the best-known example of this possibility and is the first example of resolution of a racemic compound ever reported (1848). The cases in which this possibility can be exploited are rare and unpredictable.
- (b) Spontaneous separation of one chiral crystalline form from a racemate solution has been occasionally observed, but the process is hardly reproducible. Preferential seeding of a racemate solution with the crystals of one pure enantiomer (or with enantiomorphous crystals of the appropriate morphology) can induce sometimes the selective separation of one form of crystals.
- (c) Some crystals, both from chiral (cyclodextrins) and from achiral (urea) compounds, possess chiral cavities or interstices which can occlude molecules of compatible size. When such inclusion compounds are formed from racemic products, selective incorporation of one enantiomer can occur.
2.3 Chemical methods
Chemical methods are by far the most important for the preparation of chiral non-racemic compounds. They can be divided into three main categories, each of which giving rise to a few sub-categories, as reported below:
- – reversible transformation into diastereomeric derivatives
- • isolable diastereoisomers (classical resolution),
- • non-isolable diastereoisomers (chiral phase chromatography);
- – thermodynamically controlled asymmetric transformations
- • pyramidal inversion at a trisubstituted tetrahedric stereocentre,
- • rotation around a conformationally mobile bond,
- • interconversion of chemically labile stereocentres via an achiral intermediate.
- – kinetically controlled asymmetric transformations
- • kinetic resolution,
- • asymmetric synthesis,
- • absolute asymmetric reactions.
2.3.1 Reversible transformation into diastereomeric derivatives
- (a) The classical resolution of racemic mixtures has been for a long time the unique methodology amenable for the production of optically active compounds in sizeable amounts. Even today in several industrial preparations, specific protocols resting on this technique have been assessed, where the separation of the desired enantiomer is coupled with racemization and recycle of the undesired enantiomer. In this way, a racemic mixture is led to produce more than 50% of one enantiomer.
- (b) The direct resolution of enantiomers on a chiral stationary phase is effected by fast and reversible diastereomeric interaction between the racemic solute and the chiral stationary phase. This is nothing more than a variation of the classical resolution procedure, but with the involvement of short-lived, mostly ill-defined, diastereoisomers in a continuous flow. It is expected that this methodology will expand enormously its field of application as the technology will make available cheaper instruments and chiral adsorbents which can process kilos of racemic products in a short time and with reproducible results.
2.3.2 Thermodynamically controlled asymmetric transformations
Thermodynamically controlled processes are described by an energy profile where reagents and products are connected by a pathway which allows them to be equilibrated under the conditions of the experiment (Fig. 1). An activation energy barrier ΔG# not higher than 60–70 kJ/mol is required for the equilibrium to be attained around room temperature.
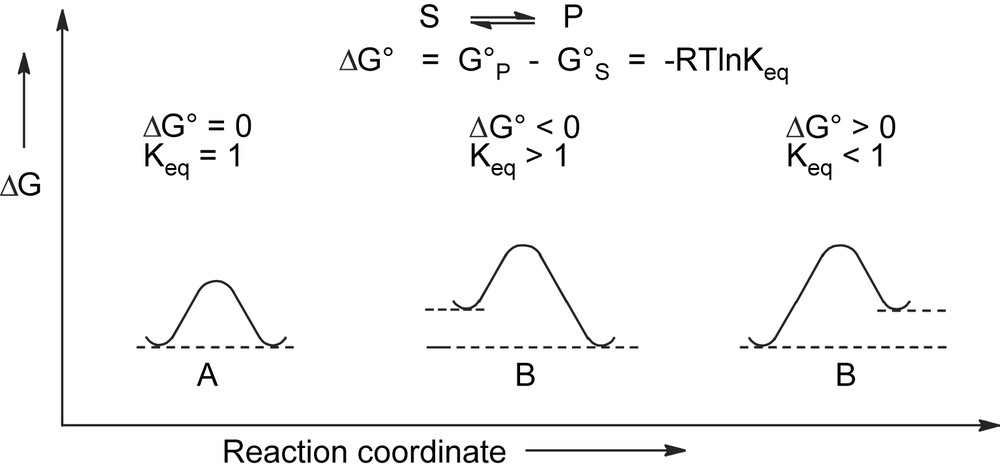
Energy plot for thermodynamically driven racemization (A) and epimerization (B).
In thermodynamically controlled asymmetric processes, the composition of the final product depends only on the differences in the ground-state free energies ΔG0 of reagents and products and is independent of the pathway. When reagents and products have the same free energy, they are enantiomers and the process corresponds to racemization. When the free energies are different, the process corresponds to the epimerization of stereochemically labile diastereoisomers.
The percent of stereoselectivity provides a quantitative measure of the efficiency of a stereoselective process. It follows from the following relation:
It corresponds to the stereoselectivity of the transformation (asymmetric induction, in an older terminology). It is most frequently expressed as enantiomeric excess (ee) or diastereomeric excess (de), whether the products of the asymmetric transformation are enantiomers or diastereoisomers.
- (a) Pyramidal inversion at the stereogenic sulfur, phosphorus or nitrogen atoms takes place more or less easily in chiral sulfoxides, sulfonium salts, phosphines or amines. The inversion process is extremely fast at room temperature for trisubstituted nitrogen compounds, while a few hours of heating at higher temperature are usually required for effecting the racemization of sulfoxides and phosphines (Scheme 1).
- (b) The hindered rotation around the σ-bond connecting the aryl groups is at the basis of the atropisomerism of 1,1′-binaphthalene and of other diaryl-core scaffolds. Their optical stability is determined by the steric hindrance of the substituents in the ortho- and ortho′-positions, which in most cases efficiently prevent the interconversion between the two atropisomeric conformations (Scheme 2). Another interesting case of racemization through rotation around conformationally mobile bonds is the case of helicenes (Scheme 2).
- (c) The best-known example of this type is the ring–chain tautomerism of glycosides which brings about the interconversion between the α- and β-anomers and is the cause of mutarotation (Scheme 3). The stereochemical lability of the anomeric carbon is a consequence of the poor stability of the protonated hemiacetalic bond towards nucleophilic attack. Upon hydrolysis, the C-1 changes its geometry from tetrahedric to trigonal, with consequent loss of its configurational integrity. The epimerization of 1-decalone takes course in a similar fashion through a keto–enol tautomerism which temporarily erases the configurational integrity of the stereogenic carbon adjacent to the carbonyl group (Scheme 3).
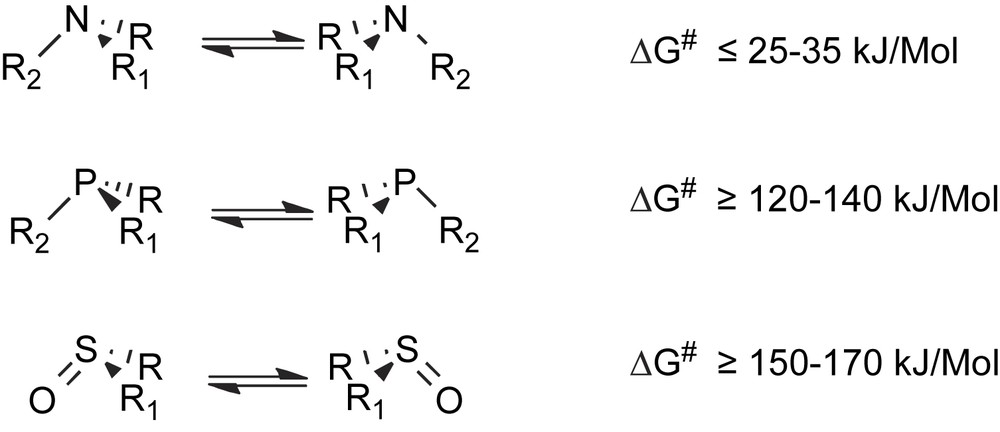
Racemization via unimolecular pyramidal inversion.
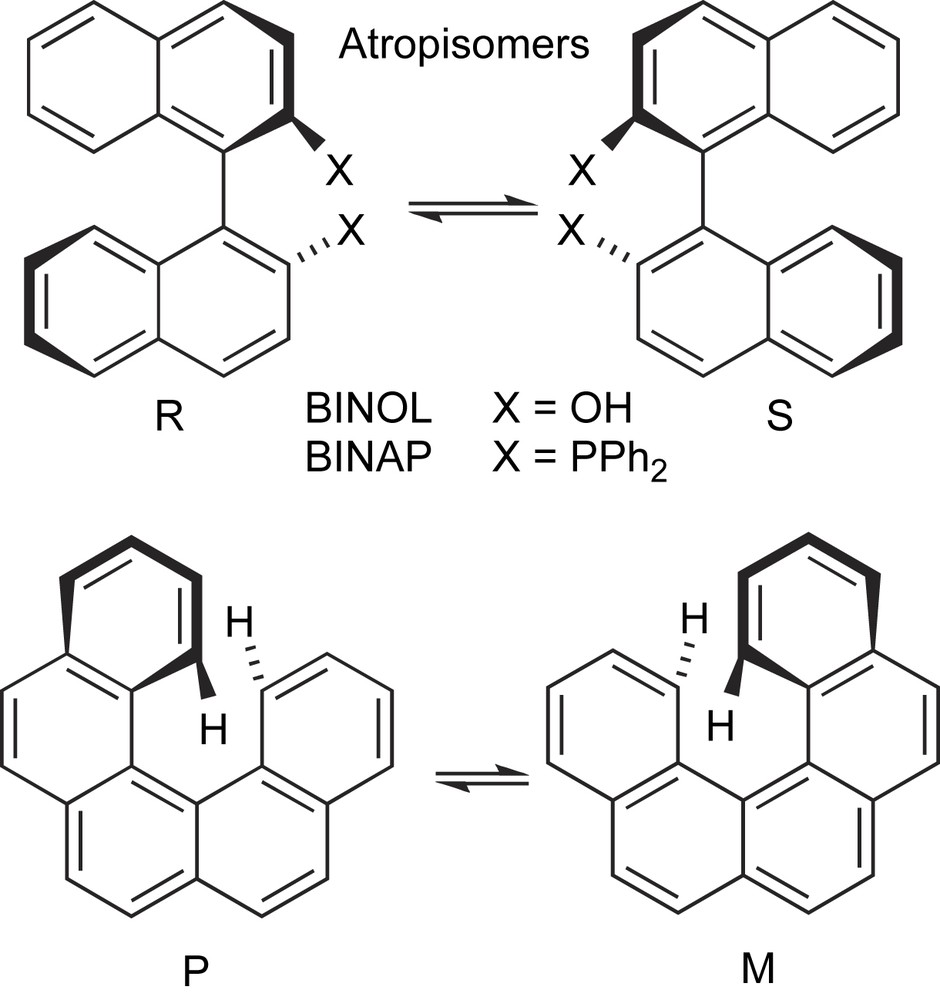
Racemization via rotation around configurationally labile bonds.
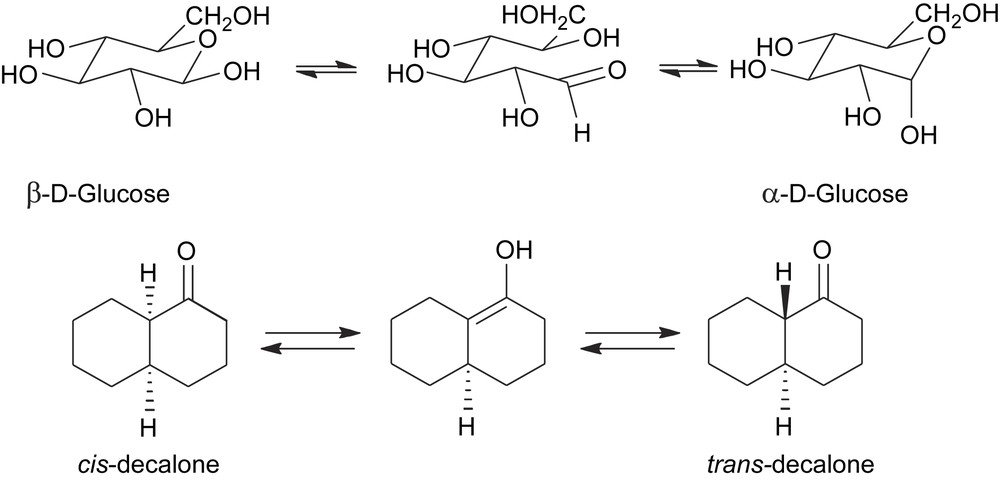
Epimerization of chemically labile stereocentres via an achiral intermediate.
2.3.3 Kinetically controlled asymmetric transformations
Kinetically controlled asymmetric processes provide the most viable and most efficient procedures for the production of chiral non-racemic compounds.
In these transformations the stereochemistry of the reaction is controlled only by the difference in the activation energy ΔΔG# of the competing reaction pathways leading to a couple of enantiomers, as in A, or to a couple of diasteroisomers, as in B and in C (Fig. 2).
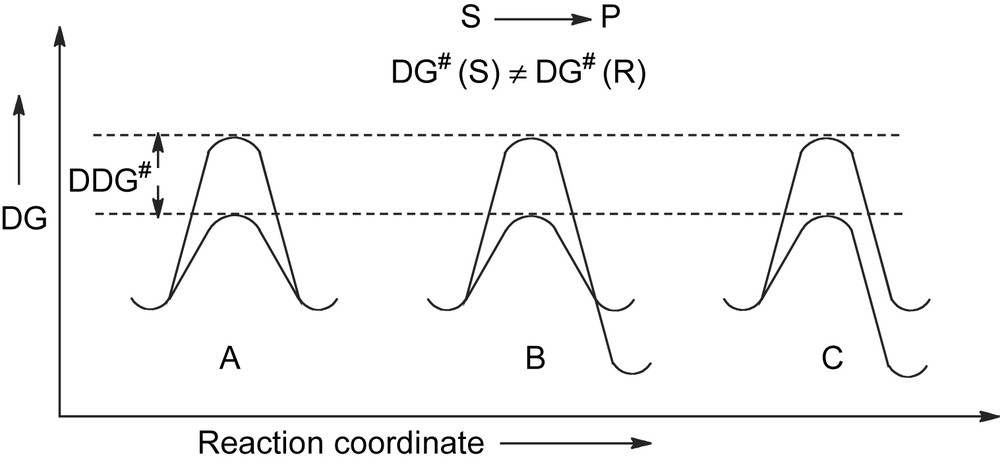
Energy profile for kinetically driven asymmetric transformations.
The condition to be fulfilled for such a process to occur is that the isomeric products are formed through diastereomeric transition states. This result can be achieved by using some chiral entity (reagent, catalyst, solvent, physical force…) which is able to discriminate the prochiral (or chiral) elements of the substrate by the rate of the reaction. The diastereotopic interaction, usually referred to as chiral recognition or stereodifferentiation, developed in the transition state is the sole factor which controls the stereoselectivity of the reaction.
A ΔΔG# of about 12 kJ/mol is required for providing a fully stereoselective reaction. Given that at room temperature a threshold of activation energy for a reaction to proceed under strict kinetic control is in the range of 100 kJ/mol, this means that the ΔΔG# is a quite small part of the whole activation energy.
It is amazing how narrow is the energy gap which separates a completely stereoselective process from a completely stereorandom process!
2.3.3.1 Kinetic resolution
Two enantiomers display a different behaviour in a chiral environment due to the development of diastereotopic interactions between the chiral elements of the substrate and the ones of the surroundings. As a consequence they must react at different rates with a chiral enantiopure reagent. This provides the rational background for a kinetic resolution (sometimes called enantiomeric enrichment) to occur.
If a racemic substrate is treated with an enantiopure reactant, one enantiomer shall be converted preferentially and shall be consumed faster than the other one. If the difference in the kinetic constants kfast and kslow is significant and the reaction is incomplete, two effects will be observed:
- – the unconverted starting material shall be no more racemic and shall contain an excess of the less reactive enantiomer;
- – the reaction product shall be mostly originated from the more reactive enantiomer.
A number of chemo- and bio-reagents in stoichiometric or catalytic amount have been successfully exploited for this purpose.
The efficiency of kinetic resolution depends not only on the relative rate constants, but also on the conversion degree, as it is apparent from the relation reported below:
As the stereoselectivity factor increases, also the chemical yield improves. Limiting cases of kinetic resolution are encountered when the stereoselectivity factor kfast/kslow is higher than 100. In this case, one enantiomer is converted almost completely before the second enantiomer starts to react. In such a case, the less reactive enantiomer can be isolated in high ee with a yield approaching the theoretical value of 50%.
This behaviour is illustrated by the plot reported below in the case of the asymmetric epoxidation of allylic alcohols by t-butyl hydroperoxide (TBHP) catalyzed by titanium–tartrate complexes (Scheme 4). This reaction is remarkable in that the (S)-enantiomer consistently reacts faster than the (R)-enantiomer, when a racemic allylic alcohol is epoxidized with (R,R)-tartrate as the chiral ligand [2].

Kinetic resolution in the asymmetric epoxidation of allylic alcohols.
The major limitation inherent with kinetic resolution is that the maximum theoretical yield of a single enantiomer cannot exceed 50%. Because of this, a suitable procedure for the second enantiomer to be racemized and recycled to resolution is mandatory for the economy of the process. If racemization can be coupled with kinetic resolution in such a way that both processes can be run simultaneously, recycle is no more required and in principle 100% of the racemic mixture can be led to the desired enantiomer. This approach, known as dynamic kinetic resolution (DKR) [3], relies on the fact that racemization is a thermodynamically favoured process due to the increase of entropy. An example of the application of this technology which exploits a lipase-catalyzed kinetic resolution of a secondary carbinol for the selective acylation of the (R)-enantiomer and a tandem Ru-catalyzed oxidation–reduction by H-transfer for the racemization of the unreacted (S)-enantiomer [4] is sketched below (Scheme 5).
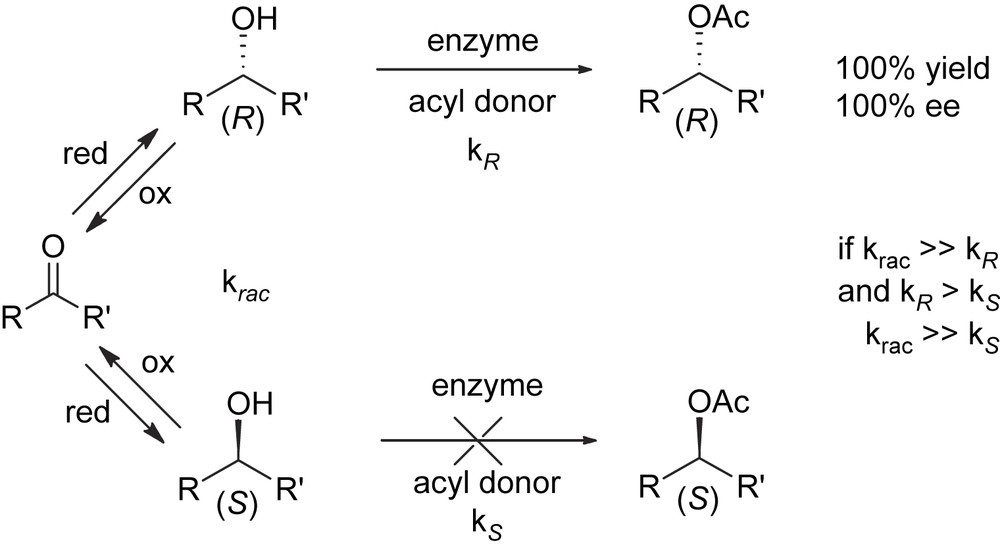
Dynamic kinetic resolution (DKR) of secondary carbinols.
2.3.3.2 Asymmetric synthesis
The birth of asymmetric synthesis dates back to 1890, when Emil Fischer recognized that the addition of hydrogen cyanide to l-arabinose affords in some 75% yield one of the two possible diastereomeric cyanohydrins [5] (Scheme 6).
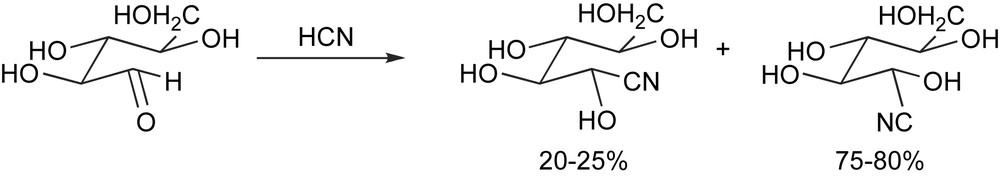
The very first asymmetric synthesis.
This discovery opened the way to the development of synthetic methodologies where a pre-existing stereogenic element of the substrate informs the chirality of the new stereogenic centre to be created in the same molecule. The diasteroselectivity of this type of reactions rests on an intramolecular transfer of chirality, which is rooted in the preferential approach of the reagent from the less hindered side of the carbonyl group.
There are cases, as above, where the stereogenic elements responsible for the stereodifferentiation are constitutional in the structure of the substrate to be transformed. Most frequently, they survive the stereoselective reaction and are retained as such until the end of the synthetic process. Sometimes they are eliminated in a subsequent step of the synthetic process after having served as chiral controllers in the stereoselective step.
There are also fairly rare cases where they go lost in the same step where they act as chiral controllers (Scheme 7). This last kind of transformations has been sometimes referred to as ‘self-immolative’ asymmetric synthesis [6].
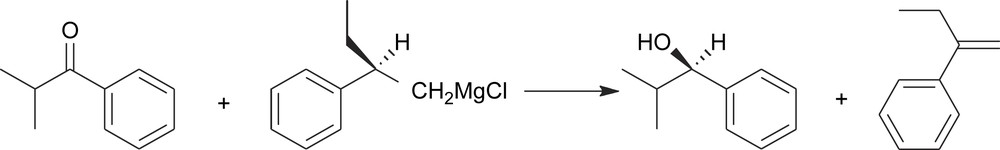
Self-immolative asymmetric synthesis.
In many other cases, the stereogenic elements required for the reaction to be stereocontrolled are appositely introduced in an originally achiral structure by binding a chiral tail to the substrate before the stereoselective step of the synthetic process is performed. The chiral tag used for this purpose is then removed once it has accomplished the task of steering the stereochemistry in the formation of a new chiral element. This methodology has been designed for expanding the utility of the intramolecular transfer of chirality even to achiral substrates and is called the ‘chiral auxiliary technique’.
The Prelog's atrolactic acid synthesis (Scheme 8) is one of the most studied and illustrative examples of this approach [7].

Prelog's atrolactic acid synthesis: an intramolecular transfer of chirality.
The second main breakthrough in this area was achieved when it was recognized that even the introduction of the first stereogenic element in an achiral molecule can be steered by a chiral element which lies outside the substrate and that the intermolecular transfer of chirality can be as efficient as the intramolecular case (Scheme 9). Thus the stereogenic elements of the chiral reagent are able to differentiate the prochiral element of the substrate, providing a diastereotopic transition state.
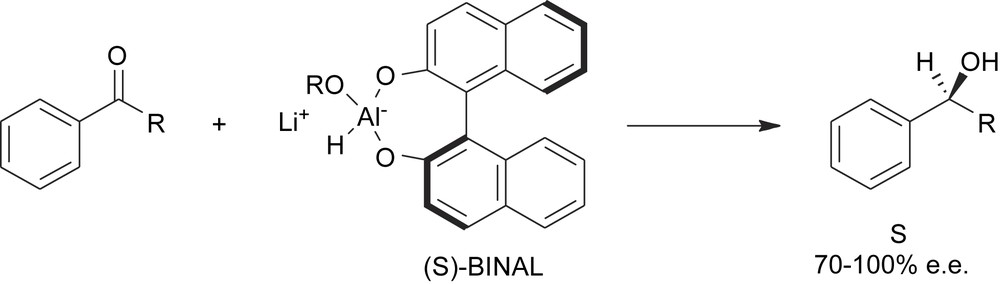
An intermolecular transfer of chirality.
This discovery has expanded enormously the scope of natural products of the chiral pool in the preparation of chiral reagents to be used in the stereo-determining step of a synthetic procedure [8].
2.3.3.3 Absolute asymmetric transformations
An absolute asymmetric transformation occurs when the chiral element responsible for the stereodifferentiation in an asymmetric transformation is a chiral physical force. The few reliable examples of asymmetric transformation of this type reported in the literature refer to kinetic resolution (or asymmetric synthesis) induced by dextro- or levo-rotatory circularly polarized light. The highest stereoselectivities obtained thus far are modest (up to 10% ee) [9]. Claims on the successful application of other chiral force fields (magnetic, gravitational) have appeared, but have been later disconfirmed.
3 Merits and drawbacks
3.1 Chemical methods
Both kinetic and classical resolutions of racemic mixtures suffer from the drawback that in principle they can give not more than 50% of the desired product. In most industrial applications, however, the unwanted enantiomer can be racemized and recycled, improving the chemical yield. Furthermore, when the conditions for a DKR transformation are met, the desired isomer can be isolated in practically quantitative yield and in very high ee, avoiding any recycle.
Transformation or derivatization of chiral substrates available in optically active form from natural sources (the chiral pool) was the first practical alternative to resolution. This methodology is, however, restricted to the preparation of chiral compounds whose molecular structure and stereochemistry correlate with a product of the chiral pool.
Intramolecular transfer and intermolecular transfer of chirality are powerful and versatile tools of asymmetric synthesis which have found a countless number of applications in asymmetric synthesis.
The main drawback of the above strategies is that the transfer of chirality occurs on a one-to-one basis. i.e., one stereogenic pre-existing element is necessary for the introduction of one new stereogenic element in the substrate. The net result is that the stereochemical information which is present in the chiral reagent is utilized in a stoichiometric basis and is exploited only once.
Asymmetric catalysis overrides this limitation because it allows a single chiral information embedded in a chiral catalyst to be reproduced a large number of times. Thus, it provides the most economic way to accomplish an asymmetric synthesis since, in principle, one molecule of chiral catalyst can create millions of molecules of chiral product (chiral multiplication). This is the same way the enzymes act in biological systems.
4 Asymmetric catalysis: a bit of history
The first demonstration that a chiral metal complex could induce a stereoselective reaction in homogeneous phase was presented in 1966 when a Japanese group reported that the reaction of ethyl diazoacetate with styrene led to optically active cyclopropane carboxylates when a chiral Cu-complex was used to catalyze the reaction (Scheme 10) [10].
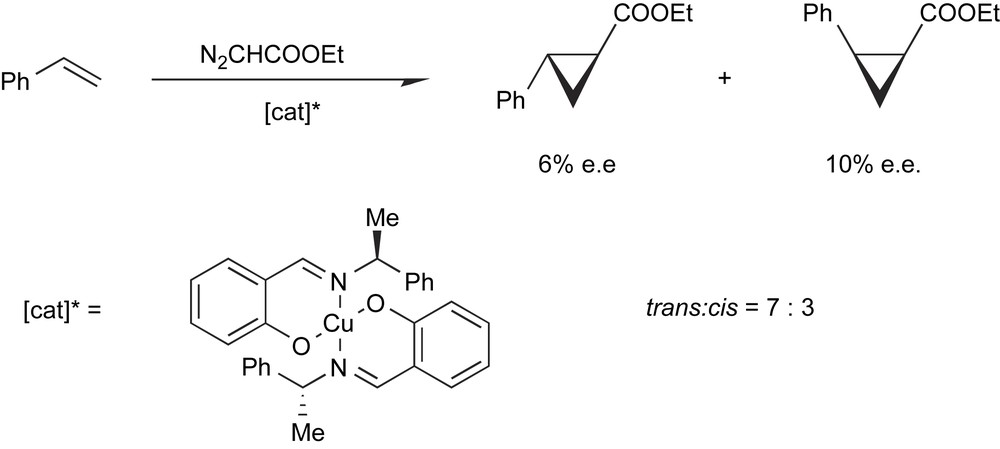
The first example of enantioselective catalysis.
This first example is quite illustrative of the basic concepts of asymmetric catalysis. In this reaction, the metal is necessary for the decomposition of diazoacetate to be accelerated thanks to the formation of Cu-carbene intermediates, while the chiral ligand plays its role in the differentiation of the enantiofaces of the incoming substrate. This separation of roles between the metal and the ligand is straightforward, but is somehow fictitious. In reality, the catalytic activity of the metal and the stereodifferentiating ability of the ligand in the complex are interdependent.
As it is apparent from the number of papers appeared in the literature, in the last thirty-five years metal-catalyzed asymmetric synthesis has grown at impressive rate to become a practical synthetic tool and now it is at the core of organic synthesis.
The strategic nature of this area of chemistry is witnessed by the enormous number of patents which have been filed and by the number of catalytic asymmetric processes which have been scaled up to pilot plant in the last ten years all over the world. Asymmetric catalysis has been successfully applied to the industrial preparation of fine chemicals. The most important industrial processes exploiting this technology are listed below.
- 1970s l-Dopa, enamides' hydrogenation (Monsanto),
- 1980s l-Menthol, CC isomerization (Takasago),
- 1990s Glycidol, Sharpless epoxidation (Arco); S-Metolachlor via CN hydrogenation (Ciba–Geigy/Novartis).
The award of the Nobel Prize for Chemistry 2001 to three pioneers of asymmetric catalysis, W.S. Knowles, R. Noyori and K.B. Sharpless, is the recognition of the central role that stereoselective catalysis has achieved in the asymmetric synthesis of pharmaceuticals, agrochemicals, flavours and essences, that is, of the most important categories of fine chemicals.
5 Asymmetric catalysis: guidelines
In asymmetric catalysis, a chiral enantiopure catalyst discriminates the enantiotopic (or enantiomeric) elements of the prochiral (or chiral) substrate by the rate of reaction. Stereoselection is then obtained from preferential binding of the substrate to the catalyst or from preferential reaction of one of the diastereomeric substrate–catalyst adducts involved as an intermediate in the catalytic cycle.
The efficiency of a catalyst can be expressed in terms of either turn-over number (TON; an adimensional number) or turn-over frequency (TOF; dimension 1/t). The first parameter, sometimes called catalyst productivity, gives the number of molecules of product produced by one molecule of catalyst. It equals the total number of catalytic cycles accomplished by the unit of catalyst before its deactivation.
The second parameter, sometimes called catalyst activity, gives the number of molecules of product produced by one single molecule of catalyst in the unit of time. It equals the number of catalytic cycles accomplished by the unit of catalyst in the unit of time. The TOF gives a sharper idea of the activity of catalyst and allows direct comparison between different catalysts. To allow correct and meaningful correlations, the reactions to be compared must be run at the same conversion, as TOF is dependent on the extent the reaction has proceeded, and the conversion must be not too low, to minimize the effects of the induction time, if any.
Asymmetric catalysis is reliant both on biocatalysts and chemocatalysts and can be delivered both in the homogeneous and in the heterogeneous phases. Chemical catalysts can be inorganic, organic or mixed metal–organic. In the last thirty years, the most significant achievements in stereoselective catalysis have been achieved with the use of chiral metal-centred homogeneous catalysts.
In metal-mediated stereoselective reactions, the source of the chiral information resides almost always in a chiral non-transferable ligand which remains bound to the metal centre in the active catalytic species. The nature of this chiral controller is crucial in determining both the catalytic activity and the stereoselectivity of the catalyst. For this reason, design and synthesis of new chiral ligands have gained a central position in this chemistry and has grown up to become itself an art.
The research efforts devoted to this subject have resulted in the preparation of a plethora of chiral ligands for asymmetric catalysis. At the end of 1993, an exhaustive survey quoted over two thousands of chiral derivatives used in asymmetric catalysis [11]. Since then, the number of chiral ligands has grown at impressive rate and by now we can confidently estimate that more than ten thousand have been prepared and tested.
From the enormous literature on this subject, it is readily apparent that for a long time bidentate chelating ligands with phosphorus donors (diphosphines and, to a lower extent, diphosphites) have constituted the most efficient and popular class of chiral modifiers [12]. Within this category, atropisomeric derivatives featuring a stereogenic axis as the unique chirogenic element have played a prominent role because they provide stereoselectivities among the highest in a wider range of reactions. The paradigmatic representative of this family is the well-known BINAP reported by Noyori in 1981, which is presently used with remarkable success in a large variety of catalytic asymmetric reactions, even on industrial scale [13].
In the course of its development, asymmetric catalysis has learned the value of imitating nature which is reliant on the catalytic activity of metal ions embedded in the active site of metalloenzymes. These highly asymmetric molecular assemblies are able to catalyze a number of specific reactions such as hydrolysis, oxidations, reductions, isomerization, nitrogen fixation and photosynthesis [14]. The chiral environment of metalloenzymes arises from the binding of nitrogen, sulfur and oxygen donors of the chiral enzyme structure to the metal centre.
Many studies have demonstrated that chiral ligands with nitrogen [15] and, to a lower extent, with sulfur donor centres, albeit of less frequent use, are sometimes capable of excellent enantioselections. In some asymmetric processes such as cyclopropanation [16], hydrosilylation [17], H-transfer reduction [18], addition of alkyl zinc to arylaldehydes [19], allylic alkylation [20] and allylic oxidation [21], metal complexes containing chiral ligands with N-donors should be considered as catalysts of choice because they usually provide stereoselectivities higher than any other category of ligands used thus far.
Following twenty-five years of investigations in stereoselective catalysis, a selected set of ligands has emerged to build up what has been sometimes called the family of ‘privileged structures’ (Scheme 11). This list was set up by the end of the 1990s and contains those ligands which with some regularity provide stereoselectivities higher than the other ones over a wide range of metals and of reactions. They can be regarded as the ligands of choice for performing a first screening of an asymmetric reaction in the aim to check for a reasonable asymmetric induction to be at hand.

Chiral ligands: privileged structures.
In spite of the differences and of the structural variety, these derivatives share some common features: (1) all but one (DHQ, DHQD) are bidentate ligands capable of chelate coordination to the metal; (2) all but the three ones in the boxes possess a C2-symmetrical structure. For sometime these two properties have been considered almost mandatory for a chiral ligand to be a good candidate for high stereoselections.
At the turn of the century, however, independent and nearly simultaneous reports from three different sources [22] have shown that the efficiency of these ‘established’ bidentate ligand can be outperformed by some selected monodentate phosphacyclic derivative such as the ones reported in Scheme 12. This discovery has dramatically shaken one of the foundational pillars of ligand design and has open new horizons in asymmetric catalysis, pushing further research in this field.
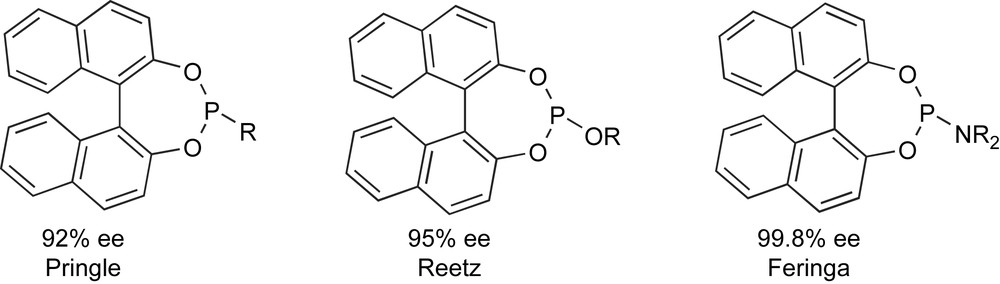
Efficient phosphacyclic monodentate ligands (2000).
Current research in ligand design is addressed in several directions (Table 1). A general issue is the development of a modular synthetic approach to a ligand lead which complies with the possibility to provide easy access to a library of derivatives with a variety of substituents of different steric and electronic demand. This is extremely useful in the optimization of a catalytic asymmetric reaction where the ‘fine tuning’ between the catalyst and the substrate is usually sought via subtle modifications of the coordination environment around the metal.
Current trends in chiral ligand design
| 1. | Moving from C2- to C1-symmetry |
| 2. | Bidentate heterodonor/hemilabile ligands |
| 3. | Chiral ligands with sulfur donors |
| 4. | Chiral tridentate arylpincer ligands |
| 5. | C2-Symmetry monodentate phosphacyclic ligands |
| 6. | ‘Chiral at metal’ catalysts |
| 7. | Cluster catalysis by inherently chiral polynuclear architectures |
Among the entries listed in the table, the most intriguing is surely the last one which calls for the use of polynuclear complexes as the catalysts. Actually, the successful use of Ru-clusters containing chiral ligands as catalysts in the asymmetric hydrogenation of unsaturated acids was reported some thirty years ago [23]. The reaction was remarkable because the stereoselectivity recorded was the highest observed with such a type of substrates at those times. Some experimental evidence was as well collected that the cluster structure was maintained intact during the catalysis, but no clear-cut demonstration of this fact could be eventually reached and the possibility that mononuclear Ru-complexes had a major role in catalyzing the asymmetric hydrogenation could not be ruled out.
The definitive proof that a cluster can perform as a chiral catalyst in stereoselective reactions and that in the meantime the cluster structure is preserved during the catalysis has been achieved many years later by a Spanish group with the use of the inherently chiral cluster derivative reported below (Scheme 13) [24].
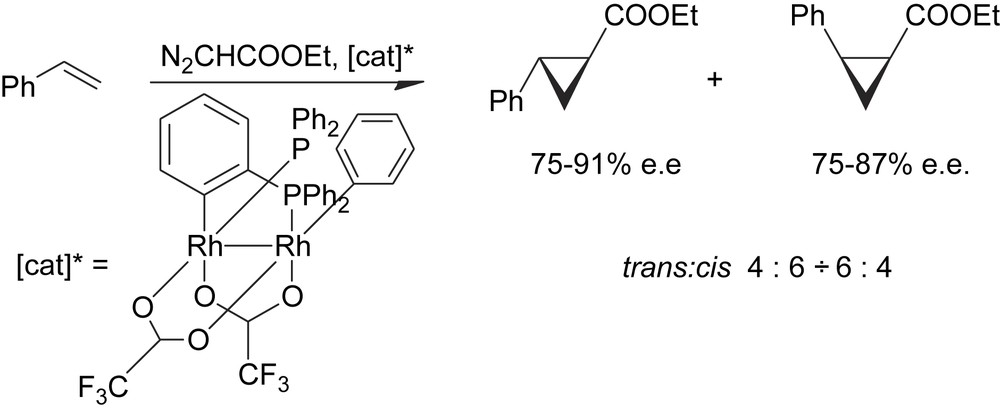
Enantioselective catalysis by an inherently chiral cluster.
This complex has given quite good stereoselectivities in several cyclopropanation/carbene insertion reactions. The high ee's observed provide a conclusive proof that the catalyst must be a dinuclear species because the chirality of this cluster is inherent to the dinuclear framework of the complex and goes irremediably lost as the cluster structure is broken down to mononuclear species.
This result opens new horizons in the design of chiral catalysts for stereoselective reactions.