

1 Introduction
The resolution of racemic mixtures has become an increasingly important field in organic chemistry due to the ease of preparing racemic substrates and the number of transformations that can be exploited to easily change one enantiomer in preference to the other [1]. Kinetic resolution has been a tool for chemists for almost 150 years and remains an important transformation for creating enantiopure samples. The one major limitation of this technique is that the maximum theoretical yield is 50% due to the consumption of only one enantiomer. Because of this, the unreacted enantiomer must be racemized and resubmitted to resolution conditions in order to increase this yield. If the racemization can occur concurrently with the kinetic resolution, known as Dynamic Kinetic Resolution (DKR), then theoretically 100% of the racemic mixture can be converted to one enantiomer [2].
Ruthenium has various valencies (0–VIII) and is a not so expensive transition metal; therefore, various useful catalytic reactions for organic synthesis have been explored [3–5]. Several attempts were made to utilize a ruthenium complex as the racemization catalyst effective for DKR [6–11]. In this review, we describe the preparation of chiral compounds by the enzymatic resolution of secondary alcohols coupled with ruthenium-catalyzed racemization. There are 10 ruthenium complexes (Scheme 1) reported as catalyst, for racemization of secondary alcohols, which on combination with the enzyme carry out asymmetric transformation, called as DKR.
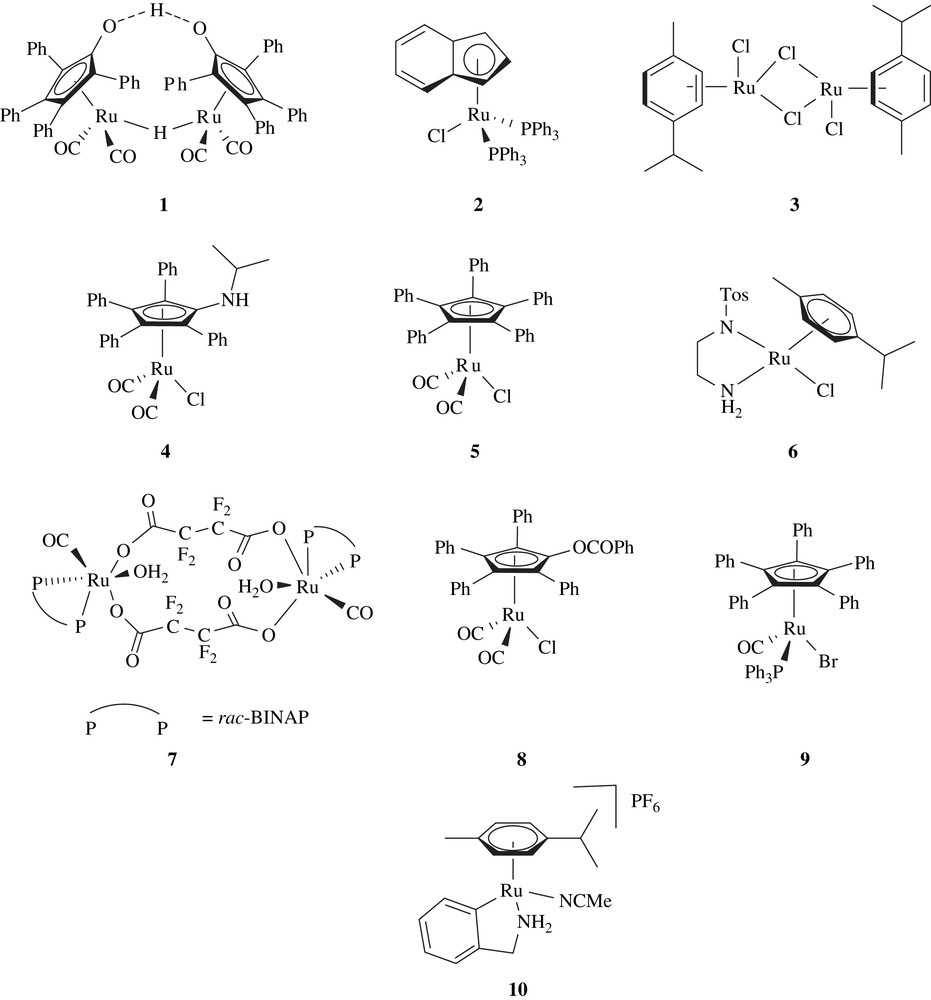
2 Shvo complex (1)
2.1 DKR of secondary alcohols
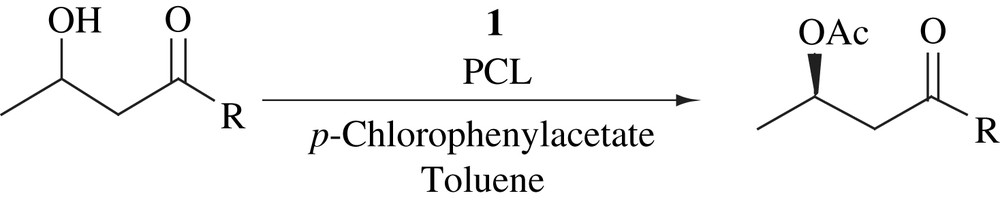
Bäckvall and co-workers studied Ru-catalyzed hydrogen transfer reactions employing catalyst 1 or [RuCl2(PPh3)3] [12–17], and observed that alcohols undergo fast isomerization at the α-carbon leading to racemization. With the aim of developing a DKR of alcohols, they studied the racemization of (R)-1-phenylethanol with catalysts 1 or [RuCl2(PPh3)3]. Both the catalysts were able to catalyze the complete racemization of (R)-1-phenylethanol, [RuCl2(PPh3)3] with a higher rate than 1. Although the racemization rate of [RuCl2(PPh3)3] is faster than 1, it required the presence of NaOH, which might interfere with an enzyme. The ruthenium-catalyzed racemization of (R)-1-phenylethanol was then combined with an enzyme-catalyzed transesterification using Novozym 435. The use of [RuCl2(PPh3)3] in combination with enzyme and different acyl donors gave poor results with decreased activity of the enzyme, and furthermore, the rapid racemization with this catalyst could not be reproduced in the presence of the enzyme. On the other hand, they observed that the combination of catalyst 1 with the enzyme and acyl donor yielded a good result (Scheme 2).
A variety of substrates were resolved using the same reaction conditions [18,19]. The yields and selectivities were high and in most cases ees were >99%. The disadvantage of this system is that acyl donor, 4-chlorophenyl acetate is not commercially available.
2.2 Conversion of ketones or enol acetates to chiral acetates
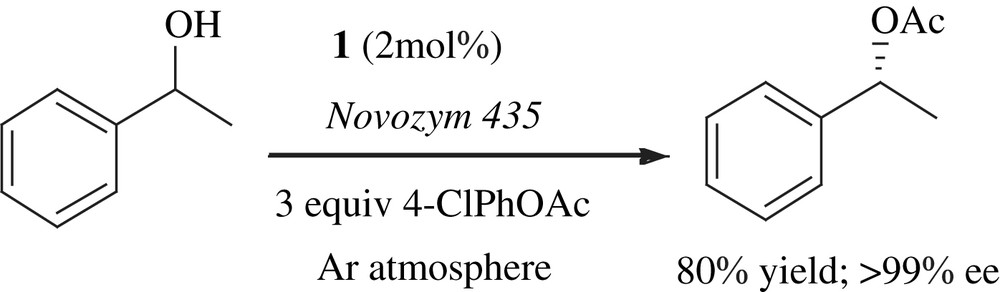
The conversion of enol acetate to chiral acetate in the presence of catalyst 1, lipase and hydrogen donor proceeded through a five-step process (Scheme 3): deacetylation of enol acetate 11 to give the corresponding enol and the acetylated lipase, keto–enol tautomerism for the formation of ketone (12), reduction of ketone 12 to the racemic mixture of (R)-13 and (S)-13 alcohols, enantioselective acetylation of (R)-13 with the acetylated lipase to produce chiral acetate (R)-14 and reversible transformation between (S)-13 and (R)-13. The scope of the ketone conversion process was investigated by using complex 1, an immobilized lipase, hydrogen donor and 4-chlorophenyl acetate as acyl donor in toluene at 70 °C [20]. 2,6-Dimethylheptan-4-ol was chosen as a suitable hydrogen donor and 4-chlorophenyl acetate was used as an acyl donor for the conversion of ketones.
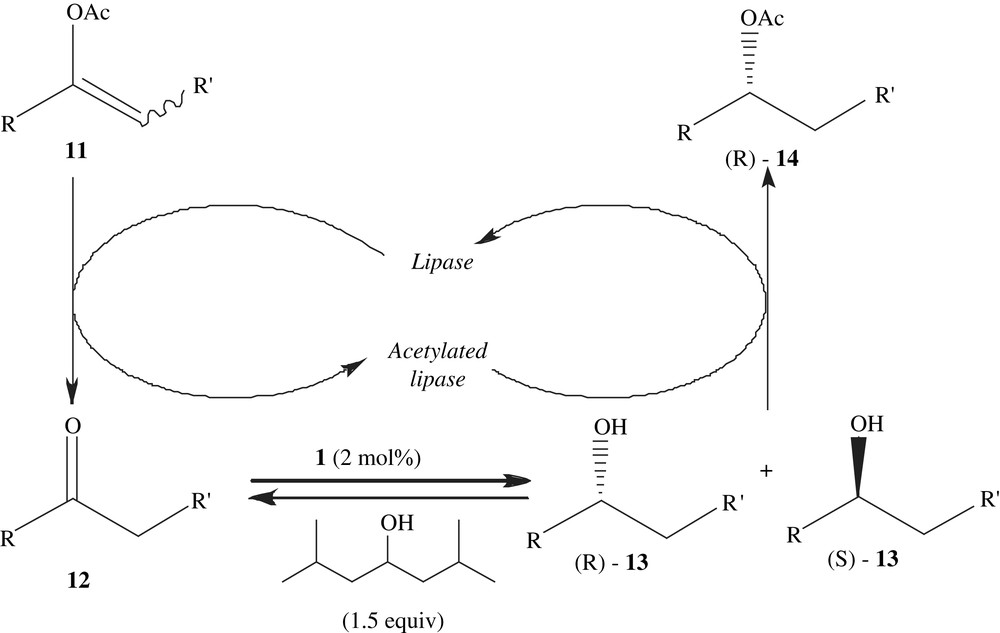
Park and co-workers demonstrated another method in which ketones were asymmetrically transformed to chiral acetates by one-pot processes using a lipase and an achiral ruthenium complex under 1 atm of hydrogen gas in ethyl acetate (Scheme 4). Molecular hydrogen was also effective for the transformation of enol acetates to chiral acetates without additional acyl donors with the same catalyst system [21].
2.3 DKR of α-hydroxy acid esters
Bäckvall and co-workers have shown that DKR of different α-hydroxy esters was carried out successfully to give enantiomerically pure compounds in good yields and in one single step (Scheme 5). It was demonstrated that the high enantioselectivity of the enzyme is compatible with the ruthenium-catalyzed racemization of α-hydroxy esters. The racemization process took place via a hydrogen transfer pathway involving dehydrogenation of the alcohol to ketone and readdition of hydrogen. This methodology was applied to a variety of substrates, although substrates with a primary carbon at the β-position of the α-hydroxy ester gave poor results [22].
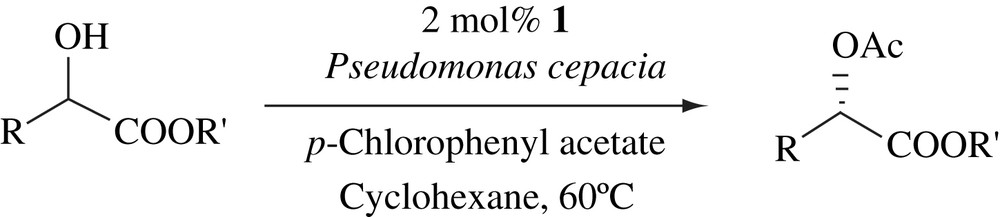
2.4 DKR of β-hydroxy esters
Recently, Huerta et al. reported a novel combination of aldol reaction and a DKR of the corresponding adduct [23]. This one-pot tandem procedure allowed the enantioselective synthesis of β-hydroxy acid derivatives from an aldehyde and a ketone without the isolation of preformed silyl enol ether (Scheme 6).
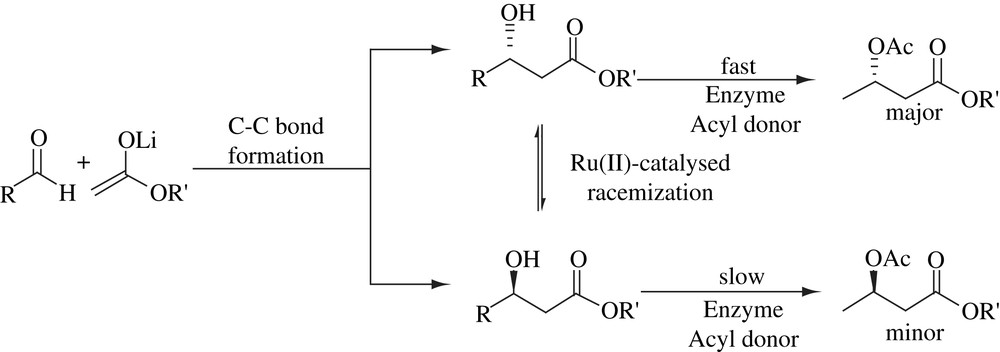
2.5 Asymmetric transformations of acyloxyphenyl ketones
A lipase/Shvo complex catalyzed multipathway process proposed for the asymmetric conversion of 3′-acetoxyacetophenone (15) to (R)-1-(3-hydroxyphenyl) ethyl acetate (16) is described in Scheme 7. The scope of the catalyst was extended to various similar substrates. The chiral products obtained are useful as intermediates for the synthesis of chiral drugs such as rivastigmine and its analogues for the treatment of Alzheimer's disease [24].
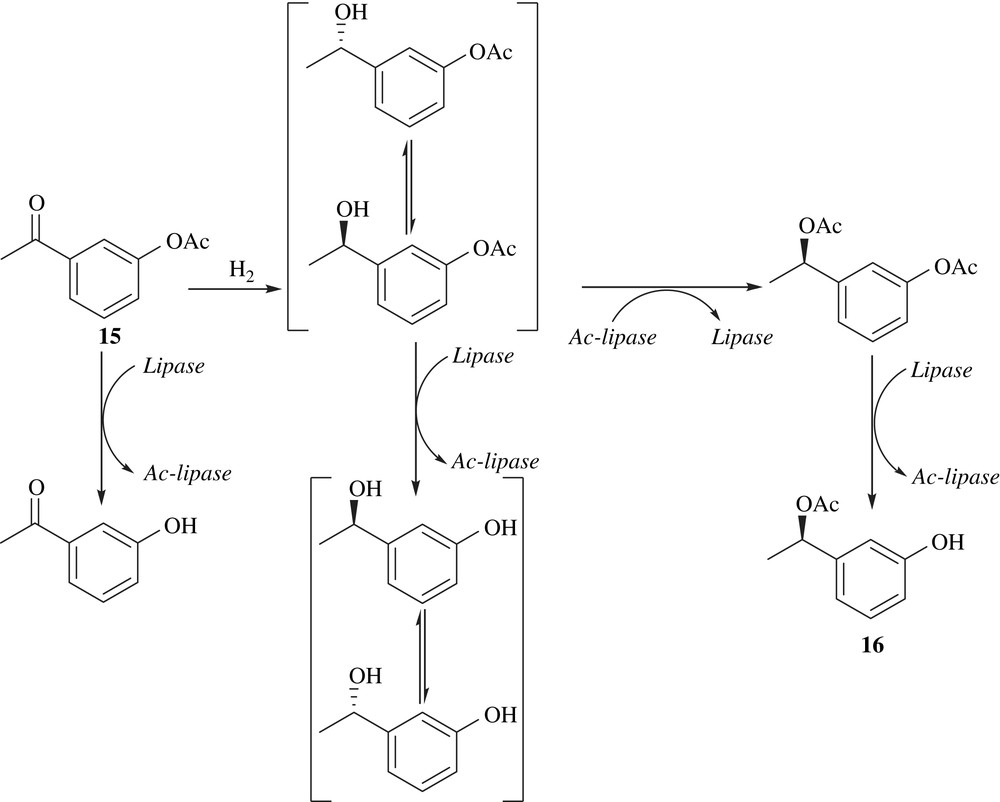
2.6 DKR of hydroxy acids, diols and hydroxy aldehydes protected with a bulky group
The DKRs of the protected β-hydroxybutyrates in which the carboxy functionality is protected with four different bulky groups including benzyl, (p-methoxyphenyl)methyl, biphenylmethyl and tert-butyl have been carried out (Scheme 8) [25]. The enantiomeric excess was the highest in the case of tert-butyl protecting group. Hence, tert-butyl group is the best protecting group as steric auxiliary for the efficient DKR of β-hydroxybutyrate.
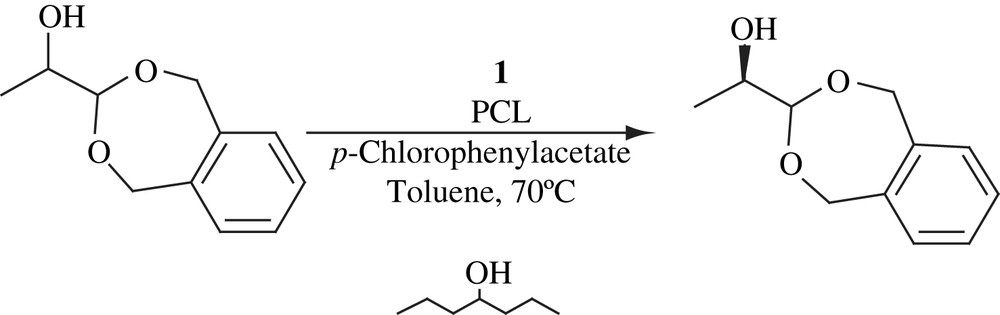
Monoprotected 1,2-diols, in which the primary alcohols were protected with the trityl group, were subjected to DKR [25] (Scheme 9). The alcohol additive (2,6-dimethyl-4-heptanol) acted as a reductant to reduce the formation of ketone. The yield and ee were good in all the cases.
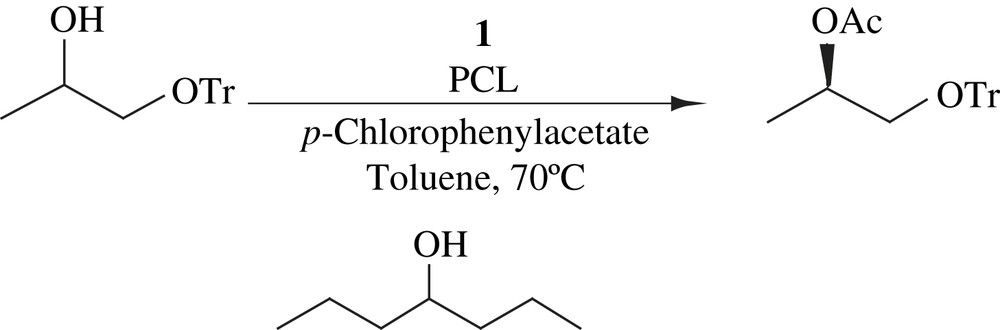
The DKR of protected hydroxy aldehyde, in which 1,2-benzenedimethanol was employed as protecting group, was performed using 1 and PCL [25]. The reaction carried out for 3 days provided 95% yield and 98% ee (Scheme 10).
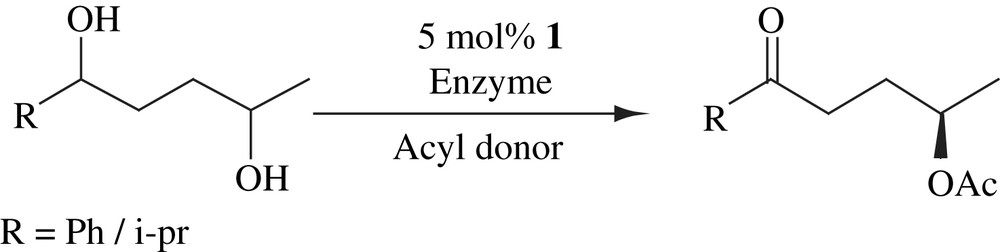
2.7 Dynamic kinetic asymmetric transformation of 1,4-diols to γ-hydroxy ketones
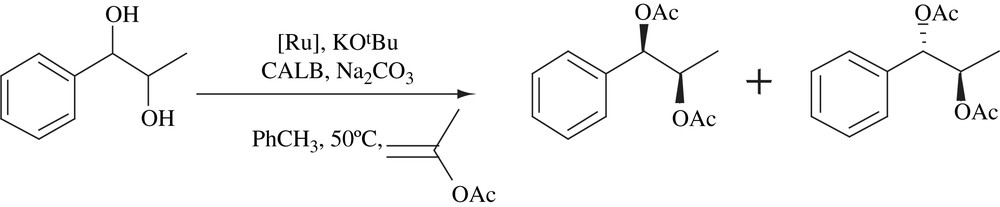
Enzymatic kinetic resolution of unsymmetrical 1,4-diols using Candida antarctica lipase B (CALB) was combined with ruthenium-catalyzed hydrogen transfer process to form least hindered γ-acetoxy ketones in high enantiomeric purity (Scheme 11). Dynamic kinetic asymmetric transformation (DYKAT) took place at least hindered alcohol and second hydroxyl group underwent oxidation under the reaction conditions. This process allowed a one-pot transformation of 1,4-diols into enantiomerically enriched γ-acetoxy ketones [26].
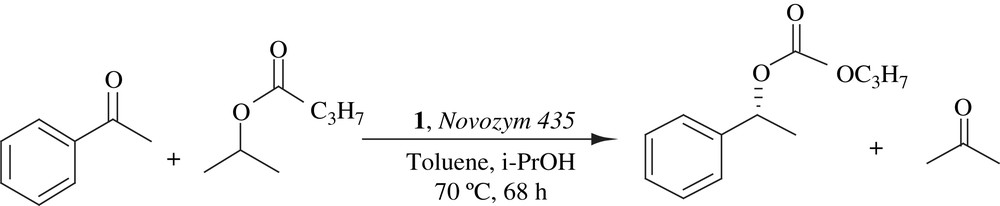
2.8 Use of simple alkyl esters as acyl donors for DKR
Verzijil et al. reported the use of simple alkyl esters as acyl donors if the alcohol or ketone residue formed during the enzymatic acylation was continuously removed during the reaction. The addition of a ketone increased the rate of the racemization process and also reduced the amounts of enzyme and catalyst 1. The scope of this method was explored and a suitable range of acyl donors found. Various benzylic and aliphatic alcohols were reacted using isopropyl butyrate or methyl phenylacetate as acyl donor and in most cases the ester was isolated in >95% yield and 99% ee (Scheme 12). Furthermore it was demonstrated that the alcohol by-products of the enzymatic resolution could be used as the hydrogen source in the asymmetric reductive transesterification of ketones. The low cost and lack of appreciable toxicity of common alkyl esters made their use as acyl donors both economically and environmentally attractive [27].
2.9 DKR of primary alcohols with an unfunctionalized stereogenic center in the β-position
DKR of primary alcohols with an unfunctionalized stereogenic center in the β-position was achieved via combined enzyme and metal catalysis (Scheme 13) [28]. The in situ racemization of the primary alcohol, required for the DKR, took place via: (i) ruthenium catalyzed dehydrogenation of the alcohol, (ii) enolization of the aldehyde formed, and (iii) ruthenium-catalyzed readdition of hydrogen to aldehyde. This method widened the scope of metal and enzyme-catalyzed DKR, which has so for been limited to α-chiral alcohol and amine derivatives.
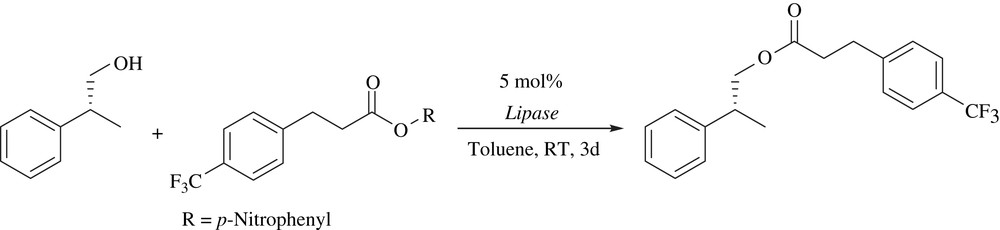
3 [(η5-Indenyl)RuCl(PPh3)2] (2)
[(η5-Indenyl)RuCl(PPh3)2] was found to catalyze the racemization of alcohols in the presence of triethylamine and oxygen. Unlike previously reported metal-catalyzed racemization, ketones were not required as hydrogen mediators in this process. The Ru-catalyzed racemization was coupled with enzymatic acetylation for the DKR of secondary alcohols to give chiral acetates in good yields (60–98%) with high enantioselectivities (82–99%) (Scheme 14). For the enzymatic resolution, Pseudomonas cepacia lipase (PCL) was employed, and two alkenyl acetates and 4-chloropheyl acetate were tested as acyl donors [29]. The drawback of this catalytic system is that amine and oxygen are needed for racemization.
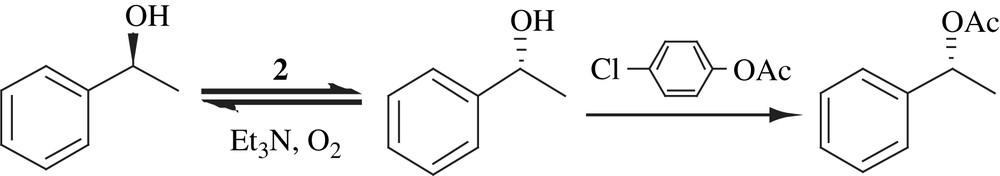
4 [RuCl2(p-cymene)]2 (3)
4.1 DKR of allylic alcohols
Kim and co-workers described an enzyme–metal combo reactions for the DKR of allylic alcohols in which racemic substrates were enantioselectively acetylated by a lipase with the in situ racemization of unreacted substrates by [RuCl2(p-cymene)]2 to give allylic acetates of high purity in over 80% yields (Scheme 15). The catalyst employed was readily available and the reactions proceeded at room temperature. But additive (amine) is required for racemization. The reaction followed by mild hydrolysis also provided a useful route to homochiral allylic alcohols [30].
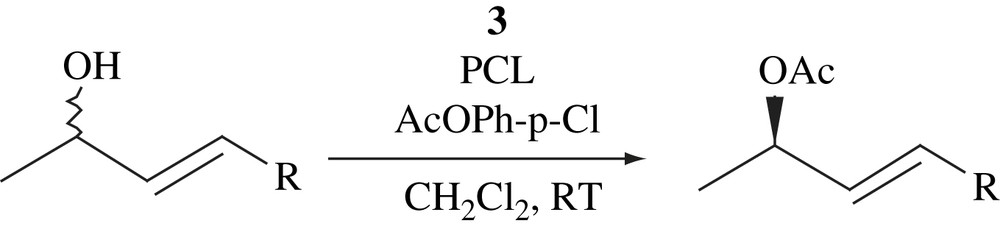
4.2 DKR of secondary alcohols by 3 in the presence of diamines
Stable racemization catalyst for secondary alcohols was obtained by an in situ mixture of readily available [RuCl2(p-cymene)]2 with chelating diamines. In particular, N,N,N′,N′-tetramethyl-1,3-propanediamine as ligand gave excellent racemization activity. This racemization catalyst was combined with lipase-catalyzed kinetic resolution using p-chlorophenyl acetate as acyl donor with good performance for the DKR of secondary alcohols (Scheme 16) [31].
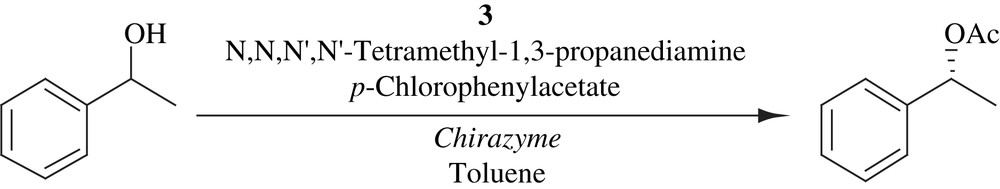
4.3 DKR of secondary alcohols in ionic liquid
Kim et al. has demonstrated that the racemization of secondary alcohols took place more efficiently in an ionic liquid than an organic solvent, leading to their successful DKRs by enzyme–metal combo catalysis at room temperature. The (R)-selective DKRs were accomplished by the combination of lipase and a cymene–ruthenium complex (Scheme 17) and the complementary (S)-selective DKRs by replacing lipase with subtilisin (Scheme 18). In most cases, the yields were good and the optical purities were good to excellent. In general lipase-catalyzed DKRs gave higher optical purities than subtilisin-catalyzed DKRs. The efficiency of the DKRs in ionic liquid was comparable to that in organic solvents. The DKRs in both the solvent systems provided similar yields and optical purities [32].
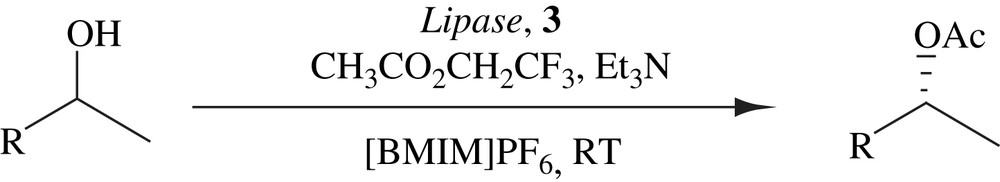
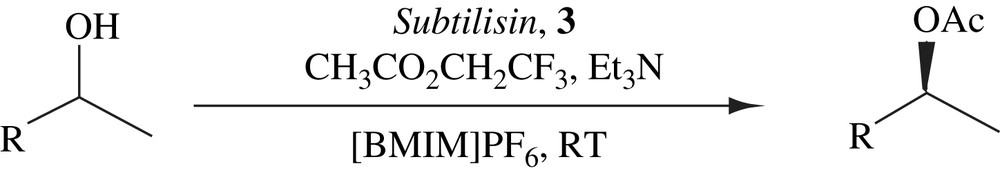
5 Aminocyclopentadienyl ruthenium chloride (4)
5.1 DKR of alcohols at ambient temperature
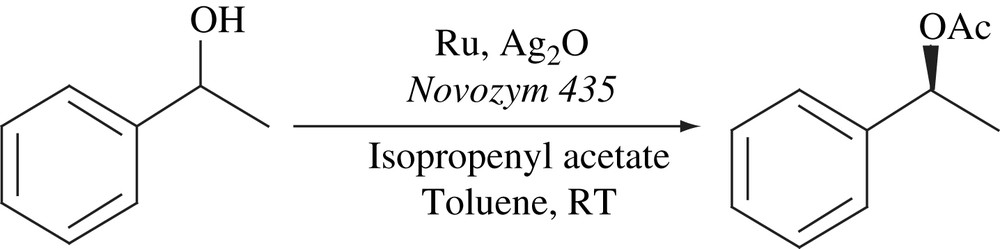
Park and co-workers have reported a novel ruthenium catalyst (4) that could racemize secondary alcohols efficiently at room temperature. Furthermore, the catalytic racemization was compatible with the use of isopropenyl acetate for the DKR of secondary alcohols at room temperature (Scheme 19). This acyl donor is commercially available.
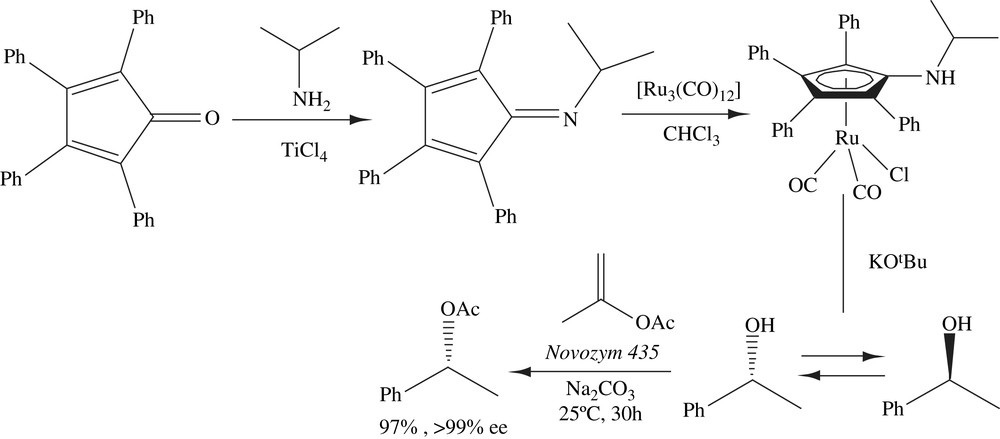
(S)-1-Phenylethanol (>99% ee) was racemized completely within 30 min at 25 °C by the treatment of 4 with potassium tert-butoxide. The DKR was successful only in the presence of sodium carbonate or 4 Å molecular sieve as an additive. Isopropenyl acetate was found to be a better acyl donor. Complex 4 was used in the DKR of various alcohols at room temperature not only simple alcohols but also functionalized ones such as allylic alcohols, alkynyl alcohols, diols, hydroxyl esters and chlorohydrins. All these racemic alcohols were successfully transformed to chiral acetates [33,34].
5.2 (S)-Selective DKR of secondary alcohols
The (S)-selective DKR of alcohols has been successfully achieved by the combination of subtilisin and an aminocyclopentadienyl ruthenium complex. The success of this DKR was based on three important factors: the enhanced activity and stability of surfactant-treated subtilisin, the high activity of the ruthenium complex at room temperature and the good compatibility between those two catalysts.
This was the first report in which (S)-enantiomer was obtained as DKR product using subtilisin as the resolving catalyst and an aminocyclopentadienyl ruthenium complex as the racemizing catalyst (Scheme 20) [35]. Synthesis of similar tetrabutylcyclopentadienyl ruthenium complex has been reported, but its racemization activity has not yet been investigated [36].

6 Pentaphenyl cyclopentadienyl ruthenium complex (5)
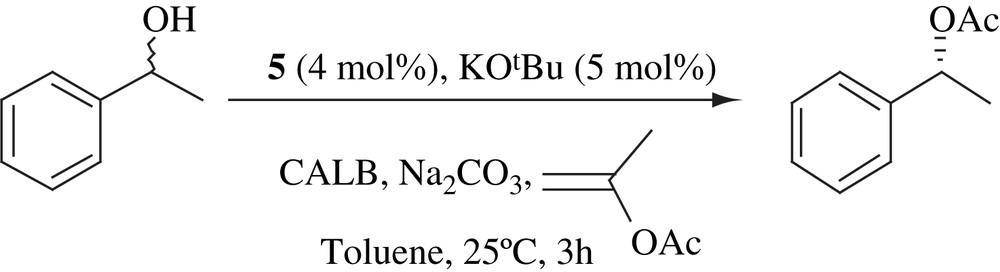
6.1 DKR of alcohols at ambient temperature
Solution of 1-phenylethanol in toluene was added to a mixture of KOtBu and 5 under an argon atmosphere. After stirring for 5 min, Na2CO3, CALB and isopropenyl acetate were added. After 4 h, only 55% yield of enantiopure (R)-1-phenylethanol acetate was obtained. When the mixture of KOtBu and complex 5 was stirred for 6 min in toluene before adding the alcohol, (R)-1-phenylethanol acetate was obtained in 99% yield with >99% ee in 4 h (Scheme 21). It has also been observed that the system was very sensitive to molecular oxygen. The scope of the system was extended to various substrates [37]. The mechanism of this process has also been studied in detail [38,39].
6.2 DYKAT of 1,2-diols
Regio and stereoselective lipase-catalyzed kinetic resolutions were investigated for some unsymmetrical, secondary/secondary syn-diols. CALB-catalyzed transesterifications of a few aryl/alkyl and alkyl/alkyl 1,2-diols were coupled in one-pot for efficient ruthenium catalyzed epimerization and intramolecular acyl migration to give a dynamic kinetic asymmetric transformation (DYKAT) affording enantio-enriched (ee upto >99%) syn-diacetates as the main diastereomers (Scheme 22) [40].
6.3 Conversion of allylic alcohols to α-methyl carboxylic acids
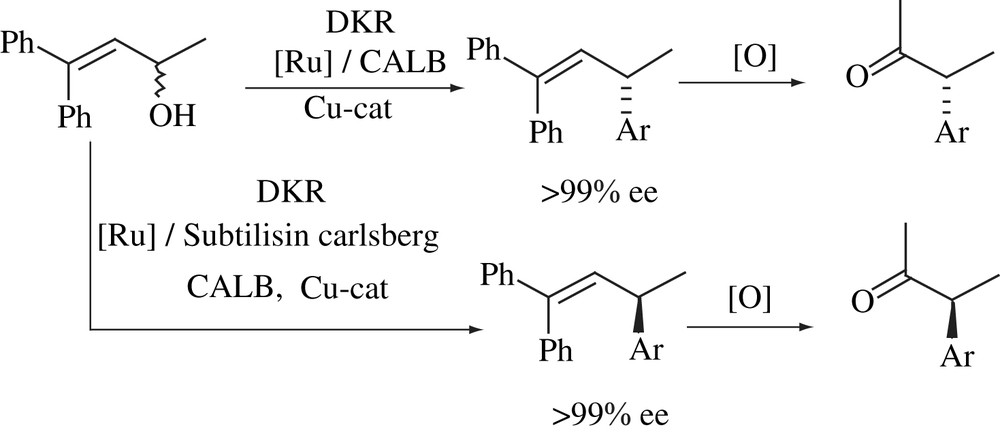
A straightforward route to α-methyl carboxylic acids via metal and enzyme catalysis has been developed (Scheme 23) [41]. DKR of allylic alcohol proceeded with an excellent yield and optical purity to produce (R)-allylic ester, and no isomerization to the corresponding ketone was observed. Likewise, DKR of allylic alcohol followed by enzymatic hydrolysis afforded (S)-allylic ester in high yield and optical purity. Copper-catalyzed SN2 substitution of the allylic esters was accomplished in high yield and high enantioselectivity, and subsequent ruthenium-catalyzed oxidation proceeded with conservation of chiral information to give pharmaceutically important α-methyl aryl acetic acids.
7 [TosN(CH2)2NH2]RuCl(p-cymene) (6)
Dijksman et al. reported that 6 in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) was able to catalyze the in situ racemization of chiral secondary alcohols during enzymatic acylation. Using this system, enantiomerically pure (>99%) 1-phenylethylacetate was obtained in 83% selectivity at 91% conversion from 1-phenylethanol in the presence of p-chlorophenyl acetate, Novozym 435 and KOH [42]. Acetophenone was formed as a side product due to the oxidation of the substrate by TEMPO.
8 Tetrafluorosuccinato ruthenium complex (7)
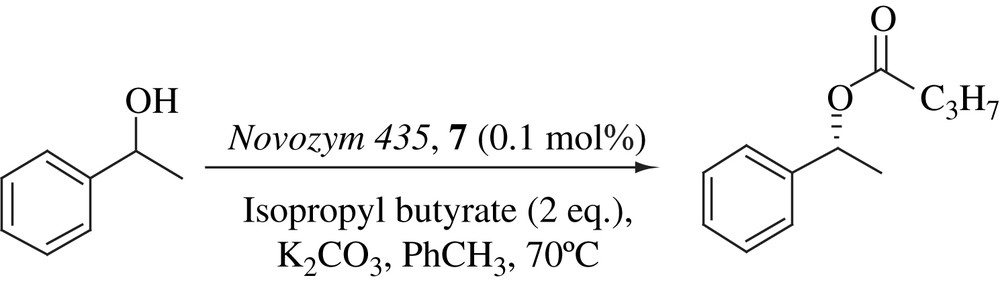
DKR of a series of secondary alcohols has been conducted with a novel dinuclear ruthenium complex, bearing tetrafluorosuccinate and (rac)-BINAP ligands as the racemization catalyst. Novozym 435 has been used as the enzyme and isopropyl butyrate as the acyl donor (Scheme 24). Five substrates underwent DKR successfully: an aliphatic and an aromatic secondary alcohol, an aromatic alcohol with an electron withdrawing substituent on the phenyl ring, an aromatic alcohol bearing an electron donating substituent on the ring and a heteroaromatic secondary alcohol. The catalyst performed optimally at 70 °C. The reaction was completed within 1 day with 0.1 mol% of racemization catalyst. The addition of the ketone corresponding to the substrate stabilized the active Ru complex and therefore increases the rate of the reaction [43].
9 [(η5-Ph4C5OCOPh)RuCl(CO)2] (8)
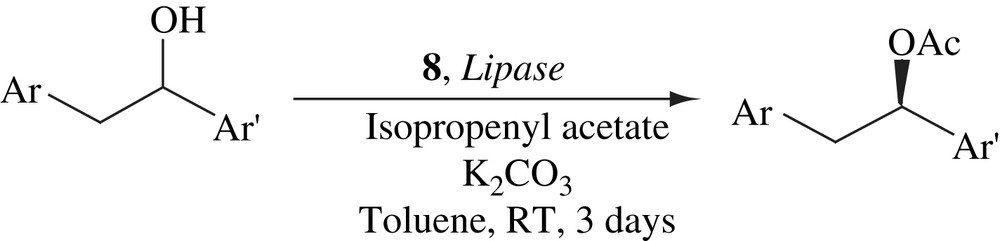
A practical procedure has been developed for the DKR of 1,2-diarylethanols (Scheme 25) [44]. This procedure employed a highly enantioselective lipase from Pseudomonas stutzeri as the resolution catalyst and a ruthenium complex as the racemization catalyst. Sixteen 1,2-diarylethanols have been efficiently resolved to provide their acetyl derivatives. In all cases, almost quantitative yields (95–99%) were obtained with excellent enantiomeric excesses (96–99%).
10 [(η5-Ph5C5)Ru(CO)(PPh3)(Br)] (9)
A new class of air stable and recyclable alcohol racemization catalysts has been synthesized and it has been demonstrated that they were applicable to the DKR of aliphatic alcohols as well as benzylic ones at room temperature (Scheme 26) [45]. The catalyst was readily synthesized from the corresponding ruthenium dicarbonyl complex through simple ligand substitution reaction. The efficiency of the catalyst in DKR was comparable to that of the parent ruthenium dicarbonyl complexes. The advantage of this catalytic system is that they do not require the strong base such as potassium tert-butoxide and catalyst is stable during the DKR under aerobic conditions.
11 Cycloruthenated amine (10)
Cycloruthenated amine was subjected to DKR in combination with CALB enzyme and potassium tert-butoxide in toluene at 70 °C [46]. For example, 1-phenylethanol was converted into 86% of enantiopure (R)-1-phenylethyl acetate (>99%) (Scheme 27).

12 Racemization of chiral secondary alcohols catalyzed by [Cp∗RuCl(COD)]
Ikariya and his group members found that the [Cp∗RuCl(COD)]/2-diphenylphosphinoethylamine/KOtBu (Cp∗ = η5-C5(CH3)5, COD = 1,5-cyclo octadiene) catalyst system carried out extremely rapid racemization of chiral non-racemic secondary alcohols, which resulted from the reversible hydrogen transfer between secondary alcohols and ketones (Scheme 28). Both tertiary phosphine and primary amine functionalities in the ligand were responsible for the high rate. The results of DKR using the same catalytic system have not yet been published [47].
13 Heterogeneous catalysts for racemization reaction
Although the DKR using homogeneous racemization catalyst, in which in situ racemization proceeds during the resolution process, has been developed for chiral alcohols and amines, its application to a large-scale production is hindered by the difficulties with the separation of the products from the reaction mixture and the catalyst recycling [48]. Thus, the development of recyclable catalysts for the racemization of secondary alcohols is an attractive research subject.
13.1 Ru–HAP catalyst for racemization of alcohols
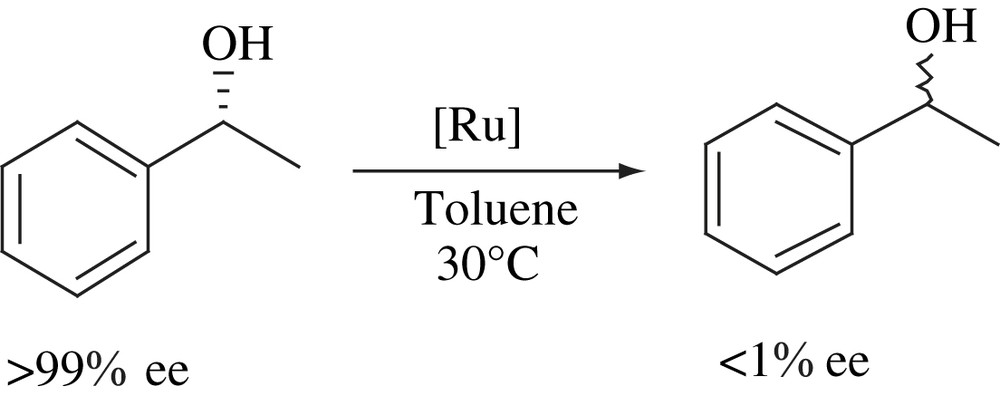
Racemization of chirally stable secondary alcohols was performed using a Ru3+ immobilized on calcium hydroxyapatite catalyst under mild reaction conditions in the absence of any additive or cocatalyst (Scheme 29). The Ru–HAP catalyst was able to racemize benzylic as well as aliphatic secondary alcohols. The catalyst scope was also investigated for racemization of several mono- and polyfunctional secondary alcohols. The heterogeneous nature of the catalyst was verified by filtrate tests [49].

13.2 Alumina-supported ruthenium catalysts for the racemization of secondary alcohols
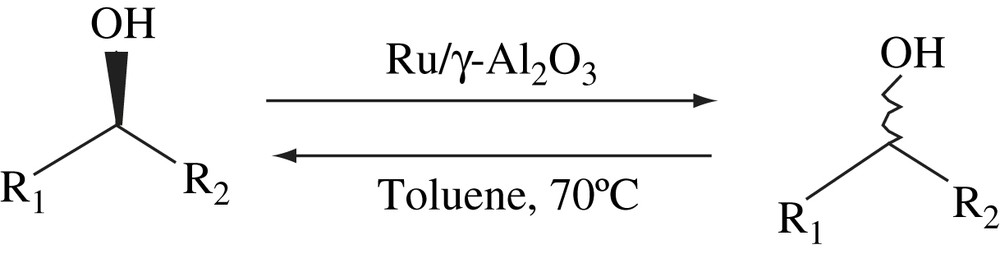
Ruthenium supported on γ-Al2O3 acted as an efficient catalyst for the racemization of secondary alcohols in toluene (Scheme 30). This catalytic system did not require any additives. Catalysts with different ruthenium content were prepared and the one with 1.25% of ruthenium exhibited high efficiency compared to other catalysts. γ-Al2O3 itself was inactive toward racemization. The racemization of (S)-1-phenylethanol was completed in 5 h at 70 °C in toluene. The racemization was faster in non-polar solvents than in polar ones. Ru/γ-Al2O3 retained more than 50% of its original activity even in the fourth use. Furthermore, washing the recovered catalyst with the 1 M aqueous NaOH solution revived the original activity almost completely. The heterogeneous nature of the catalyst was tested by a simple filtration test. The scope of the catalytic system was extended to a series of benzylic and aliphatic alcohols. A pathway for the racemization of the secondary alcohols by Ru/γ-Al2O3 was proposed [50].
The DKR results of both the heterogeneous catalysts (Ru–HAP and Ru/γ-Al2O3) have not yet been reported.
13.3 Heterogeneous racemization catalyst for DKR of secondary alcohols
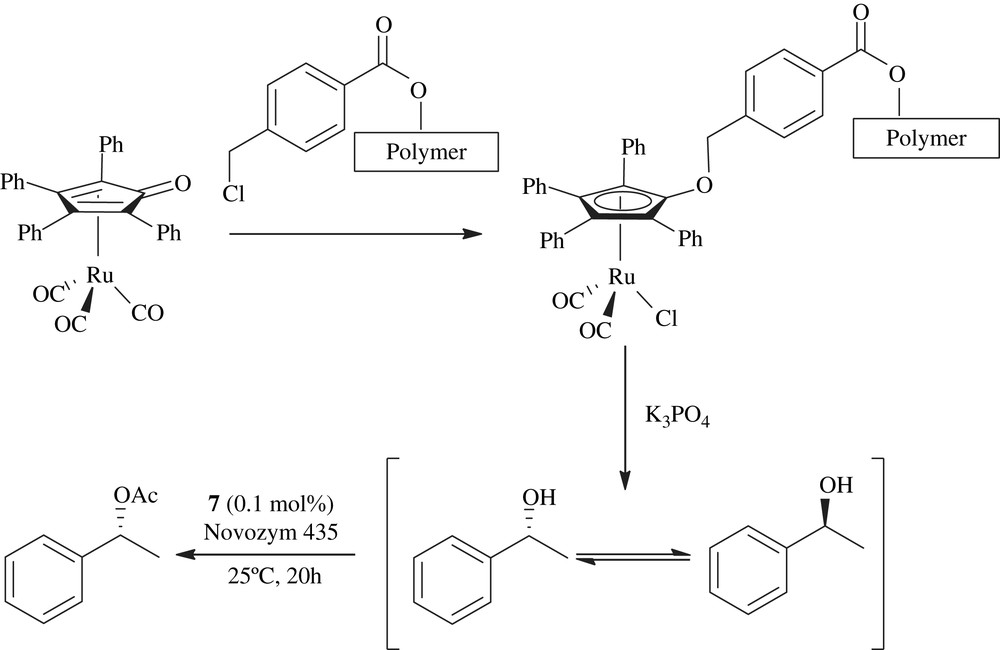
A polymer supported racemization catalyst was synthesized and tested as a recyclable catalyst for the aerobic DKR of alcohols. Novozym 435 and isopropenyl acetate were used as an enzyme and acyl donor, respectively. The scope of the catalyst was investigated in the DKR of various secondary alcohols. The catalyst displayed high efficiency toward benzylic alcohols as well as aliphatic ones (Scheme 31) [51].
14 Conclusions
The discovery of a new chemical reaction often leads to new chemical principles and new applications. DKR is one of such reaction and is used for the preparation of chiral acetates with good yields and ee. Various secondary alcohols and amines are converted into chiral acetylated compounds by using ruthenium–enzyme combo catalysis. Presently, heterogeneous catalysts have also gained importance for this purpose.
Acknowledgement
R.K. is grateful to Department of Science & Technology (DST) for financial assistance under SERC Fast Track Scheme (No. SR/FTP/CS-80/2006).