

1 Introduction
La synthèse des oxiranes constitue un thème de recherche important en synthèse organique [1]. Ils sont utilisés comme intermédiaires de synthèse. Ceci est lié aux nombreuses voies d'accès à des hétérocycles à trois chaînons et également aux possibilités offertes par l'ouverture des oxiranes par des réactifs nucléophiles [1,2]. L'utilité de ces systèmes hétérocycliques a été mise en exergue au plan biologique et pharmacologique [3]. L'action des amines primaires sur les époxydes conduit à certains β-amino alcools dotés d'activité biologique du type bêta-bloquant adrénergique [4]. Les époxydes synthétisés dans ce travail possèdent une structure similaire à celles des précurseurs du practolol (a), de l'aténolol (b), du métoprolol (c) et de l'acébutolol (d) [5]. Ces derniers constituent une classe de β-bloquants cardiosélectifs commercialisés (Fig. 1).
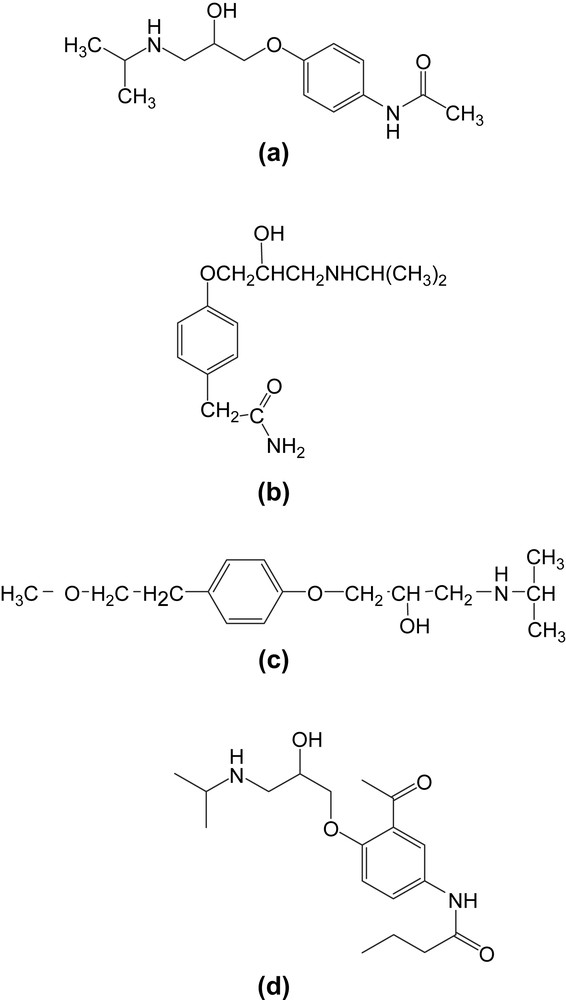
Structure des analogues d'amino-alcools, practolol (a), aténolol (b), métoprolol (c), acébutolol (d).
À cet effet, nous avons synthétisé une série de structures oxiraniques précurseurs d'arylo β-amino alcools. Les réactions sont effectuées par réaction de l'épibromhydrine avec divers composés phénoliques, 1–5 et 11–13, en milieu basique, selon des conditions de catalyse par transfert de phase (CTP) en présence d'un sel d'ammonium quaternaire, avec ou sans solvant. La base peut être NaOH en présence de bromure de tétra-n-butylammonium dans un mélange biphasé eau–benzène (méthode A). On peut également utiliser K2CO3 en présence d'une quantité catalytique d'Aliquat 336 [6], en l'absence de solvant dans un mélange hétérogène solide–liquide (méthode B). Cette dernière technique permet de réaliser simplement et efficacement des réactions difficiles en présence de solvant. Dans certains cas [7], on a couplé la méthode B avec les micro-ondes pour mettre en œuvre des réactions considérées comme virtuellement impossibles. Dans notre cas, nous avons observé un échauffement incontrôlable et très élevé sous micro-ondes, avec l'apparition des produits de dégradation non identifiés.
Dans le but de préparer de nouveaux β-amino alcools, nous avons ensuite examiné l'ouverture de ces époxydes par des amines primaires.
2 Résultats et discussion
Dans le cadre de cette communication, nous avons synthétisé divers oxiranes par o-alkylation des phénols 1–5, mettant en jeu, dans un premier temps, l'épibromhydrine comme agent alkylant, selon la méthode A [8]. Les rendements obtenus en produits 6–10 varient entre 51 et 73% après 24 h de réaction à la température ambiante, sauf pour le composé 10 (72 h). Dans un deuxième temps, nous avons amélioré la même réaction, en opérant dans un bain d'huile thermostaté à 110 °C, en l'absence de solvant [9,10], selon la méthode B. Les rendements en époxyde 6–10 varient entre 70 et 80% en un temps très court, qui n'excède pas une heure pour les phénols 1–4 ; le rendement est de 75% pour le phénol monosubstitué 5 en 6 h de temps de réaction (Fig. 2, Tableau 1).
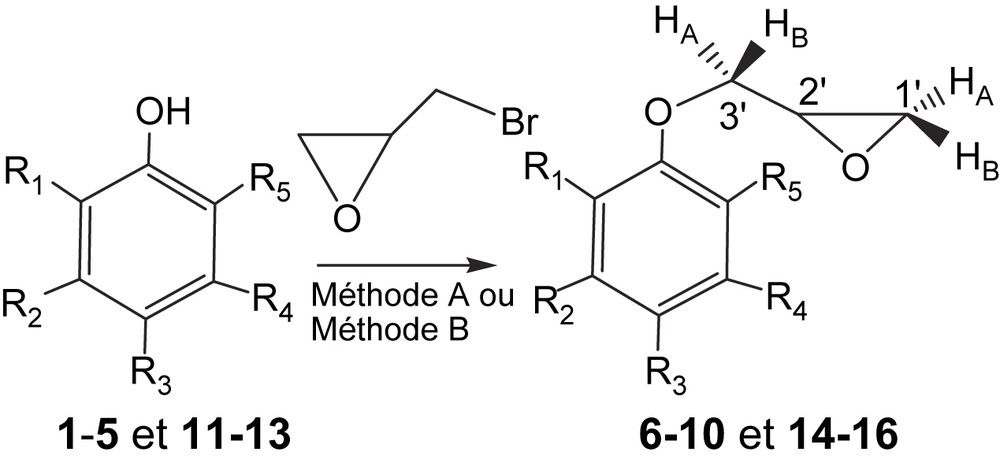
o-Épibromhydrination des phénols.
o-Épibromhydrination des phénols dans les conditions de CTP liquide–liquide et solide–liquide
| Phénols | Produits | R1 | R2 | R3 | R4 | R5 | Méthode A; T = 25 °C | Méthode B 110 °C | ||
| t (h) | Rdt (%) | t (h) | Rdt (%) | |||||||
| 1 | 6 | CO2Et | C6H5 | H | C6H5 | Br | 24 | 73 | 1 | 76 |
| 2 | 7 | CO2Et | C6H5 | H | C6H5 | H | 24 | 70 | 1 | 80 |
| 3 | 8 | CO2Et | o-ClC6H4 | H | C6H5 | Br | 24 | 71 | 1 | 78 |
| 4 | 9 | CO2Et | C6H5 | H | p-CH3OC6H4 | Br | 24 | 51 | 1 | 70 |
| 5 | 10 [11] | H | H | CO2Me | H | H | 72 | 71 | 6 | 75 |
Ce travail a été étendu à d'autres phénols tétrasubstitués (11–13) pour la réaction d'o-épibromhydrination, en choisissant les conditions de la méthode B. Les rendements obtenus en produits 14–16 varient de 72 à 76% pendant 3 à 4 h de chauffage à 80 °C. Ces rendements sont améliorés à 110 °C, pour atteindre 75 à 80% en 1 h seulement (Fig. 2, Tableau 2).
Époxydation dans les conditions de CTP solide–liquide sans solvant (K2CO3 + Aliquat, méthode B)
| Phénols | Produits | R1 | R2 | R3 | R4 | R5 | 80 °C | 110 °C | ||
| t (h) | Rdt (%) | t (h) | Rdt (%) | |||||||
| 11 | 14 | CO2Et | C6H5 | Br | C6H5 | Br | 3 | 74 | 1 | 76 |
| 12 | 15 | CO2Et | o-ClC6H4 | Br | C6H5 | Br | 4 | 76 | 1 | 80 |
| 13 | 16 | CO2Et | C6H5 | Br | (m-Br)(p-CH3O)C6H3 | Br | 4 | 72 | 1 | 75 |
Compte tenu des résultats obtenus, la technique de la catalyse par transfert de phase solide–liquide sans solvant peut jouer un rôle important dans l'o-épibromhydrination des phénols substitués. Les produits sont obtenus avec de bons rendements, dans des conditions propres sans solvant, économiques et très simples à mettre en œuvre, contrairement aux méthodes classiques.
Ces conditions sont favorisées par l'utilisation de l'Aliquat 336. Nous avons remarqué que l'o-alkylation n'est pas favorisée à une température supérieure à 120 °C, du fait d'une dégradation de l'Aliquat 336. Nous avons effectué, par la suite, l'ouverture de ces systèmes oxiraniques par l'action de quelques amines primaires, telles que la benzylamine et l'isopropylamine, en présence de LiClO4 [12], pour conduire aux β-amino alcools correspondants 17–20, avec des rendements qui varient entre 50 et 65% (Fig. 3, Tableau 3).

Préparation des β-amino alcools.
Ouverture des oxiranes 9–10 et 14–15 par des amines primaires (LiClO4/CH3CN, 24 h, TA)
| Époxydes | Produits | R1 | R2 | R3 | R4 | R5 | R | Rdt% |
| 9 | 17 | CO2Et | C6H5 | H | p-CH3OC6H4 | Br | CH2C6H5 | 50 |
| 10 | 18 | H | H | CO2Me | H | H | CH2C6H5 | 65 |
| 14 | 19 | CO2Et | C6H5 | Br | C6H5 | Br | CH(CH3)2 | 60 |
| 15 | 20 | CO2Et | o-ClC6H4 | Br | C6H5 | Br | CH(CH3)2 | 62 |
3 Conclusion
La méthode de CTP solide–liquide sans solvant décrite dans ce travail ouvre des perspectives intéressantes dans la préparation de nouveaux composés oxiraniques. Elle présente une certaine simplicité au niveau du protocole opératoire et n'est pas néfaste pour l'environnement, dans la mesure où elle fait intervenir des milieux propres sans solvant. Plus intéressant encore, les rendements en produits finaux et la durée des réactions sont tous remarquablement améliorés, grâce au couplage de la CTP et des techniques de réaction en milieu sec. Les époxydes synthétisés constituent de bons précurseurs de nouveaux β-amino alcools, qui peuvent présenter des activités biologiques et pharmacologiques.
4 Partie expérimentale
4.1 Méthodes générales
Les points de fusion ont été mesurés au moyen d'un appareil Buchi 510. Les spectres infrarouge ont été enregistrés à l'aide d'un appareil PerkinElmer 577, les produits étant dispersés en phase solide dans KBr à 5%. Les spectres RMN 1H et 13C ont été enregistrés avec des appareils Brucker, avec des solutions dans CDCl3, avec le TMS comme référence interne, respectivement à 200, 250, 300 MHz, d'une part, et 50,3, 62,9, 75,5 MHz, d'autre part. Les déplacements chimiques δ sont donnés en ppm et les constantes de couplage J en Hz. Les abréviations utilisées sont les suivantes : s, singulet ; d, doublet ; dd, doublet dédoublé ; t, triplet ; td, triplet dédoublé ; q, quadruplet ; m, multiplet et sl pour les signaux élargis. Les chromatographies sur couche mince ont été réalisées avec des plaques d'oxyde d'alumine (réf. : M. 5554), prêtes à l'emploi, avec indicateur de fluorescence, l'éluant utilisé étant un mélange de cyclohexane et d'acétate d'éthyle 8:2.
4.2 Mode opératoire général (méthode A)
À une solution de 100 ml de benzène contenant 39 mmol de phénols 1–5, on ajoute une solution aqueuse de 100 ml de soude à 50% et 10 mmol de bromure de tétra-n-butylammonium. Le mélange est agité pendant 5 min, puis une solution de d'épibromhydrine (39 mmol) dans le benzène (10 ml) est ajoutée. Le milieu réactionnel est agité à température ambiante (25 °C). L'évolution de la réaction est suivie par chromatographie sur couche mince : (support : alumine/0,2 mm; éluant : cyclohexane/acétate d'éthyle : 8:2) jusqu'à épuisement des phénols. La phase organique est lavée à l'eau (3 × 50 ml), puis séchée sur MgSO4. Après filtration sur papier, le solvant du filtrat est éliminé sous pression réduite. Les époxydes sont purifiés par recristallisation.
4.2.1 3-Bromo-2-(2-oxiranylméthoxy)-4,6-diphényl benzoate d'éthyle (6)
PF (°C) = 141 (EtOH) ; IR (KBr) 1725, 1263, 850, 762 cm−1 ; RMN 1H (300 MHz, CDCl3) δ 2,86 (dd, J = 4,2 Hz, 1H, H1′A), 2,70 (dd, J = 2,4, 5,0 Hz, 1H, H1′B), 3,38 (m, 1H, H2′), 4,15 (dd, J = 3,6 Hz, 1H, H3′A), 4,31 (dd, J = 5,9, 10,4 Hz, 1H, H3′B), 0,95 (t, 3H, Me), 4,0 (q, J = 7,1 Hz, 2H, CH2), 7,2–7,6 (m, 11HAr) ; RMN 13C (75,5 MHz, CDCl3) δ 13,6 (Me), 44,6 (C1′), 49,9 (C2′), 75,6 (C3′), 61,7 (CH2), 165,0 (), 119,3–152,0 (Car).
4.2.2 6-(2-Oxiranylméthoxy)-2,4-diphényl benzoate d'éthyle (7)
PF (°C) = 114 (EtOH) ; IR (KBr) 1730, 1268, 855, 768 cm−1 ; RMN 1H (250 MHz, CDCl3) δ 2,87 (dd, J = 4,1 Hz, 1H, H1′A), 2,60 (dd, J = 2,5, 4,9 Hz, 1H, H1′B), 3,37 (m, 1H, H2′), 4,14 (dd, J = 1,9 Hz, 1H, H3′A), 4,40 (dd, J = 5,4, 11,2 Hz, 1H, H3′B), 0,99 (t, 3H, Me), 4,09 (q, J = 6,9 Hz, 2H, CH2), 7,19–7,63 (m, 12HAr) ; RMN 13C (62,9 MHz, CDCl3) δ 13,7 (Me), 44,5 (C1′), 50,2 (C2′), 69,7 (C3′), 60,9 (CH2), 166,5 (CO), 111,3–156,1 (Car).
4.2.3 3-Bromo-6-(2-cholrophényl-2-(2-oxiranylméthoxy)-4-phényl) benzoate d'éthyle (8)
PF (°C) = 115 (EtOH) ; IR (KBr) 1725, 1262, 850, 750 cm−1 ; RMN 1H (300 MHz, CDCl3) δ 2,82 (dd, J = 4,3 Hz, 1H, H1′A), 2,67 (dd, J = 2,5, 4,9 Hz, 1H, H1′B), 3,67 (m, 1H, H2′), 4,15 (td, J = 3,7 Hz, 1H, H3′A), 4,33 (td, J = 6,3, 10,5 Hz, 1H, H3′B), 0,95 (t, 3H, Me), 4,02 (q, J = 7,1 Hz, 2H, CH2), 7,18–7,50 (m, 10HAr) ; RMN 13C (75,5 MHz, CDCl3) δ 13,5 (Me), 44,4 (C1′), 49,9 (C2′), 75,5 (C3′), 61,6 (CH2), 164,7 (CO), 119,9–151,8 (Car).
4.2.4 3-Bromo-4-(4-méthoxyphényl)-2-(2-oxiranylméthoxy)-6-phényl benzoate d'éthyle (9)
PF (°C) = 140 (EtOH) ; IR (KBr) 1730 cm−1 ; RMN 1H (300 MHz, CDCl3) δ 1,03 (t, 3H, Me), 2,88 (dd, J = 4,2 Hz, 1H, H1′A), 2,74 (dd, J = 2,6, 4,9 Hz, 1H, H1′B), 3,43 (m, 1H, H2′), 3,85 (s, 3H, OMe), 4,01 (q, J = 7,1 Hz, 2H, CH2), 4,21 (m, 1H, H3′A), 4,34 (m, J = 11,3 Hz, 1H, H3′B), 7,0–8,0 (m, 10HAr) ; RMN 13C (75,5 MHz, CDCl3) δ 13,8 (Me), 44,7 (C1′), 50,1 (C2′), 75,6 (C3′), 61,6 (CH2), 55,3 (OMe) 166,7 (CO), 110,2–159,7 (Car).
4.2.5 4-(2-Oxiranylméthoxy)benzoate de méthyle (10)
PF (°C) = 60 (sans solvant) ; IR (KBr) 1710, 1255, 840, 750 cm−1 ; RMN 1H (300 MHz, CDCl3) δ 2,88 (dd, J = 4,2 Hz, 1H, H1′A), 2,74 (dd, J = 2,6, 5,0 Hz, 1H, H1′B), 3,34 (m, 1H, H2′), 3,92 (dd, J = 2,8 Hz, 1H, H3′A), 4,30 (dd, J = 6,0, 11,3 Hz, 1H, H3′B), 3,87 (s, 3H, OMe), 6,9–8,0 (m, 4HAr) ; RMN 13C (75,5 MHz, CDCl3) δ 43,5 (C1′), 49,1 (C2′), 69,3 (C3′), 51,0 (OMe), 165,8 (CO), 113,4–161,5 (Car).
4.3 Mode opératoire générale (méthode B)
À un mélange de 10 mmol de phénols 1–5, de 12,5 mmol de K2CO3 et 2% (0,25 mmol ; 0,1 g) de l'Aliquat 336 (chlorure de méthyl trioctylammonium), on ajoute 10 mmol d'épibromhydrine sous agitation à une température de 110 (ou de 80 °C pour les phénols 11–13). L'évolution de la réaction est suivie par chromatographie sur couche mince (support : alumine/0,2 mm, éluant : cyclohexane/acétate d'éthyle : 8:2) jusqu'à épuisement des phénols. On ajoute 20 ml d'éther, puis on filtre sur Florisil (environ 2 g) sur un verre fritté selon un montage de type trompe à eau. Le solvant est éliminé sous pression réduite. Dans tous les cas, les époxydes sont obtenus sous forme de cristaux, après recristallisation dans l'éthanol.
4.3.1 3,5-Dibromo-2-(2-oxiranylméthoxy)-4,6-diphényl benzoate d'éthyle (14)
PF (°C) = 140 (EtOH) ; IR (KBr) 1725, 1250, 875, 750 cm−1 ; RMN 1H (200 MHz, CDCl3) δ 2,89 (dd, J = 4,6 Hz, 1H, H1′A), 2,75 (dd, J = 2,4, 4,7 Hz, 1H, H1′B), 3,42 (m, 1H, H2′), 4,20 (dd, J = 3,4 Hz, 1H, H3′A), 4,37 (dd, J = 5,9, 10,3 Hz, 1H, H3′B), 0,98 (t, 3H, Me), 4,04 (q, J = 7,1 Hz, 2H, CH2), 7,2–7,6 (m, 10HAr) ; RMN 13C (50,3 MHz, CDCl3) δ 13,6 (Me), 44,6 (C1′), 49,9 (C2′), 75,6 (C3′), 61,7 (CH2), 165,4 (CO), 119,3–151,7 (Car).
4.3.2 3,5-Dibromo-2-(2-oxiranylméthoxy)-4-phényl-6-(2-cholorophényl) benzoate d'éthyle (15)
PF (°C) = 130 (EtOH) ; IR (KBr) 1737, 1250, 850, 762 cm−1 ; RMN 1H (200 MHz, CDCl3) δ 2,86 (dd, J = 4,4 Hz, 1H, H1′A), 2,69 (dd, J = 2,6, 4,9 Hz, 1H, H1′B), 3,38 (m, 1H, H2′), 4,17 (td, J = 3,5 Hz, 1H, H3′A), 4,35 (td, J = 5,7, 10,3 Hz, 1H, H3′B), 0,95 (t, 3H, Me), 4,02 (m, J = 7,1 Hz, 2H, CH2), 6,9–7,6 (m, 9HAr) ; RMN 13C (50,3 MHz, CDCl3) δ 13,5 (Me), 44,4 (C1′), 49,9 (C2′), 75,5 (C3′), 61,6 (CH2), 164,7 (CO), 119,9–151,8 (Car).
4.3.3 3,5-Dibromo-4-(3-bromo-4-méthoxyphényl)-2-(2-oxiranylméthoxy)-6-phényl benzoate d'éthyle (16)
PF (°C) = 141 (EtOH) ; IR (KBr) 1737, 1250, 925, 762 cm−1 ; RMN 1H (200 MHz, CDCl3) δ 2,87 (dd, J = 4,4 Hz 1H, H1′A), 2,70 (dd, J = 2,6, 4,9 Hz, 1H, H1′B), 3,39 (m, 1H, H2′), 4,14 (dd, J = 3,2 Hz, 1H, H3′A), 4,31 (td, J = 4,5, 10,4 Hz, 1H, H3′B), 0,95 (t, 3H, Me), 4,02 (q, J = 7,1 Hz, 2H, CH2), 3,93 (s, 3H, OMe) 6,9–7,6 (m, 8HAr) ; RMN 13C (50,3 MHz, CDCl3) δ 13,5 (Me), 44,5 (C1′), 49,9 (C2′), 75,6 (C3′), 61,7 (CH2), 56,1 (OMe) 165,2 (CO), 111,4–155,6 (Car).
4.4 Synthèse des β-amino alcools
À une solution d'époxyde (1 mmol) 9, 10, 14 ou 15 dans 5 ml d'acétonitrile anhydre sont ajoutés l'amine (10 mmol) et LiClO4 (10 mmol ; 1,06 g). Le mélange réactionnel est maintenu sous agitation pendant 24 h. L'évolution de la réaction est suivie par chromatographie sur couche mince (support: alumine/0,2 mm, éluant cyclohexane/acétate d'éthyle: 8:2) jusqu'à épuisement de l'époxyde. L'éther est ajouté (20 ml) et la phase organique est lavée deux fois à l'eau (20 ml). Après évaporation sous pression réduite, les produits sont obtenus sous forme de cristaux, après recristallisation dans l'éthanol ou dans un mélange d'éther diéthylique/éther de pétrole (50:50).
4.4.1 2-[3-(Benzylamino)-2-hydroxypropoxy]-3-bromo-4-(4-méthoxyphényl)-6-phényl benzoate d'éthyle (17)
PF (°C) = 116 (éther/éther de pétrole) ; IR (KBr) 3450, 3290, 1735 cm−1 ; RMN 1H (300 MHz, CDCl3) δ 0,95 (t, 3H, Me), 2,61 (sl, 2H, NH et OH), 2,85 (m, J = 12,4 Hz, 2H, H2′), 3,85 (d, 2H, CH2–N), 3,94 (s, 3H, OMe), 4,07 (q, J = 7,1 Hz, 2H, CH2), 4,14 (m, 1H, H3′), 4,21 (dd, J = 5,9, 9,1 Hz, 1H, H4′B), 4,28 (dd, J = 3,8 Hz, 1H, H4′A), 6,9–7,6 (m, 15HAr) ; RMN 13C (75,5 MHz, CDCl3) δ 13,6 (Me), 51,0 (CH2–N), 53,9 (C2′), 56,3 (OMe), 61,68 (CH2), 69,11 (C3′), 77,5 (C4′), 167,0 (CO), 111,2–155,6 (Car).
4.4.2 4-[3-(Benzylamino)-2-hydroxypropoxy] benzoate de méthyle (18)
PF (°C) = 120 (EtOH) ; IR (KBr) 3450, 3300, 1725 cm−1 ; RMN 1H (300 MHz, CDCl3) δ 2,31 (sl, 2H, NH et OH), 2,91 (dd, J = 7,4 Hz, 1H, H2′A), 2,78 (dd, J = 3,8, 12,3 Hz, 1H, H2′B), 3,85 (d, 2H, CH2–N), 3,88 (s, 3H, OMe), 4,01–4,12 (m, 3H, H3′, H4′), 6,91–8,0 (m, 9HAr) ; RMN 13C (75,5 MHz, CDCl3) δ 51,9 (OMe), 51,1(CH2–N), 53,8(C2′), 68,3 (C3′), 70,6 (C4′), 166,8 (CO), 114,3–162,5 (Car).
4.4.3 3,5-Dibromo-6-[2-hydroxy-3-(isopropylamino)propoxy]-2,4-diphényl benzoate d'éthyle (19)
PF (°C) = 104 (éther/éther de pétrole) ; IR (KBr) 3400, 3238, 1713 cm−1 ; RMN 1H (200 MHz, CDCl3) δ 0,92 (t, 3H, Me), 1,08 (d, 6H, (Me)2), 2,31 (sl, 2H, NH et OH), 2,82 (m, 1H, CH(Me)2), 2,76 (dd, 1H, J = 6,8 Hz, H2′A), 2,86 (dd, 1H, J = 5,8, 12,4 Hz, H2′B), 3,90–4,12 (m, 3H, (CH2(ester), H3′)), 4,21 (dd, 1H, J = 4,8, 9,4 Hz, H4′B), 4,25 (dd, J = 3,4 Hz, 1H, H4′A), 7,19–7,56 (m, 10HAr) ; RMN 13C (50,3 MHz, CDCl3) δ 14,9 (Me), 24,5 (Me)2, 50,3 (C2′), 50,4 (CH(Me)2), 63,3 (CH2), 70,5 (C3′), 78,9 (C4′), 167,4 (CO), 120,5–153,3 (Car).
4.4.4 3,5-Dibromo-2-(2-chlorophényl)-6-[2-hydroxy-3-(isopropylamino)propoxy]-4-phényl benzoate d'éthyle (20)
PF (°C) = 86 (éther/éther de pétrole) ; IR (KBr) 3425, 3300, 1725 cm−1 ; RMN 1H (200 MHz, CDCl3) δ 0,97 (t, 3H, Me), 1,11 (d, 6H, Me2CH), 2,47 (sl, 2H, NH et OH), 2,72–2,95 (m, 3H, (H2′, Me2CH), 3,97–4,14 (m, 3H, (CH2, H3′)), 4,16–4,33 (m, 2H, H4′), 7,20–7,58 (m, 9HAr) ; RMN 13C (50,3 MHz, CDCl3) δ 13,5 (Me), 23,0 (Me)2, 48,9 (C2′), 49,0 (CH(Me)2), 61,8 (CH2), 69,1 (C3′), 77,5 (C4′), 165,4 (CO), 119,7–152,2 (Car).
Remerciements
Nous tenons à remercier le Dr A. Loupy (université Paris-Sud, Orsay) et le Dr R. Benhida (université de Sophia Antipolis, Nice) pour l'aide apportée à ce travail, ainsi que le CNRST–Rabat pour son soutien financier (convention CNRST–CNRS n° Chimie 04/06).