

1 Introduction
The migration of radionuclides in the environment is an important issue in the context of radioactive waste disposal [1–4]. Interaction between actinide elements and soil minerals can stem from several physico-chemical processes: for instance, sorption/desorption, precipitation or surface chemistry such as oxido-reduction processes. Therefore, a comprehensive view of the possible interactions between minerals and radionuclide elements is of strategic importance [5–15]. The inorganic portion of soils varies a lot but often contains an important part of clay minerals. Clay particles exhibit strong surface reactivity and play a key role in the mobility or retention of radionuclide contaminants [1–6]. The surface chemistry of layered alumino-silicates in aqueous solutions is rich due to the hydroxylated interlayer surfaces. Understanding the sorption of cationic contaminants on clay minerals is essential to assess the risk of nuclear waste ground storage and requires to obtain information at a molecular scale. In the present work, dickite was chosen as a reference compound for layered alumino-silicate. Dickite is a polymorph of the kaolinite group that is found in a wide range of geological environment. Dickite has a 1:1 dioctahedral structure [16–25]. One layer is formed by a tetrahedral (T) Si2O5 silicate sheet bound to an octahedral (O) gibbsite-like Al(OH)3 sheet by apical oxygen atoms. One third of all possible octahedral central positions are vacant, which leads to octahedral cavities. Each layer is bounded to its neighbouring layers by strong hydrogen bonds between the hydroxylated gibbsite-like sheet and the silicate sheet in a TO–TO arrangement. One advantage of dickite is that no interlayer cation is required to balance electrostatic charges, preventing cation exchange [16–18]. Interlayer intercalation has been evidenced for several organic molecules such as formamide [19] or dimethylsulfoxide [20]. For trivalent cation, only one case in the literature reports on the substitution of Al3+ by Cr3+ in a natural sample of dickite (Nowa Ruda, Lower Silesia, Poland), but no intercalation [21]. Intercalation of trivalent cations seems to be very unlikely for dickite minerals and can thus be reasonably ruled out. The study is thus simplified and interactions between dickite particles and cations only involve sorption/desorption or surface precipitation processes.
The europium(III) cation was chosen as an analogue to model the behaviour of trivalent actinides [3–15]. It is easy to handle and presents the interesting property to be luminescent under proper light excitation. The luminescence spectral features are very sensitive to the local environment and symmetry surrounding the cation [26–32], making Eu3+ a good probe of the sorption sites. The aim of the present study is to supplement the information traditionally obtained by analysing the solution that contains the cation. In these latter investigations, cation concentration can be low (typically, 10−6–10−4 mol L−1) and sorption isotherms can be investigated depending on the physico-chemical parameters of the solution (pH, initial cation concentration, etc.) [1,8,10–12,15]. However, the solid phase also provides interesting information on sorption sites, surface complexes and precipitation states. Spectroscopic methods are well adapted to question the solid phase with a minimum of perturbation [4]. However, due to their low sensitivity, they often require high surface concentration of cations to be deposited on solid particles, which compels to make sorption experiments with rather high cation concentrations in solution (10−3 or 10−2 mol L−1 typically). The present work is devoted to the spectroscopic investigations of dickite particles after a contact time with a solution containing 10−2 mol L−1 of Eu3+. Interaction between a well-defined clay mineral and Eu3+ species constitutes a reference system that could be generalized to give some insight into the sorption behaviour of trivalent actinides on layered alumino-silicates. The spectroscopic methods are employed to question the system at a molecular scale. The challenge is to locate the sorption sites and identify the various surface processes that may be concerned.
2 Materials and methods
The dickite powder was given by Dr. Mohamed Zamama (“Laboratoire de spectroscopie moléculaire, faculté des sciences”, Marrakech, Morocco) and comes from Genesse Tunnel, Red Mountains, Ouray, Colorado (USA). X-ray diffraction (XRD) has already been performed on this sample and revealed a well-crystallised sample [19]. The specific surface area of the sample was determined by a dynamic BET technique using N2 adsorption at 77 K (Coulter SA3100) and was measured at 0.3 m2 g−1. Pristine particles were analysed using a Phillips XL30 scanning electron microscope (SEM). Fig. 1 shows a SEM image of the dickite sample. It is composed of almost hexagonal particles with diameters in the range of 20–50 μm. Particles are stacked in a bookshelf-like structure showing basal planes (i.e. (001) crystallographic planes) and edge planes (for instance, (010) planes). This stacking of dickite particles can be responsible for the rather low specific area measured by N2 adsorption on the pristine sample. The sample is clear white, no trace of iron contamination is visible and XPS analysis confirmed the good level of purity of the dickite particles (see below).

SEM image of the dickite hexagonal particles showing basal and edge planes.
The sorption protocol consisted in immersing 100 mg of dickite powder in 2 mL of a 10−2 mol L−1 solution of Eu3+ (made by dissolving anhydrous EuCl3 salt) at pH = 5.4 and at room temperature (22 °C in an air-conditioned room). At pH = 5.4, Eu(III) is totally under the cationic Eu3+ form [1–15]. The ionic strength was not imposed by a background salt to avoid any sorption competition with europium cations. Using the specific area of 0.3 m2 g−1 determined by N2 adsorption on pristine dickite sample and assuming a sorption site density of 5 sites/nm2 (which is very high and certainly over-estimated), our working conditions assure that site saturation by europium cations and consequently thermodynamic equilibrium with the solution are reached. The sample was gently stirred during 72 h in closed flasks to avoid carbonate contamination. In a first experiment, the dickite powder was collected by filtration, dispersed on a glass cover slide and the residual solution was let to be evaporated at room temperature. This sample will be labelled SE (Sorbed, Evaporated) in the rest of the article. A second experiment was performed by separating the dickite particles from the europium solution using centrifugation at 3000g during 10 min, rinsing with 2 mL of distilled water, then centrifuging again and collecting the solid phase in a PET tube. The residual water was let to be evaporated at room temperature during one day. This sample will be labelled SC (Sorbed, Centrifuged) in the rest of the article. The reference sample was the hydrated dickite powder deposited on a glass slide. Samples were then analysed using Raman, luminescence, and X-ray photoelectron spectroscopies.
Raman confocal spectroscopy was performed using a 514-nm argon laser excitation to avoid europium excitation and luminescence. The Olympus BX41 confocal microscope coupled with a Jobin-Yvon T64000 spectrometer was used; the detector was a nitrogen-cooled CCD camera. Particles were deposited on a glass capillary tube mounted on a goniometer to control their orientation. Polarization of the excitation and analysed beams was controlled by dichroic sheet polarizers. Confocal luminescence spectroscopy used the same apparatus, but the excitation wavelength was set to the 458-nm argon laser line to excite Eu3+ cations. It was checked that pristine dickite particles were not luminescent. The spectral resolution for wavelength measurement was better than 0.1 nm.
Epifluorescence microscopy was performed on an Olympus BX51 system using a ViewColor III colour CCD camera. The 365-nm mercury line of the excitation lamp was selected with appropriate filter, and luminescence of europium cations was recorded above 430 nm to collect the maximum of emitted light.
The XPS analyses were carried out with a Kratos Axis Ultra (Kratos Analytical, UK) spectrometer with a hemispherical energy analyser using a monochromatic Al Kα source (1486.6 eV). As the delay-line detector allows a high count rate, the power applied to the X-ray anode was reduced to 90 W so that the possible X-ray induced degradation of the sample was minimised. The instrument work function was calibrated to give a binding energy (BE) of 83.96 eV for the Au 4f7/2 line for metallic gold and the spectrometer dispersion was adjusted to give a BE of 932.62 eV for Cu 2p3/2 line for metallic copper. The samples were attached to the sample holder and then evacuated overnight prior to analyses. The pressure in the analysis chamber during XPS analysis was in the low 10−9 mbar range. All spectra were recorded at a 90° take-off angle, the analysed area being currently a spot of about 700-μm width, but when necessary the analysed area was reduced to a spot of about 27-μm width. Survey spectra were recorded with 1.0-eV step and 160-eV analyser pass energy and the high-resolution regions with 0.05-eV step and 20-eV pass energy (instrumental resolution was around 0.4 eV). In both cases the hybrid lens mode was employed. Spectra were analysed using the Vision software from Kratos (Vision 2.2.0). A Shirley baseline was used for background subtraction.
3 Results and discussion
3.1 X-ray photoelectron spectra
Fig. 2 reports the XPS survey spectra of pristine dickite and SE sample particles (the same spectral features were obtained for SC sample). Table 1 collects atomic concentrations of the detected elements. Taking into account the atomic concentration of O, Al and Si atoms, results for pristine dickite sample lead to the following relative concentrations: O (70%), Al (12%) and Si (18%). The atomic formula of dickite is Al2Si2O5(OH)4, which gives O (69.24%), Al = Si (15.38%). The slight difference between the measured values and the expected stoichiometric composition might come from either a low amount of impurities at the surface of dickite particles or a surface Si enrichment associated with SiO4 terminal sheets of the particles. Indeed, XPS experiments have detected Zn, Na, and K species in very small amounts, but no iron impurity (Table 1). When Eu3+ cation is sorbed on dickite, the XPS spectrum reveals the characteristic doublet of Eu 3d levels (Fig. 2b). The Eu 3d5/2 peak is found at 1125 eV (Table 1), in agreement with the value for Eu(III) sorbed species [4,33,34]. Furthermore, one should notice that sorption experiments make the impurities to vanish, which indicates that these impurities were certainly present at the surface of dickite particles and not incorporated in the bulk. The spectrum also indicates the presence of chloride, on SE and SC samples as well. This may come from the surface precipitation of EuCl3 or a surface complex (either inner- or outer-sphere) of Eu3+ that includes chloride anions in the ligand sphere. Table 1 shows that the rinsing with water made before the centrifugation step reduces the amount of Eu3+ and Cl−. This could indicate that centrifuging and rinsing with water can wash a surface precipitate. Note that “surface precipitate” is used in the context of this study to indicate a deposition of a solid phase due to water evaporation. However, when calculating the possible amount of (EuCl3·nH2O) precipitate (using the stoichiometry of 3 Cl atoms for 1 Eu atom and the atomic concentrations of Table 1), it appears that less than 12% of the total number of Eu3+ cations can be involved in the precipitation process for the SE sample, and less than 14% for the SC sample. Consequently, a majority of europium cations are sorbed on dickite particles with less than three chloride atoms in the ligand sphere. It is tempting to postulate that surface precipitation is almost negligible in our working conditions, but caution is required. As shown below by epifluorescence microscopy, edges of dickite particles are mainly involved in the surface processes and the spectral resolution of the XPS apparatus is limited to ∼27 μm. This resolution is not sufficient to separate the respective contributions of basal and edge planes. Consequently, surface precipitation localised on edge planes cannot be excluded by XPS experiments. The second hypothesis to explain the decrease of europium and chloride amounts on particles after rinsing with water (SC sample) invokes a washing of outer-sphere complexes that are less efficiently bounded to the surface of dickite. Other spectroscopic methods are required to try to rule out the precipitation hypothesis or the outer-sphere complex formation.

Typical XPS survey spectra of (a) pristine dickite and (b) the dickite/Eu3+ SE sample.
Atomic concentrations on the surface of dickite particles before and after contact with EuCl3 solutions (pH = 5.4, 72 h contact time, [Eu3+] = 10−2 mol L−1) determined by XPS
| Pristine dickite | SE sample (no centrifugation) | SC sample (with centrifugation) | ||||
| BE (eV) | Atomic concentration (%) | BE (eV) | Atomic concentration (%) | BE (eV) | Atomic concentration (%) | |
| Eu 3d5/2 | × | × | 1134.8 | 2 | 1135.2 | 0.8 |
| Na 1s | 1071.9 | 0.3 | × | × | × | × |
| Zn 3p3/2 | 1022.1 | 0.3 | × | × | × | × |
| O 1s | 531.7 | 55 | 531.8 | 49 | 531.8 | 46 |
| N 1s | 399.9 | 1 | 399.8 | 1 | 399.8 | 1 |
| C 1s | 284.6 | 20 | 284.6 | 26 | 284.6 | 31 |
| K 2p | 293.3 | 0.3 | × | × | × | × |
| Cl 2p | × | × | 198.2 | 0.8 | 198.2 | 0.3 |
| Si 2p | 102.7 | 14 | 102.6 | 10 | 102.6 | 11 |
| Al 2p | 74.3 | 9 | 74.2 | 11 | 74.2 | 10 |
3.2 Luminescence experiments
The luminescence of Eu3+ cation comes from f–f electronic transitions. Due to the shielding of the 4f electrons by 5s and 5p electrons, the electronic transitions are almost the same for free Eu3+ and Eu3+ incorporated into host matrices or sorbed on surfaces [26–29]. Large spin–orbit coupling significantly splits the manifold of the 4f orbitals. The electronic levels of the Eu3+ cation are generally designated using the Russell–Sanders coupling scheme notation, as usual in the literature [26–29]. The lowest electronic levels are the 7Fj (j = 0–4) levels, the degeneracy of the levels given by 2j + 1. The energies brought about by light irradiation at 365 nm (for epifluorescence microscopy) and 458 nm (for luminescence spectroscopy) excite Eu3+ to high energetic levels. Next, internal conversion occurs and the luminescence emission comes from the 5D0 level to the 7Fj (j = 0–4) levels. These transitions are parity forbidden but the ligand field can relax this constraint when Eu3+ is sorbed on a solid surface [27,28]. The 5D0 → 7F2 transition is said to be “hypersensitive” to the environment since its intensity strongly depends on the site symmetry around the Eu3+ cation [26–29].
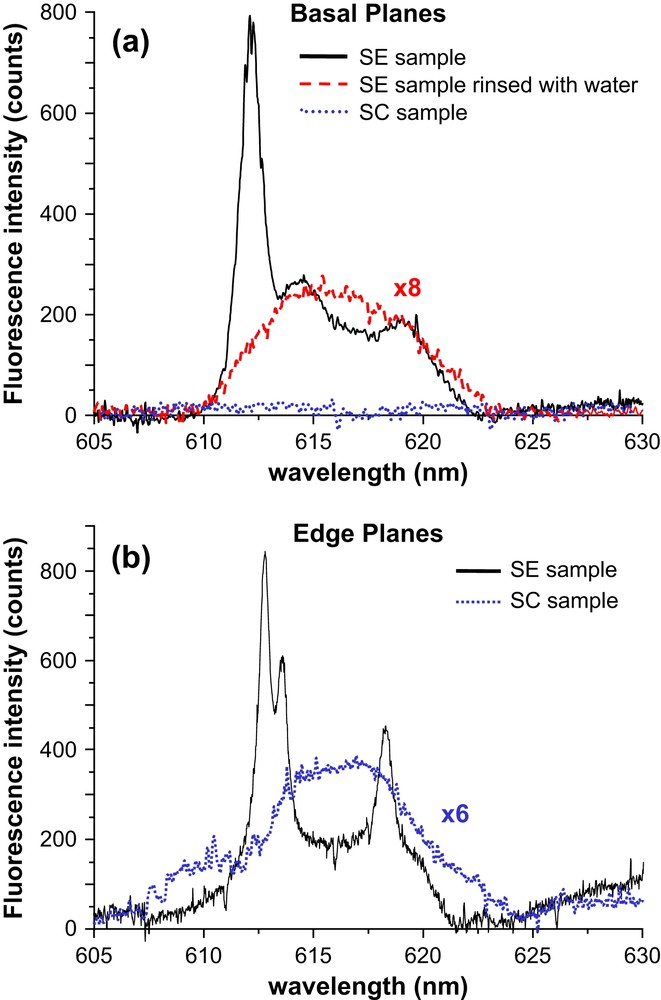
Fig. 3 shows the luminescence observed through a ×100 microscope objective after exciting dickite particles from the SE and SC samples. The excitation wavelength was the 365-nm emission line of a HBO lamp selected with an appropriate filter. The luminescence was observed above 430 nm. The image clearly shows that luminescence is mainly emitted by Eu3+ cations sorbed or precipitated on the edge planes of particles. The exposure time for the SC sample is one order of magnitude longer than for the SE sample in epifluorescence microscopy, whereas XPS spectra have indicated that Eu(III) surface concentration is only about three times lower for SC sample. This suggests a partial change of the Eu3+ environment that would decrease the luminescence quantum yield of the cations in the SC sample. For the two samples, basal planes are almost non-luminescent, contrary to edge planes. It is very unlikely that Eu3+ cations were sorbed on basal planes but gave rise to almost no luminescence. One can reasonably conclude that Eu3+ is preferentially present on edge planes of dickite particles, either in a sorption or a precipitation state. In order to try to separate these two processes, luminescence spectroscopy was performed using a confocal microscope to obtain spectral information at the adequate spatial resolution.

Epifluorescence microscopy of dickite particles after a 72-h contact time with a solution containing EuCl3 at 10−2 mol L−1, and pH = 5.4. Excitation wavelength 365 nm, fluorescence detected above 420 nm. (a) SE sample, exposure time 750 ms; (b) SC sample, exposure time 10 s.
Fig. 4 reports the 5D0 → 7F2 luminescence spectra obtained after exciting particles of the SE or SC samples with the 458-nm argon laser line. One should note that the 5D0 → 7F1 emission band was very weak for the two samples, and its structure could not be determined due to a low signal-to-noise ratio. That is why Fig. 4 only reports the 5D0 → 7F2 emissive band. For the SE sample, basal planes are featured by a 3-band emission spectrum with a maximum emission wavelength at 612.5 nm, and two broad bands around 614 and 619 nm. When this sample is rinsed with a flow of distilled water, the luminescence peak centred at 612.5 nm disappears, a very broad luminescence band is obtained and the luminescence quantum yield strongly decreases, hence the need to multiply the spectrum intensity by eight times in Fig. 4a. Finally, the basal planes for SC sample give no luminescence at all. Turning to edge planes, the SE sample leads to an emission spectrum featured by three main peaks at 612.8, 613.6, and 618.3 nm, superimposed on a broad background that gives a shoulder around 620 nm. The SC sample gives rise to a broad luminescence emission band centred around 615–616 nm and a weak band around 610 nm. The luminescence quantum yield is also lower than for the SE sample (the spectrum has been multiplied six times in Fig. 4b).
The 5D0 → 7F2 luminescence spectra of the dickite/Eu3+ samples. Excitation with the 458-nm argon laser line.
Luminescence emission measurements performed on hydrated EuCl3 crystals have shown very similar peaks to those at 612.4 nm in Fig. 4a and at 612.8, 613.6 and 618.3 nm in Fig. 4b (data not shown), depending on the hydration state of the crystals. This suggests that EuCl3 is present at the surface of the SE sample, with different states of hydration on basal and edge planes. The 5D0 → 7F2 band for the SE sample presents three well-resolved peaks in the case of edge planes, which agrees well with a high crystal field for Eu(III) species in a solid state [26–32]. Since the 5D0 → 7F1 band is poorly observed, the site symmetry of europium should be very low within this precipitated phase. This may come from the local hydration states of the precipitate. The SC sample shows a broad emission band with a considerable decrease of luminescence quantum yield, and the peaks observed with the SE sample have disappeared. The lack of emission structure indicates a very low symmetry around Eu(III) cations or a broad distribution of complexes with different ligand spheres [26,28]. Luminescence spectroscopy thus gives a good hint that the main difference observed between the SE and SC samples comes from a washing of the surface precipitate, letting surface complexes at the edge sites of dickite particles. These surface complexes have chloride atoms in the ligand sphere of Eu(III) cations. This interpretation is coherent with XPS and epifluorescence results.
3.3 Raman spectroscopy
Raman spectroscopy has been used to study the surface chemistry of the kaolinite group minerals [16–18]. The stretching of the OH hydroxyl groups of dickite or kaolinite are Raman active and interaction between OH groups and cations can be detected following the change of Raman intensity and wavenumber. Important structural information can thus be obtained after sorption of cations on dickite. The hydroxyl groups of the octahedral cage of dickite are indicated in Fig. 5, following the notation of Johnston et al. [17]. OH1 tags the less accessible hydroxyl groups that are sometimes designated as “inner-hydroxyl groups” [18]. The three other groups are more or less engaged in hydrogen bonds with the neighbouring upper layer and are designated as “outer-hydroxyl groups”. These three OH groups are the more accessible considering the basal plane (001) of dickite. Note, however, that inner groups OH1 can be accessible if the surface chemistry of the lateral planes is considered. In that case, the crystal structure is truncated and the octahedral cage cut through various crystallographic planes, which may expose the OH1 groups. Raman spectroscopy of dickite was subjected to several complementary studies in the literature, both experimental and theoretical [16–25]. Nowadays, a rather comprehensive explanation is available for the interpretation of the spectra in the OH-stretching region (i.e. 3600–3750 cm−1 for dickite). The vibrational scheme strongly depends on the polarization of the incident laser electric field and the relative orientation of dickite particles towards this field. Fig. 6 reports the Raman spectra of well-oriented pristine dickite particles in the hydroxyl stretching region. The spectra perfectly agree with those previously reported in the literature [16–18] and the attribution of the vibrational modes can be confidently performed, as indicated in Fig. 6. The band observed at 3624–3627 cm−1 is attributed to the OH1 stretching mode. At 3645 and 3656 cm−1 are, respectively, collected the in-phase TO and LO phonons of the coupled OH2 and OH4 stretching modes [18], the mode at 3645 cm−1 being the predominant one in the spectrum. Finally the OH3 stretching mode is measured at 3708–3712 cm−1.
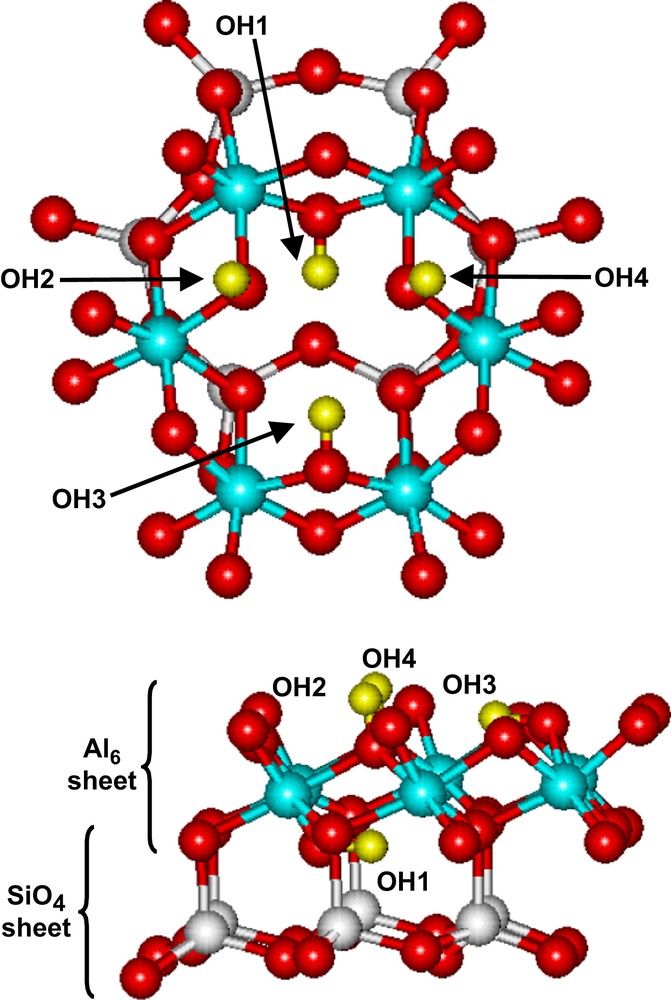
The octahedral cage of dickite and the numbering of the hydroxyl groups.
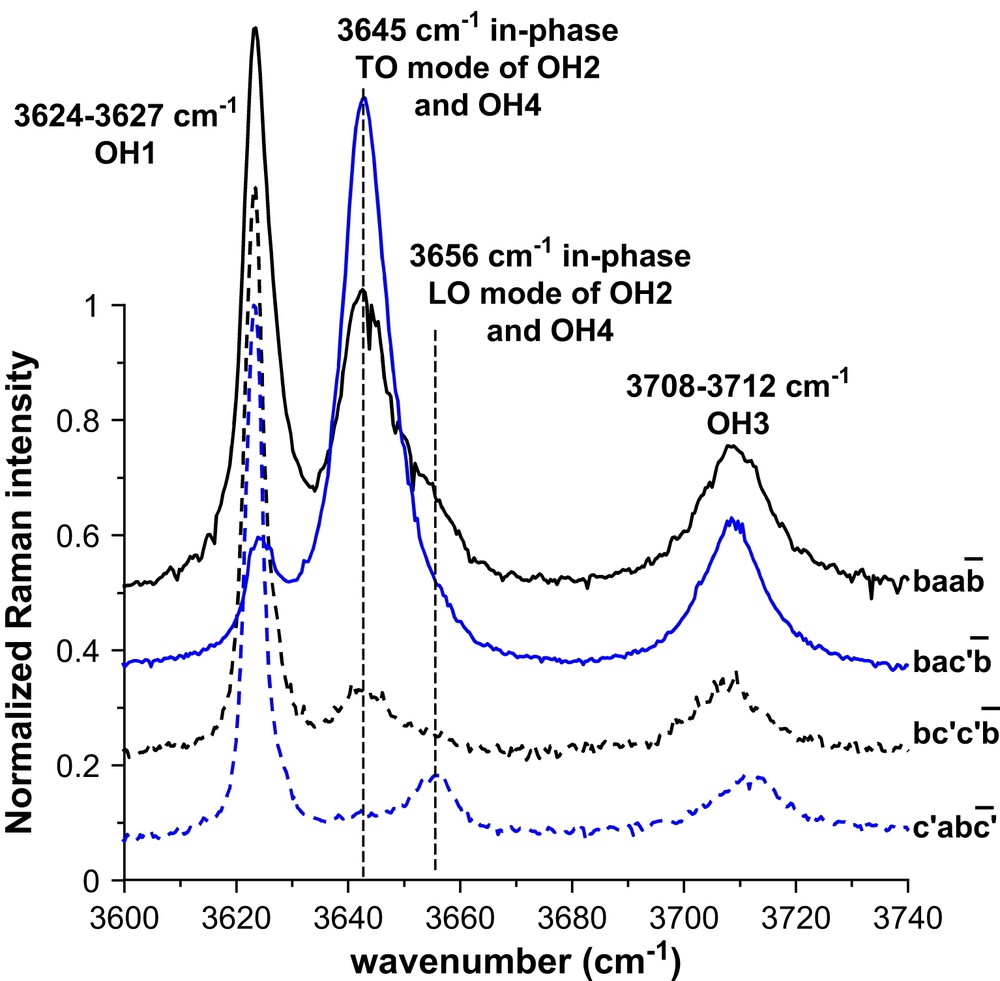
Raman spectra in the OH-stretching region for several orientations of pristine dickite particles. Orientation and polarizations are indicated following the Porto notation [16–18]. All the spectra have been normalised to their maximum, and are vertically separated for the sake of clarity.
Fig. 7 compares the Raman spectra of pristine dickite and SC sample oriented particles, both in the OH-stretching region and in the low-wavenumber region (below 1200 cm−1). This latter region was less investigated in the literature but can bring about supplemental information concerning the actual structure of surface complexes. Note that in the present study, particles before and after europium sorption were rigorously oriented to avoid any artefact coming from a slight tilt towards the electric field of the incident laser radiation. It ensures that the spectral changes noticeable in Fig. 7 are the sole consequence of europium sorption. The SE sample was not investigated by Raman spectroscopy to avoid spectral changes coming from surface precipitates. For basal crystallographic planes, there were almost no spectral changes between pristine particles and sorbed SC sample (data not shown). As a consequence, Fig. 7 only reports several spectra monitored at the (010) edge planes of particles, where epifluorescence microscopy and luminescence spectroscopy have evidenced europium sorption.
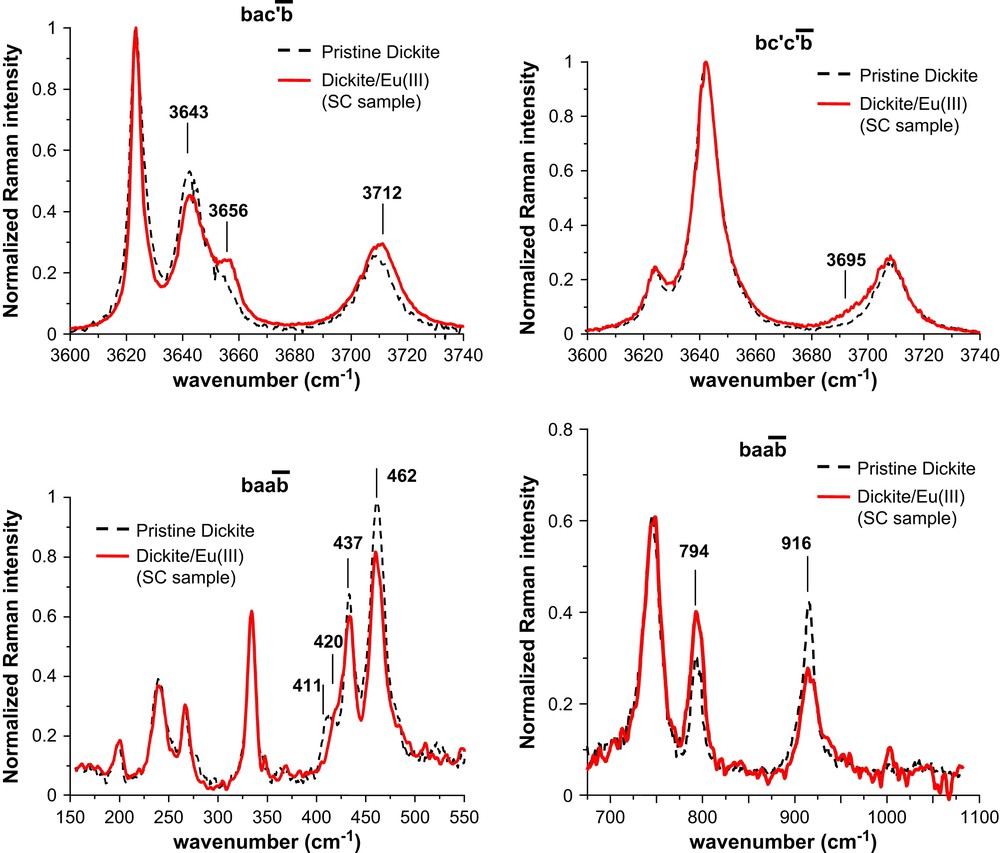
Raman spectra for pristine dickite and dickite/Eu3+ SC sample. Orientation and polarization are indicated following the Porto notation [16–18].
Fig. 7 indicates that OH-stretching modes are slightly affected by europium sorption. The changes mainly concern the relative intensity of the OH2, OH4 and OH3 stretching modes. For the spectrum, a new shoulder was found near 3695 cm−1. This observation was reproducible but the intensity of this new vibrational mode considerably changed from one sorbed particle to another. This new mode might come from OH3 groups directly engaged in the ligand sphere of Eu(III) cation for inner-sphere surface complexes. The balance between outer- and inner-sphere complexes may vary from one particle to another, depending on the presence of heterogeneities such as defects, crystallographic edges, etc. That would explain the change of this band intensity depending on the particle under investigation. The structural lattice modes in the spectra of Fig. 7 also bring about interesting information concerning the localisation of sorbed europium species. The bands at 411, 420, 437 and 462 cm−1 are all relevant to the and vibrational modes of SiO4 tetrahedrons [16]. These modes are affected by europium sorption, whereas the mode at 200 cm−1, attributed to the vibrational mode of AlO6 octahedron [16], seems to remain unaffected. Other spectral changes are observed at 794 and 916 cm−1, which are attributed to the OH translation and libration modes [16].
These facts confirm the presence of inner-sphere complexes of europium species at the edge planes of dickite particles for the SC sample. They suggest that hydroxyl groups are involved in these inner-sphere complexes and that europium species disturb more the SiO4 tetrahedrons than the AlO6 octahedrons. The results also confirm the crucial importance of edge site for the sorption and/or surface precipitation phenomenon [35–40]. This distinctiveness of edge sites arises from the cut of the crystallographic lattice and the corresponding creation of accessible hydroxyl groups and change of protonation state of lateral oxygen atoms [35,36]. Any surface complexation model should then explicitly incorporate the reactive area formed by edge planes of clay minerals [37].
4 Conclusion
The specific reactivity of the edge sites of dickite particles related to the surface precipitation and complexation of Eu(III) species has been evidenced by XPS, Raman, and luminescence spectroscopies. Surface complexes are more probably inner sphere and involve chloride atoms in the ligand sphere of Eu(III). They are localised at the edge sites of dickite particles, and mainly disturb the OH2, OH4, and OH3 hydroxyl groups, and the silica tetrahedrons of the dickite lattice. The symmetry around sorbed europium species is found to be low and the distribution of surface complexes wide, corresponding to rather heterogeneous surface processes. In order to precise more accurately the structure of these surface complexes, site-selective excitation with a tunable laser source is required. Such work is currently in progress in our laboratory. Moreover, in order to collect signals more specific from the surface of particles, optical non-linear spectroscopies such as second harmonic [41,42] or sum frequency generation [43] will be employed. The sensitivity of these techniques will be useful to decrease the europium concentration in solution, with the hope to simplify the distribution of observed surface complexes.