

1 Introduction
The aim of this review is to show how the migration of long-lived radionuclides (which are potentially harmful for Man and its environment) is strongly correlated with the action of microorganisms. The latter induce oxido-reduction reactions on numerous elements, and this is and will be an evermore frequent case in the future. This is directly correlated with life on Earth since its beginning. This review being intended for physico-chemists, the first part will be devoted to the origin and the diversity of microorganisms on Earth, with a particular interest on how life, which emerged on Earth several billion years ago, was implicated in the evolution of the original Earth crust. From this, we will infer on a geological timescale (the order of magnitude is the half-life of the radionuclides synthesized in nuclear processes) how and to what extent the microorganisms interact with nuclear wastes.
The first traces of life on Earth date from 3.5 to 4 billion years ago, when free oxygen did not yet exist in the atmosphere. As these microorganisms diversified, adapting to the conditions then encountered, they contributed to the formation of the environment of the primitive Earth. Using present techniques of micro- and molecular-biology, the number of microorganisms per gram of earth or water is estimated to be in the range from 106 to 109. They are classified in three categories, Archaea, Bacteria and Eucarya, which developed from a common cell LUCA (Last Unicellular Common Ancestor). The microorganisms developed various strategies in order to obtain the elements (water, mineral salts, carbon, etc.) required to assure their growth and multiplication, and they are able to survive in media hostile to more complex forms of life (extremes of pH, salinity, radioactivity, temperatures > 100 °C). The activity of microorganisms concerns nearly every field of nature and human endeavor: e.g., agronomy (assimilation of nitrogen and phosphor by plants), geochemistry, formation of ore deposits, oxidation–reduction reactions and chemical transformations of all kinds, biotechnologies, etc.
The ability of the microorganisms to chemically transform numerous elements during oxidation–reduction reactions is independent of the nature (stable or radioactive) of the element involved. On a geological timescale and in spite of the heat and the radioactivity, a number of interactions between radionuclides and microorganisms could occur on nuclear waste storage sites after corrosion of concrete and containers, corrosion which, in addition, could be initiated and facilitated in certain cases by the microorganisms or their by-products.
The present review, after dealing briefly with the source and diversity of our planets' population of microorganisms, will present their effects on metals and metalloids, analyzing particularly the consequences of their activity on key radionuclides in the environment (actinides, fission products from the electronuclear cycle).
2 Origin and diversity of microorganisms
The Precambrian (or cryptozoic) period extends from the birth of the Earth, −4.55 Ga till −0.540 Ga. It is subdivided into the Hadean (−4.55 to −4 Ga), the Archaen (−4 to −2.5 Ga) and the Proterozoic (−2.5 to −0.54 Ga) time lapses.
The evolution of the lithosphere [1–4] and the other external envelopes of the Earth occur simultaneously: the hydrosphere [5] appears and its physicochemical characteristics (temperature, salinity) evolve [6]; the atmosphere, reducing or neutral at first, slowly becomes enriched in free oxygen [7,8]. Three major hypotheses have been presented to account for the origin of life on Earth [9–12]:
- • life already present in the interstellar medium arrived on Earth on asteroids and/or comets [13–16];
- • life was created on the Earths' surface under conditions similar to the simulation experiments of Miller and his successors. In 1953, Miller [12] synthesized amino acids comparable to those of living systems. Although conditions similar to those which then existed were simulated, the appearance of life (and its autoreplication) in the mineral world remains to be demonstrated;
- • life was created in the ocean depths in hot vents [17–20]. Recent work, however, does not support that hypothesis [21].
In 1969, Woese [22] studied the small ribosomal subunit RNA sequence of archaeabacteria, eubacteria, and eukaryotes. It is excluded that one of these lines gave rise to the others. The three groups descended from a common ancestor later called LCA or LUCA [23,24] and they co-existed precociously. The passage from a simple eukaryote to a hierarchized, then pluricellular, eukaryotic organism took place over time through the effects of bacterial endosymbiosis and oxygen. The association between prokaryotes and eukaryotes appears as one of the keys to the evolution of our bacterial universe [25,26]. The three biological groups according to Woese et al. [27] are shown in Fig. 1.
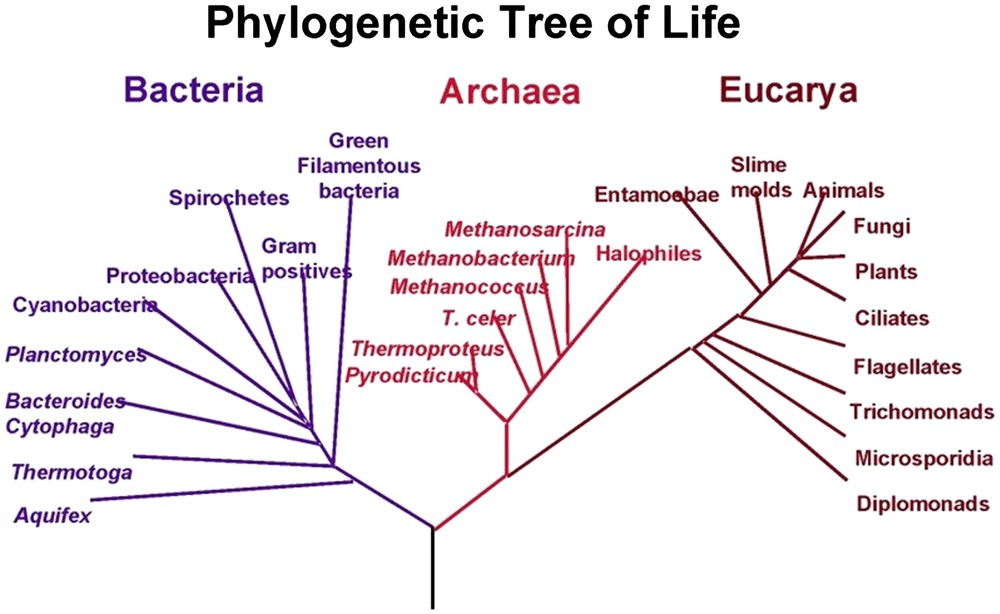
The three biological groups according to Woese et al. [27]. Reprinted from Wikipedia.
The primitive atmosphere of the Earth arose from the degassing of the magmas, then, from reductive, it probably evolved through the action of photosynthetic cyanobacteria [28–30] towards one progressively enriched in free oxygen to a level which will permit a fantastic adaptation [31] of the eukaryotic cell at the limit of the Precambrian and the Paleozoic (Cambrian). The next paragraphs will examine the first steps of life on Earth and its evolution. A valuable review on this topic is to be found in Ref. [32].
When the Earth is being formed, it receives a mass of organic matter from asteroids, meteorites and comets, the last of which is also the source of some terrestrial water. Volcanic activity is intense and tectonic plates begin to move as the first continents are formed. During the first four billion years, life forms consist mainly of bacteria. The image of the Precambrian Earth from space showed red periods due to iron minerals and sandstones, and white periods during global glaciations. Sudden changes of color occurred due to bacterial ‘blooms’, then continents and ocean margins become progressively green during the proterozoic chlorophyllian algae and the planet then becomes slowly blue. Life was present during the period 4.55–3.9 Ga, as testified to by the study of the carbon isotopes in the most ancient rocks presently known (Isua series, Greenland) dated 3.9 Ga [33–41]. From the beginning, the bacteria secrete varieties of mucilage and combine to form bacterial mats [42] which trap particulates or favor the precipitation of iron salts and carbonates, gradually laying down successive biomineral layers to form structures called stromatoliths (Warrawoona series, Australia, 3.5 Ga). In the different series corresponding to more recent periods, spherical bacteria, cyanobacteria, ferruginous bacteria, eukaryotic algae, etc., have been observed [43]. The principal accumulations of iron are found in the banded sedimentary layers. These thick, regular layers of iron oxide appear from 3.9 Ga (Isua series); 95% are formed between 2.6 and 2 Ga as Fe2O3 and Fe3O4 (the magnetite). The mats of photosynthetic cyanobacteria [29,42] produced the oxygen which was fixed by the iron dissolved in water. A major crisis in the biosphere probably occurred around 2 Ga. As the supply of FeIII disappeared, the cyanobacteria adapted and the banded ferriferous stromatoliths gave way to carbonated ones. Recent studies [44] show that the Archaea and Bacteria close to LUCA are capable of reducing FeIII to FeII. LUCA was probably the first micro-organism capable of assuring respiration by the reduction of FeIII to FeII.
The oxygen level begins to grow from 1.8 Ga on. Certain bacteria, including the cyanobacteria, consume carbon dioxide by their photosynthetic activity, lowering the partial pressure of carbon dioxide in the aquatic media and enhancing the precipitation of calcium carbonate.
The progressive increase of the oxygen level in the atmosphere revolutionizes the biosphere. During the major part of the Precambrian, the biosphere is a bacterial world functioning without sexuality. Cellular division by mitosis can take place only if the level of free oxygen is at least 1% of its present-day value. That date was reached at about 2 Ga, which is the date of the appearance of eukaryotes. An oxygen level of at least 7% of its present value is necessary for oxygen to diffuse through membranes, creating the first condition enabling the formation of a multicellular organism. Bacteria have proliferated on Earth since at least 3.9 Ga and the Precambrian biosphere has not disappeared.
At present, the Precambrian and the most recent organisms coexist [28–30]:
- • the prokaryotes (Bacteria and Archaea) are of the size of a micron and are composed of a single compartment surrounded by a membrane, inside which are the cytoplasm and the chromosome made of DNA;
- • the eukaryotes (mean dimension 10 μm) are defined by the presence of a nucleus containing the genetic material of the DNA, surrounded by a cytoplasm which can have the specialized organites (mitochondria, plasmids, peroxisomes, etc.), surrounded itself by a membrane defining the limits of the cell.
Among the Bacteria are found the photosynthetic organisms such as the cyanobacteria, as well as those living at high temperatures (thermophilic) and below 0 °C (psychrophilic). Some Archaea live in extreme environments: halophiles require high salt concentrations, thermoalcaliphiles grow only at temperatures of at least 70 °C and pH between 10 and 11, whereas the thermoacidophilic ones grow at 80 °C and pH below 2 (hot sulfurous source); the methanogens in absence of oxygen in swamps or marshes generate methane by reduction of carbon dioxide.
The recent discovery of microorganisms in deep marine sediments [18,45], as well as those living at the heart of the Earth's crust in deep basalt, considerably increases the extent of the biosphere. The message of the latest discoveries is very clear: we know but a small percentage of the species presently living in the biosphere (8 × 105 species are known and it is estimated that 106 species are present in the biosphere) [46].
3 Microbial colonization of radioactive environments
As seen above, microbial life in the biosphere is ubiquitous and fairly well known, but the level of microbial colonization of radioactive environments has only recently been investigated. Landa [47] studied mill tailings comprising uneconomic residues from mining and milling operations. Other environments that have received radioactive wastes have been explored for microbial content. Characterization of microorganisms attached to the walls of a pool storing nuclear materials from the Spanish nuclear plant was reported by Chicote et al. [48]. Several isolates were obtained and six different bacteria (affiliated to β-Proteobacteria, Actinomycetales and the Bacillus/Staphylococcus groups) as well as fungus (Aspergillus species) were identified.
The sediments contaminated in 1962 by a leak from a waste tank (USA DOE Hanford Site, Washington State) containing high concentrations of alkali, nitrate, aluminate, chromate, 137Cs and 99Tc have been studied by Fredrickson et al. [49]. Viable aerobic heterotrophic bacteria were found in most of the samples. Gram+ bacteria most closely related to Arthrobacter species were the most common among all isolated samples, but other phyla such as Rhodococcus and Nocardia were also found. Even the most radioactive samples (>10 μCi 137Cs g−1) yielded active bacteria and those from some isolates from samples with >20 μCi of 137Cs g−1 were closely related to Deinococcus radiodurans and were able to survive acute doses of ionizing radiation ∼20 kGy. These organisms were highly resistant to desiccation.
At the Severnyi repository for low-level liquid radioactive wastes in eastern Siberia, Russia, water samples collected from a depth of 162–405 m below sea level [50] contained anaerobic denitrifiers, fermenters, sulfate-reducers and methanogens. A wide range of functional groups of microorganisms was detected and rates of several respiratory processes were higher in the zone of dispersion of radioactive waste than in the background areas. The authors noted that microbial gas production could result in a local increase in pressure in the repository and affect the consequent discharge of wastes, although key microbial activities (for example formation of insoluble sulfides from sulfate reduction) could also result in the immobilization of some radionuclides. Such processes have recently been shown to occur in microcosms prepared from sediments in two types of predwater lakes of the Moscow Oblast [51]. These results show that the radioactivity of several nuclear waste types is not necessarily inhibitory to all microbial life.
Nevertheless a recent study [52] using pure cultures of bacteria (D. radiodurans, Pseudomonas putida and Schewanella putrefaciens) in tests concerning the toxicity of actinides, metals and chelators showed that actinide toxicity is primarily chemical, not radiological, and that radiation resistance (for example in Deinococcus) does not ensure radionuclide tolerance.
In conclusion, the radioactivity does not eradicate the microbial populations in storage sites for nuclear wastes; at most it effects a selection, assuring the survival of several species among the multitude of microorganisms initially present.
4 Effects of microorganisms on radionuclides
The fate of the radionuclides in the environment will depend on pH, Eh, ligands and the presence of microorganisms. The environmental chemistry of actinides and some fission products (particularly Tc and Se) is very complex due to the multiple oxidation states [53]. Microbes will affect radionuclides directly by changing the oxidation state or indirectly by producing reducing compounds (H2S, FeII, ligands). Microbial activity will deeply affect the solubility of radionuclides by the following mechanisms: biosorption, metabolism-dependent bioaccumulation, enzymatic biotransformations, biomineralization by ligands.
4.1 Biosorption
Sorption may be responsible for the accumulation of radionuclides on the surface or interface. Living cells and dead biomass can biosorb; dead biomass often sorbs more metal(loid)s than does the living one. Ligands such as carboxyl, amine, hydroxyl, phosphate and sulfhydril groups are involved. Biosorption is rapid and unaffected by temperature and can be described by the Langmuir, Freundlich and Brunauer–Emmett–Teller (BET) isotherms [54,55]. An extensive review of radionuclide biosorption is given by Lloyd and Macaskie [56].
4.2 Metabolism-dependent bioaccumulation
It is well known that it is possible for radionuclides and numerous stable elements to penetrate the cell by a classical transport system, as would an ion habitually involved in the physiology of the cell (Cs+ instead of K+, Sr2+ and Ra2+ instead of Ca2+). Once in the cell, the toxic metal (radioactive or not) is sequestrated by cystein-rich metallothioneins [57] or in the vacuole in the case of fungi [58]. This uptake by a metabolism-dependent transport mechanism is poor for the heavier radionuclides, like actinides. Other explanations can be assumed: a recent review [59] explains intracellular accumulation of uranium by increased membrane permeability due to uranium toxicity and not to metabolism-dependent transport mechanisms.
4.3 Enzymatic biotransformations: bioreduction, biomethylation, biodegradation
Microorganisms can catalyze the direct transformation of toxic metals and metalloids to less soluble or more volatile forms via enzymatic mechanisms. Bioreduction can precipitate solid metal(loid)s by enzymatic reduction [60,61], or produce volatile derivatives (by biomethylation, for example, with Se, Te, Hg [62,63]); alkylated actinides are instable and few experiments have been carried out [64].
Under anaerobic conditions, several microorganisms can respire by the reduction of alternative electron acceptors. These oxido-reduction reactions are well recognized for nitrate, sulfate, and carbon dioxide in presence of microorganisms and in absence of oxygen and are called bioreduction or dissimilatory reductions. More recent studies [65] show that higher valence metal(loid)s, radioactive or not, also function as alternative electron acceptors during anaerobic respiration of specialist organisms (FeIII, MnIV, UVI, SeVI, TcVII). Biomethylation can produce volatile methyl derivates, as for Se, Tc, Hg, I, and so decrease the concentration of soluble contaminants in water or soil under the action of microorganisms.
Biodegradation of associated organic compounds has been observed with citrate. Citrate can form with highly soluble radionuclide citrate complexes, then be in some cases degraded by microorganisms to give a precipitation of the radionuclide [66], as observed with Pseudomonas aeruginosa and P. putida [67].
4.4 Biomineralization by ligands produced by microbes
Ligands enzymatically generated, such as phosphate, carbonate and sulfide, are able to precipitate metals (stable or radioactive), and provide an appreciable way to eliminate radionuclides from solution.
The microbial biomineralization by phosphate involves Citrobacter sp. (now Serratia sp.), since molecular methods identified the phoN phosphatase gene [68]. The role of a phosphatase in metal accumulation has been confirmed by the fact that the expression of the phoN gene clones in Escherichia coli conferred the ability to bio-precipitate uranyl phosphate [69].
Carbonates are ligands produced with Ralstonia eutropha (formerly Alcaligenes eutrophus), Pseudomonas fluorescens [70–72] for precipitation of strontium carbonate, whereas the formation of uranyl carbonate species enhances the dissolution of uranyl [72].
Sulfides produced by sulfate-reducing bacteria, ubiquitously distributed in acid, neutral and alkaline conditions [73], have the potential to play a critical role in controlling the migration of radionuclides in a range of environments. The liberation of gas (H2S) will accelerate a precipitate of high-valence element or its reduction.
5 Key radionuclides of environmental importance
The presence of radionuclides in the environment is the result, initially, of significant quantities of radioisotopes released as a consequence of nuclear weapons' testing in the 1950s and 1960s. Then accidental contamination, coming from nuclear activities, can arise, like Chernobyl catastrophe. Moreover, burden of anthropogenic radioactivity continues with the controlled discharge of process effluents produced by industrial activities bound to nuclear power. Wastes generated during uranium mining concern low- and high-level liquid and solid wastes produced from reactor operation and fuel reprocessing. Finally, some nuclear wastes have to be stored either in surface or in subsurface.
Thermal fission of 235U generates more than 100 products in the range 72Zn to 161Tb with long or short half-lives. Interaction of the uranium nuclear fuel with α-particles and neutrons produced during fission, moreover, will generate the transuranic elements Np, Pu and Am in the reactors. Radionuclides present in some nuclear wastes arise from activation, corrosion of fuel cladding or assemblies (60Co, 59Ni, 93Zr). The environment can be at risk from radionuclides with significant activity, long half-lives, large quantities and bioavailability. In the fission products and actinides produced, there are about a dozen elements with key radionuclides of environmental importance [74].
Table 1 gives some characteristics of key radionuclides of environmental importance: fission products have few oxidation states compared to actinides with multiple oxidation states for Am, Np, Pu; nonetheless Th is essentially present as ThIV, and uranium most frequently as UVI and UIV.
Some characteristics of key radionuclides of environmental importance: fission products have few oxidation states compared to actinides with multiple oxidation states for Am, Np, Pu
| Element | Isotope | Half-life (a) | Oxidation states |
| Cobalt | 60Co | 5.27 | II,III |
| Selenium | 79Se | 4.8 × 105 | −II,0,IV,VI |
| Strontium | 90Sr | 29.1 | II |
| Technetium | 99Tc | 2.15 × 105 | IV,VII |
| Iodine | 129I | 1.57 × 107 | −I,0,V |
| Cesium | 137Cs | 30.17 | I |
| Thorium | 229Th | 7.34 × 103 | IV |
| 230Th | 9 × 104 | ||
| Uranium | 238U | 4.47 × 109 | II,IV,V,VI |
| Neptunium | 237Np | 2.14 × 106 | III,IV,V,VI,VII |
| Plutonium | 238Pu | 87.7 | III,IV,V,VI,VII |
| 239Pu | 2.41 × 104 | ||
| 240Pu | 6.55 × 105 | ||
| 241Pu | 14.4 | ||
| Americium | 241Am | 432.7 | II,III,IV,V,VI,VII |
6 Fission products
The redox chemistry of the fission products is in general limited to some valencies. Both Cs and Sr exist in only one oxidation state and their environmental behavior is therefore not dependent on any redox chemistry. Their oxidation states account for their high bioavailability. Cs+ is analogous to K+ and enters cells via pathways for K+. Ca2+, as other alkaline earth metals, Ba2+, Ra2+, forms complexes and insoluble precipitates. Technetium has, in particular, two environmentally stable oxidation states, VII, as the anion TcO4− (soluble), and IV with soluble and insoluble forms. For iodine, −I, 0, V are most frequently found as I−, I2, IO3−. Selenium has four environmentally important oxidation states, VI and IV as mobile and soluble forms in oxic media, and 0, elemental selenium. −II is very often bound to proteins or methylated.
6.1 Technetium (group VII)
Technetium is an artificial radioelement with an atomic number of 43. It belongs to the Mn group and has no stable isotope. The isotope 99Tc is of great practical importance because it is one of the fission products with a relatively high yield in the thermal-neutron fission of 235U and 239Pu (about 6%) and it has a long half-life (2.1 × 105 a) [75]. Technetium-99 has been released to the environment via nuclear weapons testing and nuclear facilities such as nuclear fuel reprocessing plants, nuclear power plants and waste disposal facilities. But it is also produced by the use of 99mTc (T½ = 6.01 h) in nuclear medicine and decays directly to 99Tc. Medical uses of 99mTc will continue to increase in the future. For those reasons, 99Tc in the environment requires special consideration.
Technetium is known to exist in all valence states from +7 to −1. The dominant species in natural aqueous solutions in equilibrium with the atmosphere is the pertechnetate (TcO4−), which has a high geochemical mobility [76] and availability for plants [74,75]. However, through a combination of factors [51,77–79] such as redox conditions and microbial activity [80–82], TcVIIO4− can be reduced and immobilized in insoluble forms in soils, in or around microorganisms. The redox chemistry of Tc is crucial in governing its mobility [75]. Under environmental conditions, TcVIIO4− can be transformed to lower oxidation states as TcIV species, amongst which the insoluble forms TcO2 under anaerobic conditions, as predicted by the Eh–pH diagram of Tc [83], while at valence 4 numerous soluble forms exist, such as TcO2+, TcO(OH)+, TcO(OH)20 aq, Tc(OH)3+, Tc(OH)(CO3)2−, and Tc(OH)2(CO3)22−. Phosphorimager is of great help for the determination of Tc species [84]. The reduction of Tc can be achieved by abiotic mechanisms using zerovalent iron, or FeII-containing minerals under anoxic conditions; magnetite is particularly a efficient reductant for TcVII [85]. X-ray photoelectron spectroscopy XFS has confirmed that the Tc was precipitated on the surface of the magnetite as TcO2. The retention of Tc as Tc2S7 on stibnite Sb2S3 has been studied by proton X-ray emission microbeam [86].
An example of TEM images of S. putrefaciens CN32 after enzymatic reduction of Tc is shown in Fig. 2 [87].
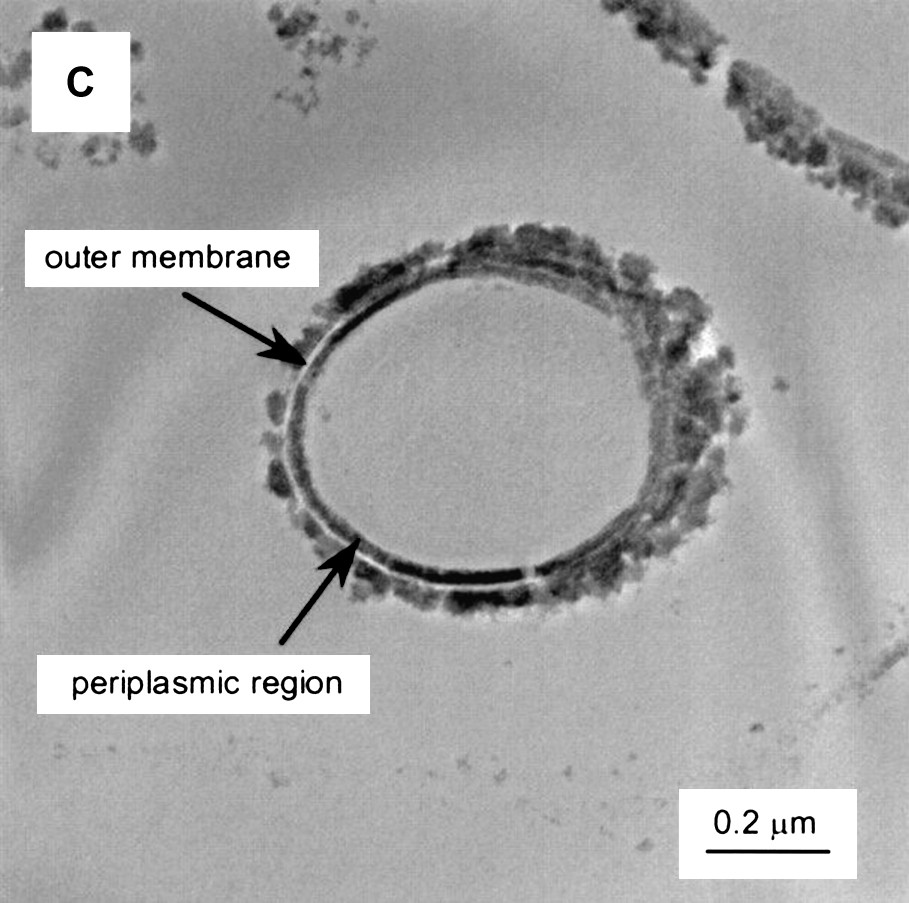
TEM images of S. putrefaciens CN32 after enzymatic reduction of Tc with different electron donors and solution compositions (bicarbonate-H2). The cells are unstained, and the images result because the presence of reduced Tc. Reprinted by permission from the American Society for Microbiology [87], copyright 2000.
Microbial metabolism may affect Tc speciation by indirect (chemically mediated by either sulfide or FeII) and direct (enzymatic) mechanisms [61,80,87,88]. Microbial reduction of technetium has been studied very intensively. Sulfate-reducing bacteria, Desulfovibrio desulfuricans [89,90], metal-reducing bacteria, Shewanella putrefaciens [87] and Geobacter sulfurreducens [61] reduce TcVII at neutral pH. Under acidic conditions, Thiobacillus ferrooxidans and Thiobacillus thiooxidans reduce TcVII to low valency forms [91]. It was shown for the first time by Khijniak et al. [60] that haloalcaliphilic bacteria isolated from soda-lake environments are capable of reducing TcVIIO4− to TcV, TcIV and TcIII at pH 10. Dissimilatory Metal-Reducing Bacteria (DMRB) include Shewanella putrefaciens [87], Shewanella alga, Geobacter metallireducens [87,92,93] and the sulfate-reducing bacterium D. desulfuricans [88,89]. These microorganisms are capable of gaining energy for maintenance and growth by coupling the oxidation of organic C, from lactate, acetate or H2, to the reduction of TcVII.
| TcVIIO4−(aq) + 3e− + 4H+ → TcIVO2, ·nH2O(s)+(2−n)H2O |
A direct enzymatic reduction of TcVII has been demonstrated by the FeIII-reducing bacteria G. metallireducens and also with the widespread E. coli [81], and the sulfate-reducing bacteria D. desulfuricans [88]. Tc reduction was catalyzed by the hydrogenase component of the Formate Hydrogen Lyase complex (FHL). Precipitation of TcO2 was at the periphery of the cell; these results were consistent with direct, hydrogen-dependent reduction of TcVII by a periplasmic hydrogenase. The identification of hydrogenase 3 of FHL as the TcVII reductase of E. coli opened the way for a program to screen organisms with naturally-enhanced activities against TcVII.
The addition of sulfate-reducing bacteria to mixed cultures of anaerobically grown soil bacteria increased Tc removal. This was mediated by an indirect mechanism utilizing microbially-generated H2S. This metal sulfide formation also explained Tc accumulation by mixed anaerobic bacteria isolated from marine sediment [82].
Several extremophiles resistant to temperature, salinity or radiation have shown ability to reduce TcVII. D. radiodurans, the most radiation-resistant organism, capable of surviving acute exposures up to 15,000 Gy without lethality [94] is described as having a strictly aerobic respiratory metabolism, but under strict anaerobic conditions reduces FeIII, coupled to the oxidation of lactate to CO2 and acetate and TcVII in combination with humic acid or synthetic electron shuttle agents [95]. Thermoterrabacterium ferrireducens and Tepidibacter thalassicum (Gram positive thermophilic bacteria) precipitate Tc as TcIV with some amounts of soluble TcV at 65 °C (hydrogen, glycerol and lactate served as electron donors) [96].
In conclusion, numerous microorganisms are capable of immobilizing technetium in the environment under the most diverse conditions, thus favoring the retention of this long-lived fission product, produced in large amounts in the uranium electronuclear cycle.
6.2 Selenium (group VI)
Among the key radionuclides that must be taken into consideration with regard to environmental impact, 79Se, a long-lived isotope (T½ = 0.65 × 105 a) produced in very small amounts in uranium fission, has a special importance because of its role as an essential trace element for humans and animals. As an integral part of glutathione peroxidase, selenium contributes to the elimination of free radicals produced by oxygen or other inducers. Selenium is implicated in seleno-amino acids (the new 21st amino acid), selenoproteins, gaseous methyl-seleno compounds and contributes to the maintenance of homeostasia [63]. Moreover, selenium is definitely an essential element for many eukaryotes, bacteria and archaea [97–100]; its major role is in selenium-substituted amino acids, although it is also found in the active sites of some metalloenzymes.
In aerated environments, selenium can exist in several redox forms, including the elemental form Se0, which is solid, and the oxidized forms: selenate (SeVIO4)2− and selenite (SeIVO3)2−. Since microorganisms are involved in the geochemical cycle of selenium, soil bacteria may transform the soluble forms Se6+ and Se4+ to insoluble Se0 or to volatile compounds (Se2− selenide, dimethyl selenide…).
Because of their proximity as group VIA elements, sulfur and selenium share many chemical and biochemical properties. In this regard, sulfate has been identified as an antagonist for selenate transport or toxicity in various microorganisms [101]. In the biogeochemistry of selenium [102,103], a selenium cycle has been proposed with reduction of selenate to selenite, selenium zero or sometimes selenide. The Se cycle [104–106] differs from the S cycle because S0(s) is neither a product nor an intermediate in sulfate reduction, but arises from partial oxidation of sulfide [107].
Thin-section micrographs of Rhodobacter sphaeroides IL-106 grown under anaerobic photosynthetic conditions in the presence of 1 mM SeO32− are shown in Fig. 3 [108].
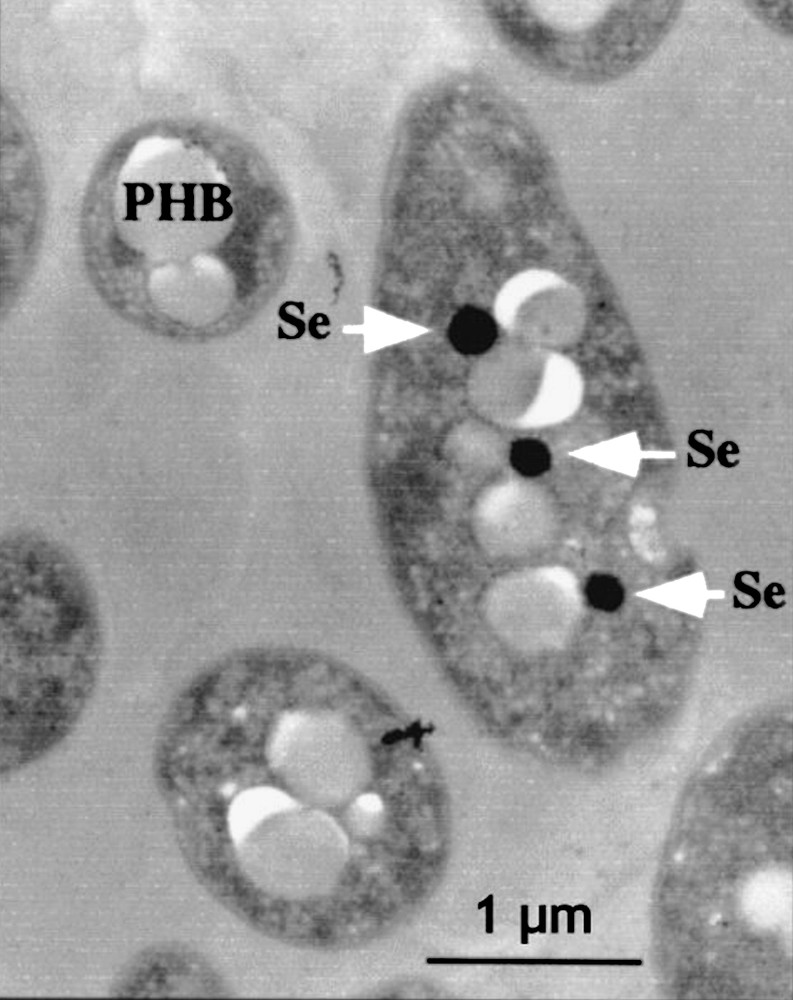
Thin-section micrographs of Rhodobacter sphaeroides IL-106 grown under anaerobic photosynthetic conditions in the presence of 1 mM SeO32−. Arrows indicate the presence of electron-dense particles of selenium (Se). White particles correspond to polyhydroxybutyrate (PHB) granules. Reprinted by permission from American Society for Microbiology [108], copyright 2001.
Selenate and selenite seem to be the most abundant forms of bioavailable selenium in the environment and both can serve as electron acceptors. Like nitrate (denitrification), sulfate (sulfate reduction), and bicarbonate (methanogenesis), it is possible that selenate, selenite, and other oxidized metal or metalloids (TcVII, heavy metals, UVI, Fe3+…) can oxidize deposited organic matter by dissimilatory processes and be implicated as electron acceptors in bacterial mineralization of organic matter in anoxic sediments. In bacterial anaerobic respiration, the organic matter concerned, usually acetate, succinate, lactate, is oxidized to CO2 and is considered as an electron donor, as with, for example, the selenite or selenate electron acceptors:
| (1) |
| (2) |
Several observations of Se0(s) precipitation from selenate (as in Eq. (1)) in various bacterial cultures have been reported [109–113]. Selenate reduction to Se0(s) by anaerobic bacteria growth on acetate electron donors with formation of carbon dioxide as the respiratory end product is the major sink for removal of selenium oxyanions in anoxic sediments and takes place with Proteobacter [114] and some species of Pseudomonas [111].
Bacterial Se0(s) precipitation from selenite by several microbes, including algae [115] and bacteria [116], has been frequently reported. Microorganisms can reduce selenite to selenium, as is the case for the common aerobic soil bacterium Bacillus subtilis [117]. The accumulation of selenium by R. sphaeroides has also been reported [118]. Ralstonia metallidurans CH34 [119] reduces large amounts of selenite to elemental red selenium.
Macy et al. [120,121] isolated two organisms from a site with a high level of selenium; a Thauera selenatis reducing selenite to elemental Se0 and a Pseudomonas species which transforms selenate to selenite as in Eq. (2). Cells grown anaerobically in minimal medium on acetate plus selenate oxidized 14C-acetate to 14CO2 with concomitant reduction of selenate to selenite and small amounts of Se0.
Multiple detoxification processes may occur during selenite reduction by microorganisms, since elemental selenium has been described as being deposited in the cytoplasm of Wolinella succinogenes, D. desulfuricans and Pseudomonas alcaligenes [122,123], in the periplasmic space with E. coli [124], and outside the cell for Clostridium pasteurianum and E. cloacae [125–128]. According to Tomei et al. [123], the particles containing elemental selenium found outside are related to cell lysis, while Losi and Frankenberger [126] suggested that the reduction reaction occurs close to the membrane of E. cloacae, possibly as a result of membrane-associated reductase(s), and that the precipitate is rapidly expelled by a membrane efflux pump. On the other hand, elemental selenium deposited inside or outside cells has been described as being in spherical or spherical to oval-shaped structures [123,126], fibrillar and granular structures [129], amorphous aggregates [127] or selenium nanoparticles [128]. In E. coli, elemental selenium deposition has been observed both in the periplasmic space and in the cytoplasm. Kessi et al. [130] described the ability of Rhodospirillum rubrum to reduce selenite to Se0 and showed that elemental selenium was observed in the cytoplasm and could be expelled across the plasma membrane and the cell wall. R. metallidurans [119] can resist high concentrations of selenite (6 mM) and reduce selenite to elemental selenium, which accumulated essentially in the cytoplasm.
Several microorganisms, among which T. ferroxidans, can transform Se0 into volatile selenide. With selenate and selenite metabolized by R. sphaeroides, Corynebacterium [131], volatile products (as dimethyl selenide or dimethyl diselenide) account for only a small proportion of the accumulated selenium, most of which was present in organic form (selenomethionine) and as the red, elemental Se0 retained in or on the cell.
Several types of bacteria convert selenium compounds to volatile methylated forms and, in soils amended with organic or inorganic selenium, one observes the formation of volatile dimethyl selenide or dimethyldiselenide or of methyl selenite. This volatilization depends on microbiological activity, temperature, hygrometry, weather and the seasonal concentration of soluble selenium [132].
The immobilization of selenite and selenate by reduction to the much less bioavailable red elemental form of selenium may provide a useful approach for remediating selenium contamination [133]. The selenate or selenite bacterial respiration constitutes the major sink for selenium removal in anoxic sediments by sequestrering Se0(s). The dissimilatory reduction of selenate to selenite could be responsible [107] for the relatively high levels of selenite in oxygenated ocean waters.
6.3 Cesium
137Cs accounts for 65% of all isotopes activity in low-level wastes from the reprocessing plant at Sellafield, United Kingdom. 137Cs persisted in a mobile form in the environment after the Chernobyl accident in 1986. It is an alkali metal, easily accumulated by microorganisms; 137Cs reached a variety of trophic levels. Experiments on Cs uptake in E. coli, cyanobacteria, eukaryotic algae and fungi are detailed by Lloyd and Macaskie [56]. But currently 137Cs is decontaminated using zeolithes (aluminosilicates), since microbial biosorbent offers low Cs uptake and no improvement compared to zeolithes.
6.4 Strontium
Biomineralization of 90Sr has been demonstrated after exclusion from P. fluorescens [71] or Citrobacter and precipitation in the growth medium as crystalline strontium carbonate or insoluble phosphate, mediated via phosphatase activity. Several microorganisms can show Sr uptake (Penicillium chrysogenum, Saccharomyces cerevisiae, Rhizopus) [55,134]. Strontium is an alkaline earth metal like radium, from which 226Ra can be biosorbed by Arthrobacter [135] much more than by activated carbon.
6.5 Zirconium (group IV)
93Zr (T½ = 1.5 × 106 a) is present in some nuclear wastes and arises from fission, activation and corrosion of fuel cladding, but is considered to be biologically inert. In unreprocessed fuel, 93Zr activity is about 20% of that of 99Tc. Biosorption has been observed by microalgae and cyanobacteria [136,137].
6.6 Cobalt and nickel (groups IX and X)
60Co (T½ = 5.27 a) is generated by neutron activation. Iron-reducing bacteria are able to reduce from CoIII to CoII. This valence is better absorbed by soils and offers possibility for in situ immobilization in contaminated areas [138].
59Ni (T½ = 7.6 × 104 a) arises from activation of fuel assemblies; microbial interactions have been almost unexplored, but stable nickel has been considerably studied and biosorption is poor with Pseudomonas algae, Citrobacter [70,139].
6.7 Iodine
The halogen 129I (T½ = 1.57 × 107 a) is a key radionuclide from nuclear wastes, with an important environmental impact because of its half-life, bioavailability and mobility. Little is known about its biogeochemistry. The species in the environment are iodide, iodine and iodate. Several microorganisms convert iodide I− to volatile methyl iodide [140]. Enzymatic reduction of iodate (IO3−) to iodide is possible with sulfate and FeIII-reducing bacteria, and also by indirect mechanisms with the reducing compounds produced by bacteria (FeII, sulfide, iron monosulfide FeS) [141,142].
7 Actinides
Microbial interactions with actinides have been discussed in reviews by Banaszak et al. [143] and by Lloyd and Macaskie [56,74,144,145]. If 238U is the priority pollutant in medium and low-level radioactive wastes, other actinides can also be present (230Th, 237Np, 241Pu, 241Am). Actinide chemistry is complicated by the multiple oxidation states of most of the radionuclides, which govern their behavior in the environment. Fig. 4 shows the expected oxidation states for key actinides as a function of redox potential at pH 7.0. The standard redox potential of ferrihydrite/Fe2+ (approximately 0 V) [75] is more electronegative than the potentials for PuV/PuIV, NpV/NpIVand UVI/UIV. Thus FeIII-reducing bacteria have the metabolic potential to reduce these radionuclides enzymatically or via FeII produced from the reduction of FeIII oxides.
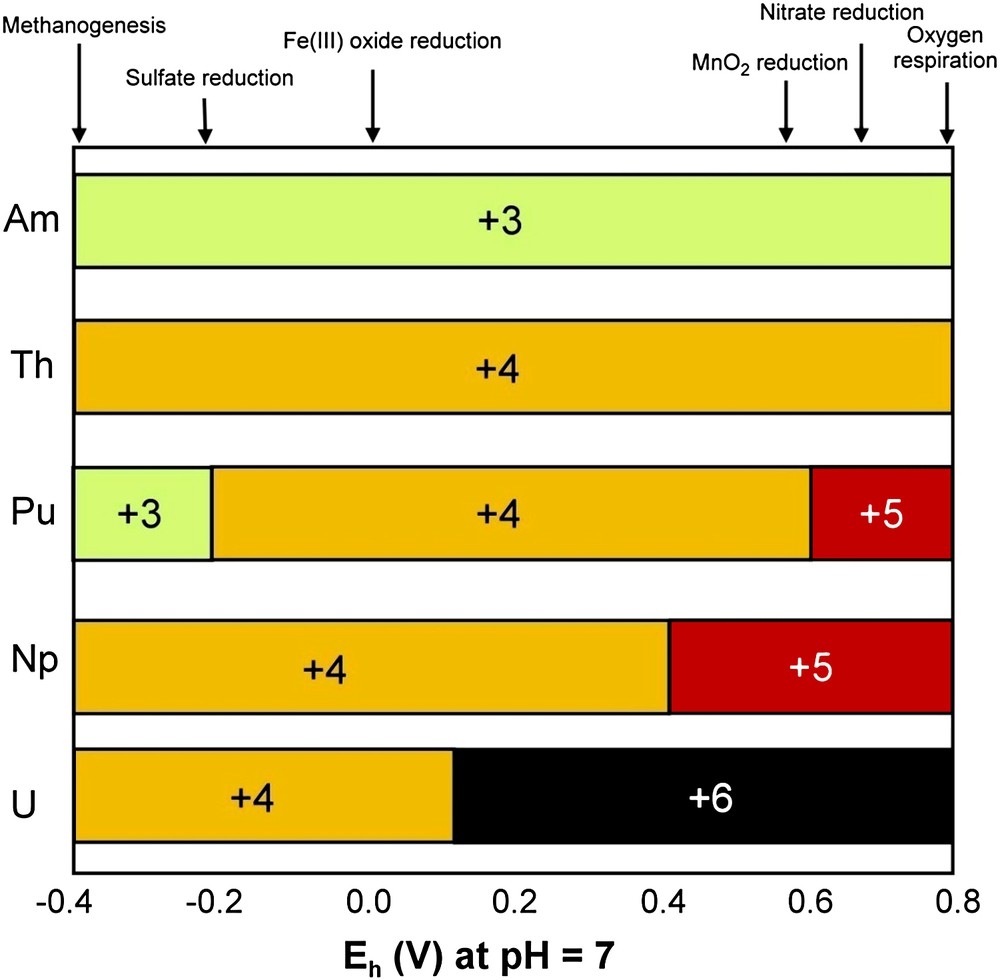
Expected oxidation states for key actinides as a function of redox potential at pH 7.0. Adapted by permission from Springer Verlag UK [143], copyright 1999.
Tetravalent actinides have high ligand complexing abilities [56] and are also immobilized in sediments containing active biomass [146]. FeIII-reducing bacteria are able to transform actinides to cations more amenable to remediation, or to insoluble uraninite (UO2) for the uranyl cation UO22+. This is illustrated by the transformation of the highly soluble NpV (NpO2+), which is reduced to soluble NpIV by Shewanella putrefaciens, with the NpIV removed as an insoluble phosphate biomineral by a phosphate-liberating Citrobacter [147]. FeIII-reducing bacteria can reduce PuIV to PuIII, leading to the solubilization of sediment-bound PuIV, but yield a trivalent actinide that is amenable to bioremediation with several microbially produced ligands [56]. In Fig. 4, one sees that ThIV and AmIII are stable across most Eh values encountered in radionuclide-contaminated waters, but are removed easily through biosorption or biomineralization by several ligands, including biogenic phosphate [56].
7.1 Uranium
Uranium-bearing wastes from nuclear weapons production [148], mining [149], electronuclear production, and nuclear waste treatment have generally been disposed of in near-surface environments. Dispersion of toxic aqueous uranium species through groundwater is of great environmental concern. Consequently, uranium is a widespread groundwater contaminant at many sites where uranium was processed [150]. UVI, the oxidized, soluble form of uranium, is mobile in groundwater. To prevent the spread of uranium contamination in the subsurface, possible methods could involve the reduction of mobile UVI to UIV, which is insoluble and precipitates [151–153]. The proposed reductive immobilization of uranium would be analogous to the formation of some subsurface uranium ores in which UVI dissolved in aerobic groundwater enters an anaerobic zone, enriched in organic matter; UVI is then chemically or biologically reduced to UIV, which precipitates, forming the ore body [1,154]. Thus, uranium-contaminated subsurface environments receiving special enrichment in organic matter, chemical products of bacteria or microorganisms, could be places for precipitation of UIV [155,156]. Although biooxidation of several metals including FeII, MnII, AsIII is well documented [62], microbial oxidation of UIV is not well explained and oxidation of PuIV or NpV is not identified. Ehrlich [62] gives a detailed discussion concerning biosorption of various biomasses for different metals. There are also some examples of microbially-generated mineral deposits that have been known to sorb UVI; for example, ferric (and manganese) oxides formed by a variety of microorganisms can sorb UVI [62], like sulfide minerals, but with concomitant partial reduction of UVI to UIV in this case [157].
Geobacteraceae is frequently present among the natural populations near sites contaminated by uranium. Geobacteraceae have simple nutritional requirements and are able to fix atmospheric nitrogen. Even if abiological mechanisms for UVI reduction can take place involving sulfides, FeII or hydroquinones [158,159], they seem limited compared to the naturally occurring, microbially-catalyzed UVI reduction to UIV in the subsurface [1,160,161].
Both FeIII-reducing and sulfate-reducing microorganisms have been shown to precipitate uranium from contaminated waters [160,162]. The following equations show the reactions of various electron donors coupled to FeIII and UVI reduction under geochemical conditions containing 130 mM HCO3− [163].
| CH3COO− + 15H+ + 8Fe(OH)3 → 2HCO3− + 8Fe2+ + 20H2O |
| CH3COO− + 4H2O + 4UVIO2(CO3)22− → 10HCO3− + 4UIVO2 + H+ |
| CH3COO− + SO42− → 2HCO3− + HS− |
| AH2QDS + UVIO2(CO3)22− → AQDS + UIVO2 + 2HCO3− |
Several other microorganisms can use UVI as an electron acceptor (e.g., Shewanella putrefaciens) and, in anaerobic conditions, acetate or hydrogen is oxidized with the concomitant reduction of UVI to UIV [151].
The black precipitate of UIV as UO2 (uraninite) is very insoluble. Precipitate suspension after UVI reduction indicates an extracellular amorphous mass of electron-dense material. Cells appear in some cases to be coated with enzymetically-reduced uraninite or produce, as for Spear et al. [164], amorphous uraninite particles. These amorphous and finely grained materials with nanometer-sized particles (Fig. 5) aggregated to form micrometer-sized particles easily disassociated from the cells [158]. This electron-dense precipitate is entirely extracellular with Shewanella [165] and with many other organisms [152–154,158–161]. No intracellular uranium was detected [75]. A uranium precipitate is absent from heat-killed suspensions. See an instance of this result in Fig. 6.
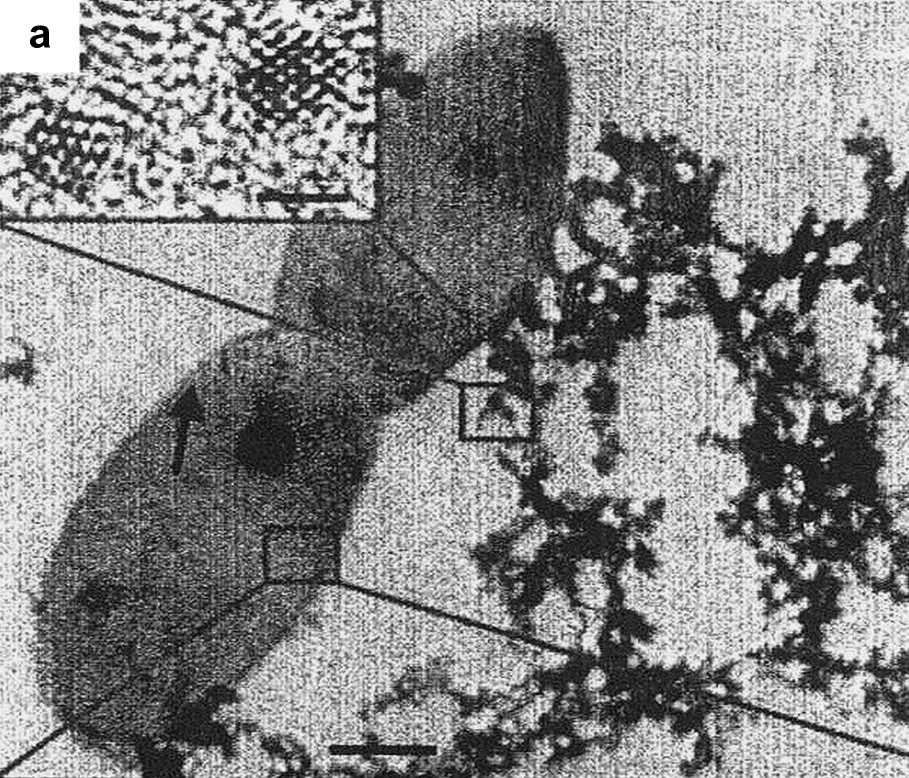
Characterization of bioreduced uraninite (UO2) nanoparticles. Transmission Electron Microscopy (TEM) image of flocculated UO2 nanoparticles associated with Desulfosporosinus spp. bacteria (arrow). Inset: high-resolution TEM image of isolated particles. Reprinted by permission from Macmillan Publishers Ltd [178], copyright 2002.
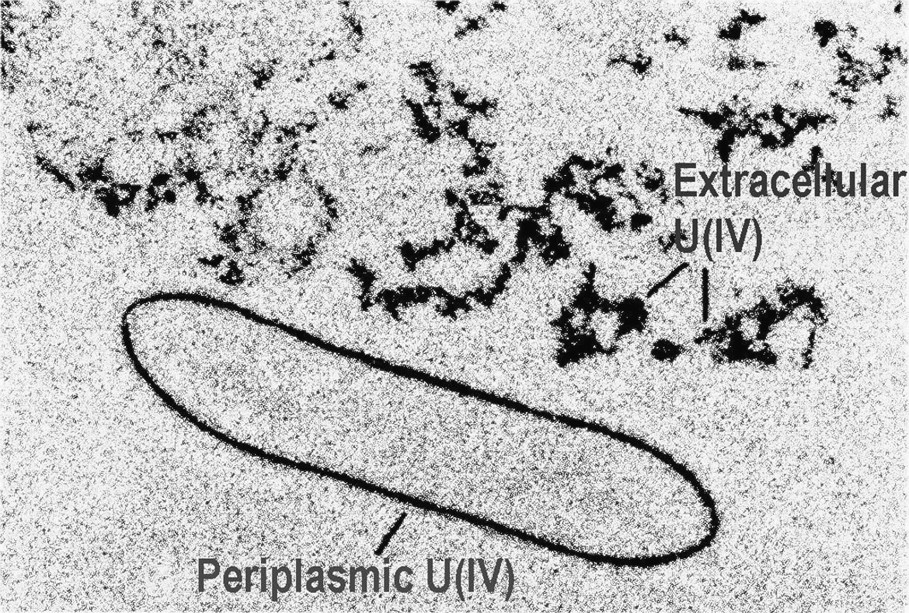
Transmission Electron Microscopy (TEM) showing the U(IV) precipitates formed by G. sulfurreducans via enzymatic reduction of U(VI). TEM of thin sections of cells containing electron-dense U(IV) precipitated outside the cell and within the periplasm. Reprinted by permission from Taylor and Francis Group [75], copyright 2002.
To date Desulfovibrio vulgaris is the only organism in which the enzyme system has been characterized: a tetraheme cytochrome C3 functions as a UVI reductase in combination with hydrogenase, its physiological electron donor [161]. It has been postulated that electron transfer enzymes including hydrogenase are implicated in the UVI reduction of some bacteria. As reduced UIV is often observed in the space between the cytoplasmic membrane and cell wall, it is suggested that the terminal enzyme that donates electrons to UVI is located at the outer surface of cytoplasmic membrane, or in a soluble form [166].
Microorganisms that reduce UVI in pure culture include a hyperthermophilic Archae [167], a thermophilic bacterium [168,169], mesophilic FeIII and sulfate-reducing bacteria [151,160,161,164,170–172] and fermentive bacteria [66].
UVI is reduced by microbes in solutions containing organic or inorganic ligands or other cations [173,174], or that contain other electron acceptors such as FeIII oxides, sulfate, selenate [95,175,176]. A few studies have evaluated the impact of inorganic ions found in waste waters on uranium removal by bioreduction. Generally, high concentrations of bicarbonate, sulfate and nitrate ions appear to negatively impact the enzymatic removal of uranium [174,177]. Moreover, organic ligands influence the bioreduction of uranium. Organic compounds, through carboxyl, hydroxyl and other functional groups, interact spontaneously from coordinate-covalent bonds with metal cations, resulting in organic-metal complexes. Metal availability to assimilatory metal reduction bacteria is often affected by the presence of organic-metal complexes. Ganesh et al. [173] have evaluated four aliphatic ligands (acetate, malonate, oxalate, and citrate) and an aromatic one. The trends in uranium reduction varied with the nature and the amount of UVI-organic complex. Sulfate-reducing bacteria rapidly reduced uranium from a monodentate aliphatic acetate complex. The reduction from multi-dentate aliphatic complexes (malonate, oxalate and citrate) was slower. Uranium from an aromatic complex was rapidly available for reduction. Selection of bacteria for rapid uranium reduction thus depends on the organic composition of the waste streams [179].
7.2 Thorium
Thorium is widely present in the environment at valency IV, so bioreduction is not possible, but, in presence of microorganisms, biosorption may occur. Biosorption of ThIV on fungal masses has indeed been reported by several authors. The cation Th4+ is available only at low pH (∼2). If the pH increases, the sorbed species will not be Th4+, but species with one to four hydroxyl groups. At pH 4, hydrolyzed ThIV forms colloidal Th(OH)4, increasing its uptake by some microorganisms. Andres et al. [180] studied thorium biosorption by Mycobacterium smegmatis at pH 1: the sorbed amount was 4% of the biomass dry weight and comparable to values for UO22+ with that strain. Some thorium removal as an insoluble biogenic phosphate has been reported [181], affording an interesting method for the precipitation of thorium phosphate in presence of NH4+.
7.3 Neptunium
Neptunium-237 is an alpha-emitting radionuclide produced in ton quantities in nuclear reactors. It will eventually predominate in wastes after 104–107 years. It arises via decay of 241Pu (T½ = 14.9 a) and 241Am (T½ = 433 a) to 237Np (T½ = 2.1 × 106 a). Neptunium-V forms complexes with ligands poorly and the α-emitting NpO2+ cation is highly mobile, has high biological toxicity and is very difficult to remove from solution by physicochemical or biotechnical methods [182]. Np uptake is negligible with marine algae and microorganisms. So the solubility of Np does not have a seasonal cycle linked to microbial activity in a West-Cumbrian salt marsh [183], unlike Pu and Am. Nevertheless, biological reduction and removal of NpV can be effected with Shewanella putrefaciens for reduction to NpIV in conjunction with simultaneous precipitation as phosphate, produced by Citrobacter [56]. This preliminary study illustrates the potential for the biotechnological removal of NpV, which is not easily achieved by chemical means.
7.4 Plutonium
Studies on the biogeochemistry of plutonium are far more challenging than those for the other actinides, due to its high radiotoxicity and complex redox chemistry. The most stable oxidation state of Pu is PuIV, but PuIII,V,VI can also be found in the environment. Several studies attempted to understand the potential role of microorganisms in controlling Pu solubility. Peretrukhin et al. [182] utilized a stock solution of 239Pu of 2 mg/ml as the carbonate and added it to natural sediments. All the Pu mass was removed after 4 months, with only 34% of the removal being attributed to biosorption using heat-killed controls, implicating additional biochemical mechanisms. The reduction of PuIV to PuIII was suggested by the authors and could be achieved by FeIII-reducing bacteria. PuIV reduction may lead to solubilization of sediment-bound PuIV, and yield a trivalent actinide that reacts with a range of microbially produced ligands [56].
Pure cultures of Rhizopus arrhizus can cause bioreduction of PuVI and biosorption of PuIV at pH 4 and 9. The removal of PuIV as Pu(OH)4 has been tested with P. aeruginosa immobilized on a plasma-treated polypropylene web [184]; from 1.7 nCi of Pu solution, up to 95% of the Pu was removed, but Pu removal (possibly as PuO2) was particle-size dependent. A metal phosphate accumulating Citrobacter removed 50% of the 238/239Pu from a 60 nM solution [185].
Although microorganisms have the potential to control the solubility of Pu in a range of environments, the precise mechanism of Pu–microbe interactions remains unclear [145].
7.5 Americium
Americium is stable in the trivalent oxidation state for a large range of environmental conditions. Studies of seasonal variations of actinides in water and sediments show that microbes play an important role in controlling AmIII solubility [79]. The summer solubility minima for AmIII correlate with maximal microbial biomass levels.
241Am can be efficiently sorbed onto various algal biomasses at three orders of magnitude greater than for Pu. Biosorption of 241Am by R. arrhizus was 40-fold greater than that for PuIV at pH 2, comparable at pH 4 and less than PuIV at pH 7 [136]. Removal of 90% of 241Am from a solution of 299 nCi/ml was observed with Candida utilis [145].
Enzymatically mediated biodeposition has been observed by Macaskie et al. [185] where phosphate biodeposition of 241Am, with 100% removal from solution, was possible by Citrobacter sp.
Am is the most extensively studied of the trivalent actinides; some data for CmIII show that it is partially removed by sterilized sediments and completely removed after 4 months from natural sediments [146].
8 Conclusion
The release of radionuclides from nuclear weapons' testing and from nuclear power plants in the environment is a subject of intense public concern. Low- and high-level chemical treatment methodologies for fuel reprocessing are well controlled operations, but nevertheless some nuclear wastes have to be stored in surface or subsurface deposits. From these deposits, in long timescale, radionuclides can migrate to the neighboring environment. The fate of an element, be it radioactive or not, depends greatly on microorganisms as it was revealed by the microbial diversity during Earth's history and their chemical effects on valencies of encountered metals(oids). The interaction of numerous microbial species with the key radionuclides coming from the fission of uranium constrains, most of the time, soluble products into non-soluble compounds. These products are easily isolated by man through bioremediation processes or are trapped into the geochemical environment. Their solubility and mobility properties are hopefully to be supervised by enzymatic transformation of radionuclides (biotransformation): uranyl UO22+ ion into UIV as uraninite UO2, pertechnetate TcO4− into TcIV as TcO2, selenite or selenate into elemental selenium Se0 or gaseous forms of selenium. When reduced species are not intrinsically insoluble, appropriate ligands can change that. Therefore microorganisms, some acting under extreme environmental conditions, should help to confine or at least limit the spreading of radioactivity in the environment.
Acknowledgements
The authors hereby wish to acknowledge the academic and financial support for this interdisciplinary research field from: CNRS, “Ministère de l'Éducation nationale, de l'Enseignement supérieur et de la Recherche”, GdR PACE, “Programme international de coopération scientifique” PICS 2730, “Région Aquitaine”, “Université Bordeaux-1”, ANDRA. A special mention goes to Dr Liza Bonch-Osmolovskaya for her collaboration and many fruitful discussions. Many thanks to Dr Arthur MacKenzie Peers for his help with the manuscript.