1 Introduction
Through the development of highly efficient well-defined homogeneous catalysts, olefin metathesis has been recognized as an important reaction to build carbon–carbon bonds whether it is used in fine chemical or polymer synthesis [1–4]. In contrast, classical heterogeneous catalysts based on silica or alumina supported Mo, W or Re oxides have been limited by their incompatibility with functionalized olefins. Because heterogeneous catalysts have some advantages in terms of developing chemical processes (easy separation of products from the metal residue, continuous flow process), a large effort has therefore been devoted at developing supported homogeneous catalysts derived from Schrock or Grubbs type catalysts [5–8]. However, currently none of these supported systems out-performed their homogeneous equivalents. Another approach can also be undertaken, called Surface Organometallic Chemistry, which consists in using silica as a solid siloxy ligand: this has the advantage to heterogenize organometallic complexes [9], but also to activate the metal center [10,11] and to stabilize the system through site isolation so that highly active well-defined olefin metathesis catalysts can be obtained based on Mo, W and Re (Scheme 1) [12–19]. All the characterization data (IR, NMR, EXAFS, DFT calculation) show that well-defined monosiloxy species, with spectroscopic properties very close to their corresponding molecular species, are obtained when prepared by reacting well-defined metal complexes having at least one neopentyl ligand on a silica partially dehydroxylated at 700 °C (SiO2-(700)). Overall, one M–C bond has been cleaved and one M–O bond has been formed during grafting, but detailed mechanistic study of the reaction of [Re(CtBu)(CHtBu)(CH2tBu)2] have shown that a simple electrophilic cleavage of the M–C bond through σ-bond metathesis does not account for the isotopic labelling experiment [20]. Notably, [Mo(NAr)(CHCMe2R)(CH2tBu)2] [21] (1-Me, R = Me or 1-Ph, R = Ph) reacts with alcohols and phenols either via direct σ-bond metathesis or addition of the O–H on the alkylidene double bond depending on the size and the pKa of the reagent [22,23]. In order to gain more insight into the (molecular) property of the isolated silanols present at the surface of silica, we have investigated in detail the grafting mechanism of [Mo(NAr)(CHCMe2R)(CH2tBu)2] (R = Me or Ph) (1-R) on SiO2-(700).
Highly active well-defined olefin metathesis catalysts.
2 Results and discussion
First, grafting of [Mo(NAr)(CHtBu)(CH2tBu)2] (1-Me) on deuterated SiO2-(700) (75% deuterated) gives 1.0 equiv. of 2,2-dimethylpropane, 75 ± 5% monodeuterated. In principle, the deuterated silanol group can react as follows (Scheme 2):
- - in one step, directly via the reaction of the Mo–C σ-bond leading to 2-Me and to 100% mono-deuterated 2,2-dimethyl propane (mechanism i),
- - in two steps, with first the reaction of the MoC leading to 3-Me, followed by its decomposition into 2-Me and a one-to-one mixture of non- and mono-deuterated 2,2-dimethylpropane (mechanism ii),
- - in two steps, with first reaction with the MoNAr leading to 4-Me, followed by its decomposition into 2-Me along with 100% mono-deuterated 2,2-dimethylpropane (mechanism iii).
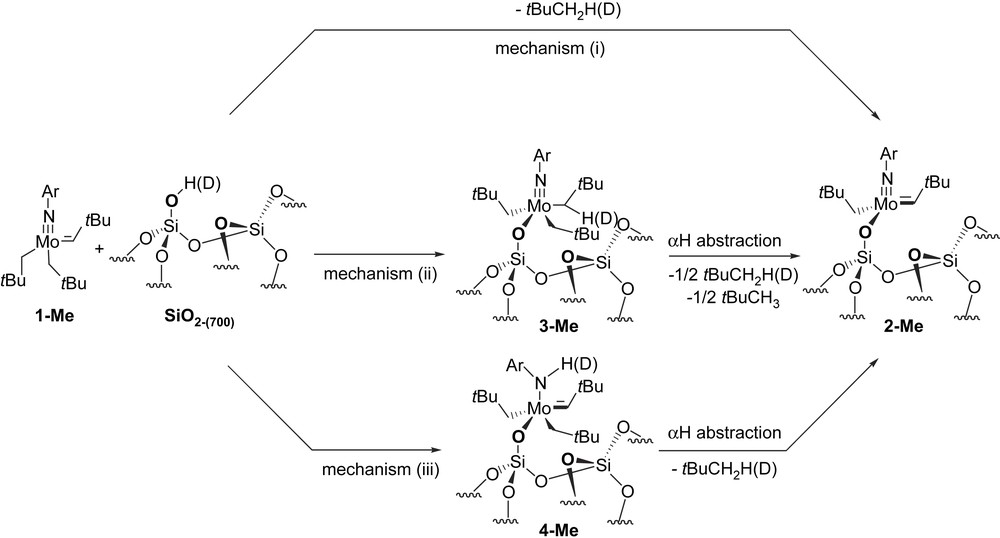
Possible mechanism for the grafting of [Mo(NAr)(CHtBu)(CH2tBu)2] (1-Me) onto a silica or a deuterated silica partially dehydroxylated at 700 °C.
The labelling is therefore consistent with mechanisms i and/or iii, and in order to further understand the grafting mechanism, we have also investigated the grafting of 1-Me selectively labelled on the methylene of the neopentyl carbons [Mo(NAr)(CHtBu)(∗CH2tBu)2] (1∗-Me) and that of the bisneopentyl neophylidene complex (1-Ph).
Firstly, it is worth mentioning that the synthesis of the metal carbon single and double bonds in the preparation of [Mo(NAr)(CHtBu)(CH2tBu)2] (1-Me) are introduced in two different alkylation steps: (1) the MoC double bond is generated during the treatment of [Mo(NAr)2(CH2R)2] with triflic acid (TfOH) in dimethoxyethane (dme), which yields the triflate derivatives, [Mo(NAr)(CHR)(OTf)2(dme)2] [24], and (2) the singly bonded neopentyl ligand is introduced by reaction of the Grignard reagent [tBuCH2MgCl] on the aforementioned triflate derivatives. This strategy has been used to prepare selectively [Mo(NAr)(CHCMe2R)(CH2tBu)2] (R = Ph) and one isotopomer, 1∗-Me, labelled on the neopentyl ligand, [Mo(NAr)(CHtBu)(∗CH2tBu)2]. In the latter case, no H-transfer occurs over a one-month period for a 0.01 M C6D6 solution of 1∗-Me maintained at 25 °C according to 1H NMR [25]. Upon grafting of 1∗-Me on SiO2-(700), 0.9 equiv. of 2,2-dimethylpropane, 93 ± 5% 13C mono-labelled was formed, which is consistent with a selective cleavage of the Mo–C σ-bond. The analytical data on the resulting solid are all similar to those of 2-Me [18], but for the 13C solid-state CP MAS NMR spectrum (Fig. 1), which displays only one intense signal at 56 ppm, corresponding to the 13C labelled methylene carbon of the neopentyl ligand attached to Mo, along with the expected low-intensity signals of the unlabelled carbon of 2-Me. The formation of 13C labelled 2,2-dimethylpropane (93 ± 5%) and the absence of the carbenic signal at 279 ppm [18] (Scheme 3) is consistent with a selective grafting through the electrophilic cleavage of the Mo–C σ-bond (Scheme 2, mechanism i).
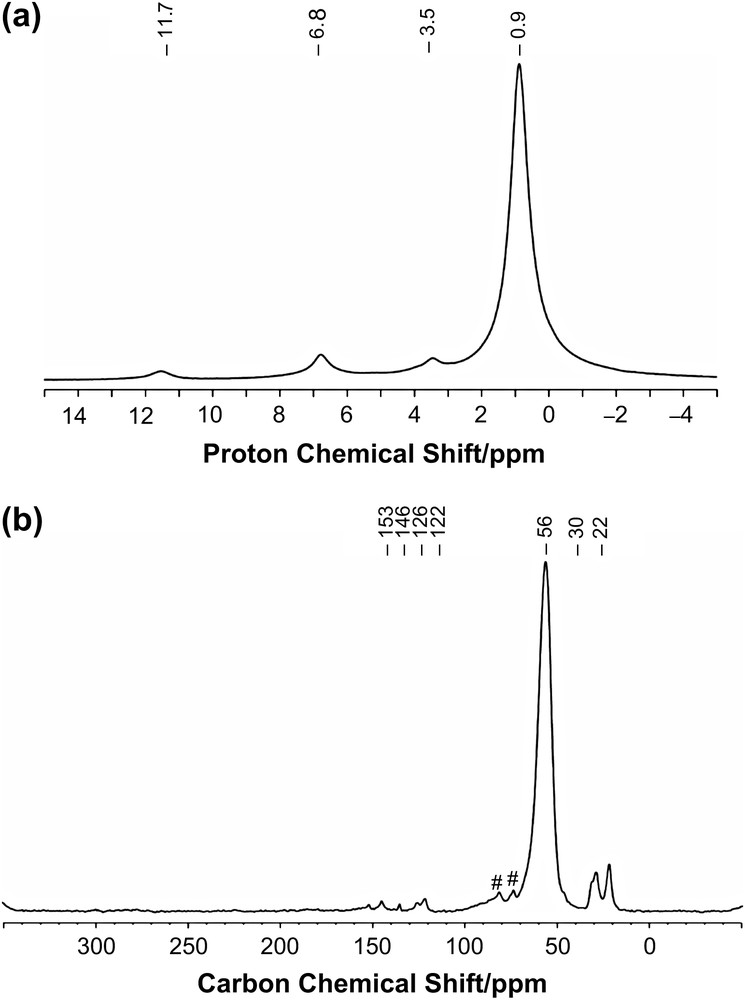
Solid-state NMR spectra of [(SiO)Mo(NAr)(CHtBu)(∗CH2tBu)] (1∗-Me) recorded under MAS at 12.5 kHz. (a) Proton single-pulse spectrum. (b) 13C CP spectrum. The contact time for CP was 1 ms. A total of 20 000 scans was collected. Small impurities are marked with #.
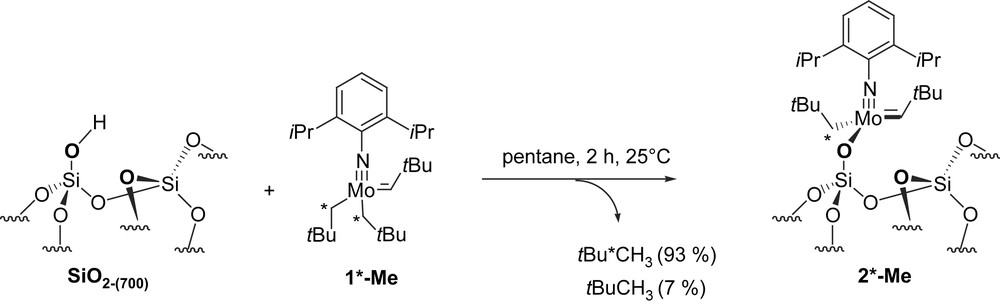
Grafting of 99% 13C labelled on CH2 positions [Mo(NAr)(CHtBu)(∗CH2tBu)2] (1∗-Me) onto a silica partially dehydroxylated at 700 °C.
Secondly, grafting of 1-Ph also gives 0.9 equiv. of 2,2-dimethylpropane per grafted Mo, as observed for 1-Me. Monitoring the reaction by IR spectroscopy shows the disappearance of surface silanols, and the appearance of bands associated with aromatic and alkyl ligands still bound to Mo (Fig. 2). The resulting solid contains 2.14%wt Mo (0.22 mmol/g), which is close to that expected for a quantitative grafting on all surface silanols (maximum density: 0.26 mmol/g), which is consistent with what has been observed by IR spectroscopy. Furthermore, the solid contains 7.27%wt C (27 C/Mo), in agreement with the formation of 2-Ph (expected 27 C/Mo). Moreover, the 1H MAS NMR spectrum (Fig. 3a) displays the following signals: 11.4 (MoCH), 6.9 (aromatic C–H), 3.5 (CHMe2), 2.0 (CH2tBu) and 0.9 ppm (Me) (Table 1), while the 13C solid-state CP MAS NMR spectrum (Fig. 3b) shows signals at 153–122 (Ar), 56 (CH2tBu), 48 (CHC(CH3)3, 30 (CHMe2), 29 (Me, tBu) and 22 ppm (CHMe2) (Table 1), in agreement with the structure 2-Ph (Scheme 4), the alkylidene carbon (MoCH) at 283 ppm being only a weak signal at natural abundance, as previously observed for other complexes [12,13,18,19]. We have also investigated the reactivity of 1-Ph with molecular silanols, [(c-C5H9)7Si7O12Si–OH] (POSS–OH) [26] or [(tBuO)3SiOH]. Monitoring the reaction by solution NMR spectroscopy shows that no intermediate such as 3 or 4 was formed, and that 2m-Ph and 2n-Ph were directly and selectively obtained via σ-bond metathesis according to 1H and 13C NMR spectroscopy (Scheme 4).
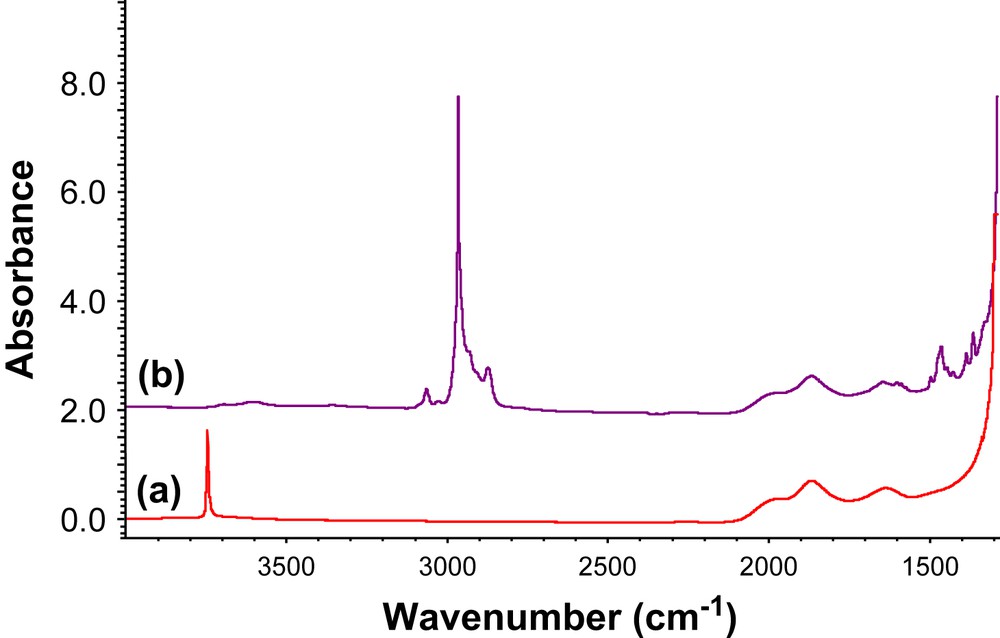
Grafting of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) on a silica partially dehydroxylated at 700 °C, SiO2-(700), monitored by IR spectroscopy. (a) SiO2-(700) pellet (38 mg). (b) After impregnation of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (20 mg) in pentane (12 mL) (2 h, 25 °C), followed by three washings (30 min, 25 °C) in pentane and a drying step under vacuum (1.34 Pa, 1 h, 25 °C).
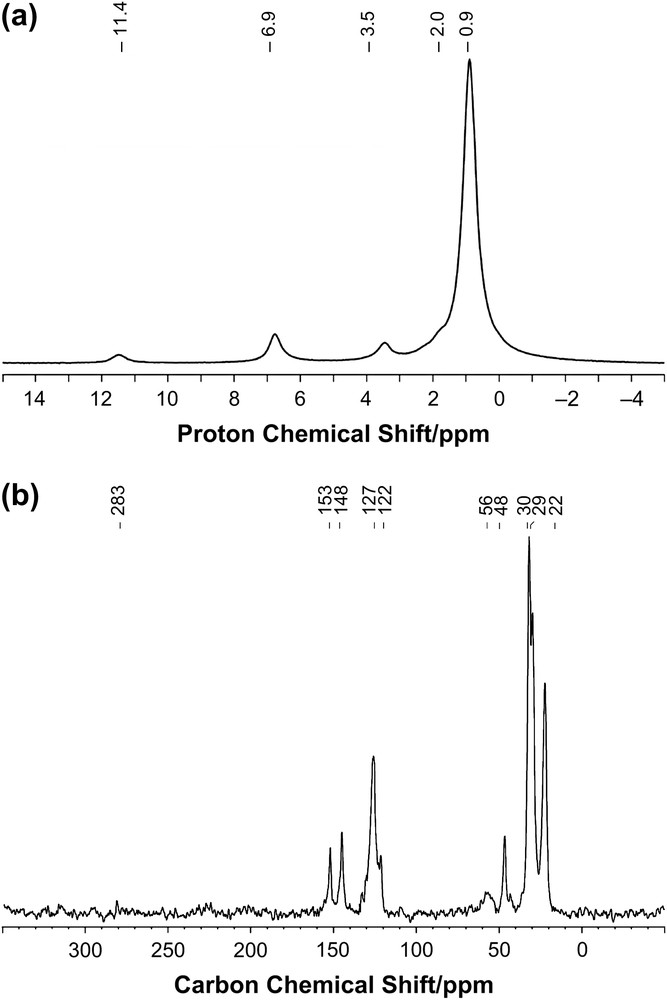
Solid-state NMR spectra of [(SiO)Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2-Ph) recorded under MAS at 12.5 kHz. (a) Proton single-pulse spectrum. (b) 13C CP spectrum. The contact time for CP was 10 ms. A total of 80 000 scans was collected.
Discernable 1H and 13C NMR signals of [(SiO)Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2-Ph), [(POSS)OMo(NAr)(CHCMe2Ph)(CH2tBu)] (2m-Ph) and [(tBuO)3SiOMo(NAr)(CHCMe2Ph)(CH2tBu)] (2n-Ph)
| Complex | 1H NMR (ppm)b | Assignment | 13C NMR (ppm)c | Assignment |
| 2-Ph | 0.9 | CH3 | 22 | CH3, iPr |
| 2.0d | CH2C(CH3)3 | 29 | CHC(CH3)2Ph | |
| 3.5 | CHme2 | 30 | CH, iPr | |
| 6.9 | ArH | 30 | CH2C(CH3)3 | |
| 11.4 | CHC(CH3)2Ph | – | CH2C(CH3)3 | |
| 48 | CHC(CH3)2Ph | |||
| 56 | CH2C(CH3)3 | |||
| 122 | Cmeta | |||
| 127 | Cpara + Ph | |||
| 148 | CriPr | |||
| 153 | NCipso | |||
| 283 | CHC(CH3)2Ph | |||
| 2m-Ph | 1.14–1.25 | c-CH(CH2)4 | 22.6, 22.7 | c-CH(CH2)4 |
| 1.29 | CH(CH3)2e | 23.7 | CH(CH3)2e | |
| 1.30 | CH2C(CH3)3 | 24.2 | CH(CH3)2e | |
| 1.35 | CH(CH3)2e | 27.4, 27.5 | c-CH(CH2)4 | |
| 1.61 | CHC(CH3)2Ph | 27.8, 28.2 | ||
| 1.35–1.96 | c-CH(CH2)4 | 29.5 | CH(CH3)2e | |
| 30.8 | CH(CH3)2e | |||
| 2.32 | CHaHbC(CH3)3 | 31.4 | CHC(CH3)2Ph | |
| 2.59 | CHaHbC(CH3)3 | 33.6 | CH2C(CH3)3 | |
| 3.86 | CHMe2 | 33.7 | CH2C(CH3)3 | |
| 7.04 | ArH | 51.4 | CHC(CH3)2Ph | |
| 7.19 | CHCMe2Ph | 58.4 | CH2C(CH3)3 | |
| 7.33 | CHCMe2Ph | 123.4 | Cpara | |
| 12.19 | CHC(CH3)2Ph | 125.6 | CHCMe2Ph | |
| 125.9 | CHCMe2Ph | |||
| 126.1 | CHCMe2Ph | |||
| 126.5 | Cmeta | |||
| 145.6 | Cortho | |||
| 149.9 | CHCMe2Ph | |||
| 150.6 | CHCMe2Ph | |||
| 152.9 | NCipso | |||
| 278.7 | CHC(CH3)2Ph | |||
| 2n-Ph | 1.29 | CH2C(CH3)3 | 24.1 | CH(CH3)3e |
| 1.31 | CH(CH3) | 24.4 | CH(CH3)3e | |
| 1.39 | OC(CH3)3 | 29.0 | CH(CH3)3 | |
| 1.67 | CHC(CH3)2Ph | 31.8 | CHC(CH3)2Ph | |
| 2.38 | CHaHbC(CH3)3 | 32.3 | OC(CH3)3 | |
| 2.69 | CHaHbC(CH3)3 | 32.8 | CH2C(CH3)3 | |
| 3.92 | CHMe2 | 34.0 | CH2C(CH3)3 | |
| 7.05 | ArH | 53.0 | CHC(CH3)2Ph | |
| 7.18 | CHCMe2Ph | 56.8 | CH2C(CH3)3 | |
| 7.40 | CHCMe2Ph | 72.7 | OC(CH3)3 | |
| 12.46 | CHC(CH3)2Ph | 123.4 | Cmeta | |
| 126.0 | CHCMe2Ph | |||
| 126.2 | CHCMe2Ph | |||
| 126.8 | Cpara | |||
| 145.6 | Cortho | |||
| 149.9 | CHCMe2Ph | |||
| 152.7 | NCipso | |||
| 279.9 | CHC(CH3)2Ph |
b 1H MAS solid-state NMR for 2-Ph 1H solution NMR in benzene-d6 for 2m-Ph and 2n-Ph.
c 13C CP MAS solid-state NMR for 2-Ph 13C{1H} solution NMR in benzene-d6 for 2m-Ph and 2n-Ph.
d Weak signal at natural abundance.
e Not equivalent.
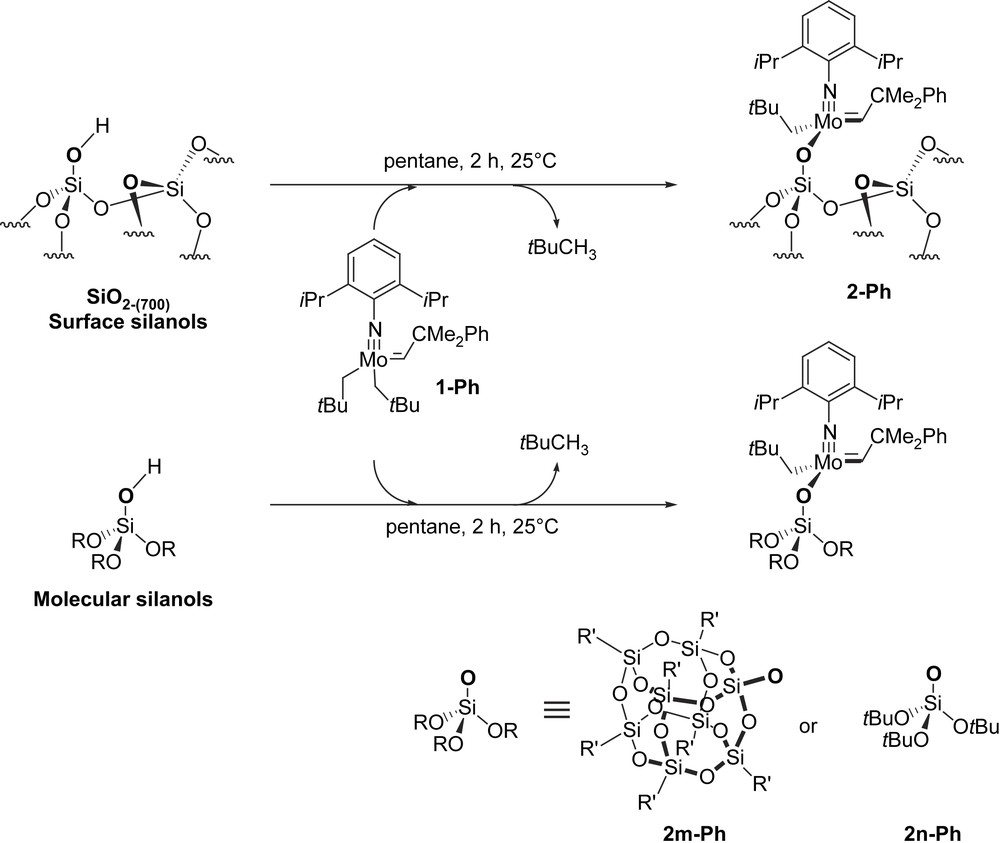
Reaction of 1-Ph with silica and molecular silanol derivatives and formation of the surface complex 2-Ph and its soluble analogues 2m-Ph and 2n-Ph (R′ = c-C5H9).
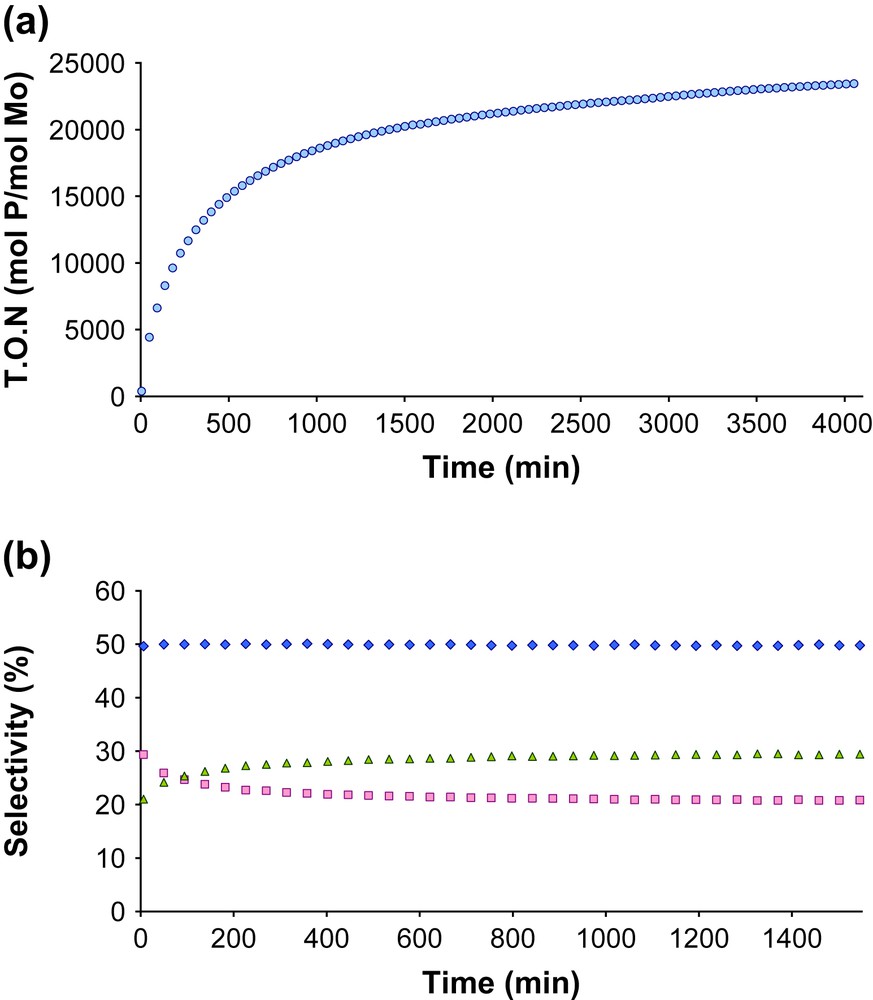
Notably, 2-Me and 2-Ph display very high activity in olefin metathesis. Whether starting from 2-Me and 2-Ph, similar initial rates (1 s−1) are obtained, and they equilibrate 500 equiv. of propene in 20 min. In contrast, while 0.87 equiv. of 3/1 mixture of 2,2-dimethylbutene and Z 3,3-dimethyl-2-pentene, the initiating product resulting from the cross-metathesis of propene with the neopentylidene ligand, are detected for 2-Me, less than 0.02 equiv. is detected for 2-Ph, which is consistent with the formation of 2-Ph (presence of the neophylidene ligand) as the major surface species. We have also tested these catalysts in a flow reactor at 30 °C using propene (320 mL/min). Using 2-Me (49 mg, 2.1 %wt Mo, 1240 mol propene/mol Mo/min), the initial conversion (at 6 min) corresponds to a turnover frequency of 120 mol propene/mol Mo/min, and the productivity decreases with time on stream (deactivation). After 1500 min, ca. 20 000 mol of propene per Mo have been transformed through metathesis (Fig. 4). The selectivities slowly evolve with time1, and after 400 min they are nearly constant: ethylene (50%), E 2-butene (ca. 22%) and Z 2-butene (ca. 28%) along with small amounts of 1-butene (0.3%) and 2-pentenes (0.06%). 1-Butene is probably formed via rearrangement of the intermediate metallacyclobutanes, and the 2-pentenes via cross-metathesis of propene or 2-butenes with 1-butene. The E to Z 2-butenes ratio is 0.8, which shows that the catalyst is slightly Z selective. As observed in the batch reactor, using 2-Ph in place of 2-Me shows exactly the same performances in a flow reactor (similar initial rates, TON and selectivities).
3 Conclusion
Grafting of 1-R (R = Me or Ph) on the surface silanols of SiO2-(700) occurs selectively through the electrophilic cleavage of the Mo–C σ-bond bond to give 2-R according to labelling (GC/MS and solid-state NMR), reactivity studies (monitoring initiation) and molecular models. This shows that surface or molecular silanols have reactivities similar to those of alcohols and phenols having pKa ≥ 9 [23], which is consistent with the proposed pKa for the silanol, ca. 9, determined through the use of POSS–OH [27]. Moreover, we have shown that 2-R are highly efficient olefin metathesis catalysts (R = Me or Ph). These new generations of heterogeneous catalysts already display reactivity and stability greater than their isoelectronic homogeneous equivalents, and further development is currently underway. This also shows that it is now possible to develop well-defined heterogeneous catalysts through a structure–reactivity relationship.
4 Experimental procedures
4.1 General procedure
All experiments were carried out under dry and oxygen-free Ar using either standard Schlenk or glove-box techniques for the organometallic synthesis. For the syntheses and the treatments of the surface species, reactions were carried out using high vacuum lines (1.34 Pa) and glove-box techniques. [tBuCH2MgCl] [28], [Mo(NAr)(CHtBu)(OTf)2(dme)2] [24], (Ar = 2,6-iPr2C6H3) [Mo(NAr)(CHCMe2R)(CH2tBu)2] (1-R) (R = Me or Ph) [22], were synthesized according to literature procedure. 99% 13C labelled [Mo(NAr)(CHtBu)(∗CH2tBu)2] (1∗-Me) was synthesized according to a published procedure from [Mo(NAr)(CHtBu)(OTf)2(dme)2] and [(1-13C 99%) tBu13CH2MgCl] [20]. Silica (Aerosil Degussa, 200 m2g−1) was compacted with distilled water, calcined at 500 °C under air for 2 h and treated under vacuum (1.34 Pa) at 500 °C for 12 h and then at 700 °C for 4 h (support referred to as SiO2-(700)). [(c-C5H9)7Si7O12Si–OH] (Aldrich, 99%) was dried under vacuum (1.34 Pa) at 60 °C for 16 h prior to use. C6D6 (SDS) was distilled from sodium benzophenone ketyl. Pentane and toluene were distilled from NaK under N2. Propene (N, L'Air Liquide) was purified over MS 4 Å prior to use. Elemental analyses were performed at the University of Bourgogne, Dijon (C and N) and at the Service Central Analyse in Solaize (Mo). Gas phase analyses were performed on a Hewlett Packard 5890 series II gas chromatography (GC) apparatus equipped with a flame ionization detector (FID) and a KCl/Al2O2 column (50 m × 0.32 mm). Liquid-phase analyses were performed on a Hewlett Packard 6890 series II GC apparatus equipped with a FID detector and an HP1 column (30 m × 0.32 mm). Products were identified by GC/MS (HP G1800A) equipped with a KCl/Al2O2 or an HP1 column. Liquid-state NMR spectra were recorded in C6D6 using a Bruker AC 300 spectrometer or Bruker DRX-500 spectrometer and referenced to the residual protonated solvent peaks (1H δH = 7.15 ppm, 13C δC = 128.0 ppm). Coupling constants are reported in Hz. The H and C assignments were established by DEPT 135, hsqc and hmbc 2D NMR experiments. All solid-state NMR spectra were recorded under MAS on a Bruker Avance 500 spectrometer with a conventional double resonance 4-mm CP-MAS probe operating at 12.5 kHz of MAS for all the spectra. The samples were introduced in a 4-mm zirconia rotor in the glove box and tightly closed. Chemical shifts are reported in ppm downfield from SiMe4 (± 0.1 and 1 ppm for 1H and 13C NMR spectra, respectively). All the proton spectra were obtained by accumulation of eight scans and a recycle delay of 3 s. All the carbon spectra were acquired under TPPM-15 1H decoupling at ν1H = 83 kHz. The recycle delay was set to 1 s. An exponential line broadening of 80 Hz was applied before Fourier transform.
4.1.1 Preparation of deuterated silica (SiO2-D-(700))
When deuterated silica was used, after partial dehydroxylation under vacuum (1.34 Pa) at 500 °C for 12 h, the silica was exposed to D2O vapor for 10 min at room temperature, followed by reaction at 60 °C for 1 h and partial dehydroxylation at 500 °C for 5 h (4–5 cycles). After partial dehydroxylation at 700 °C for 4 h (support referred to as SiO2-D-(700)), the silica was 75% deuterated according to titration with [tBuCH2MgCl] and quantification by GC/MS of the d0-2,2-dimethylpropane (25%) and d1-2,2-dimethylpropane (75%) released during the reaction.
4.1.2 Grafting of [Mo(NAr)(CHtBu)(CH2tBu)2] (1-Me) on SiO2-D-(700): preparation of [(SiO)Mo(NAr)(CHtBu)(CH2tBu)] (2-Me)
Representative procedure: Grafting of [Mo(NAr)(CHtBu)(CH2tBu)2] (1-Me) (25 mg, 52 μmol) and SiO2-D-(700) (170 mg, 35 μmol SiOD) was performed as described previously [18]. 1H MAS NMR: 11.7, 6.8, 3.5, 0.9. 13C CP MAS NMR: 153, 145, 126, 122, 56, 46, 31, 29, 22. Analysis of the 2,2-dimethylpropane by GC/MS evolved during the grafting gave the following isotopomeric composition: d0-2,2-dimethylpropane (25 ± 5%), d1-2,2-dimethylpropane (75 ± 5%), d2-2,2-dimethylpropane (< 1%).
4.1.3 Preparation of 99% 13C labelled on CH2 positions [(SiO)Mo(NAr)(CHtBu)(∗CH2tBu)] (2∗-Me) by impregnation of [Mo(NAr)(CHtBu)(∗CH2tBu)2] (1∗-Me) onto SiO2-(700)
This compound was prepared from 99% 13C labelled [Mo(NAr)(CHtBu)(∗CH2tBu)2] (1∗-Me) (50 mg, 0.10 mmol) and SiO2-(700) (325 mg, 0.07 mmol SiOH) as previously described [18]. 1H MAS NMR: 11.7, 6.8, 3.5, 0.9. 13C CP MAS NMR: 56 (labelled 13C). Analysis of the 2,2-dimethylpropane by GC/MS evolved during the grafting gave the following isotopomeric composition: C(CH3)4 (7 ± 5%), C(CH3)3(13CH3) (93 ± 5%), C(CH3)2(13CH3)2 (< 5%).
4.1.4 Preparation of [(SiO)Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2-Ph) monitored by in situ IR spectroscopy
Silica (38 mg) was pressed into a 18-mm self-supporting disk, put into a sealed glass high-vacuum reactor equipped with CaF2 windows. After calcination at 500 °C under air for 2 h, the silica disk was treated under vacuum (1.34 Pa) at 500 °C for 12 h and then at 700 °C for 4 h. The silica support thus obtained, referred to as SiO2-(700) (38 mg, 9 μmol SiOH), was then immersed into a pentane (12 mL) solution of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) (20 mg, 37 μmol, 4 equiv.) at 25 °C for 2 h, followed by washing three times with pentane (12 mL) and drying under vacuum (1.34 Pa) at 25 °C for 1 h. IR: 3690, 3606, 3090, 3064, 3026, 2968, 2936, 2913, 2875, 1600, 1587, 1573, 1495, 1463, 1446, 1425, 1385, 1364 cm−1. Elemental analysis: 2.13%wt Mo, 5.89%wt C, 0.40%wt N, 22 ± 3 C/Mo (27 expected), 1.3 ± 0.3 N/Mo (1 expected).
4.1.5 Preparation of [(SiO)Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2-Ph) by impregnation of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) onto SiO2-(700)
Representative procedure: A mixture of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) (164 mg, 0.30 mmol) and SiO2-(700) (1.00 g, 0.26 mmol SiOH) in pentane (15 mL) was stirred at 25 °C for 30 min. After filtration, the yellow solid was washed three times with pentane, and all volatiles were condensed into another reactor of known volume (>6 L) in order to quantify 2,2-dimethylpropane released during grafting. The resulting yellow powder was dried thoroughly under vacuum (1.34 Pa) at 25 °C for 1 h to yield [(SiO)Mo(NAr)(CHCMe3Ph)(CH2tBu)] (2-Ph). Analysis by gas chromatography indicated the formation of 0.24 mmol of 2,2-dimethylpropane during grafting (0.9 tBuCH3/Mo). No tertiobutylbenzene could be detected during grafting by neither gas chromatography nor NMR. 1H MAS NMR: δH: 11.4, 6.9, 3.5, 2.0, 0.9. 13C CP MAS NMR: 283, 153, 148, 127, 122, 56, 48, 30, 29, 22. Elemental analysis: 2.14%wt Mo, 7.27%wt C, 27 ± 3 C/Mo (27 expected).
4.1.6 Synthesis of [(c-C5H9)7Si7O12Si–O–Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2m-Ph)
A suspension of [(c-C5H9)7Si7O12Si–OH] (312 mg, 0.34 mmol) in pentane (10 mL) was added dropwise at 25 °C to a red solution of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) (185 mg, 0.34 mmol) in pentane (10 mL). The color changed from deep red-orange to orange over 30 min at 25 °C. After 4 h at 25 °C, all volatiles were then condensed into another reactor of known volume (>6 L) in order to quantify 2,2-dimethylpropane released during grafting. Analysis by gas chromatography indicated the formation of 0.20 mmol of 2,2-dimethylpropane during grafting (1.0 tBuCH3/Mo). Moreover no tertiobutylbenzene was observed by gas chromatography. The resulting oily solid was extracted with a minimum amount of pentane (2 mL) and an orange powder (279 mg, 0.20 mmol, 59%) crystallised out upon removing the solvent under vacuo. 1H NMR: δH = 12.19 (s, 1H, CHCMe2Ph), 7.33 (d, 2H, CHCMe2Ph, 3JHH = 8 Hz), 7.19 (m, 3H, CHCMe2Ph), 7.04 (m, 3H, ArH), 3.86 (sept, 2H, CHMe2, 3JHH = 6 Hz), 2.59 (d, 1H, CHHCMe3, 2JHH = 13 Hz), 2.32 (d, 1H, CHHCMe3, 2JHH = 13 Hz), 1.96–1.35 (m, 56H, CH2, c-C5H9), 1.61 (s, 1H, CHC(CH3)2Ph), 1.35 (d, 12H, CH(CH3)2, 3JHH = 6 Hz), 1.30 (s, 9H, CH2C(CH3)3), 1.29 (d, 6H, CH(CH3)2, 3JHH = 6 Hz), 1.25–1.14 (m, 7H, CH, c-C5H9). 13C NMR: δC = 278.7 (CHCMe2Ph, 1JCH = 110 Hz), 152.9 (Ar Cipso), 150.6 (Ph), 149.9 (Ph), 145.6 (Ar Cortho), 126.5 (Ar Cpara), 126.1 (Ph), 125.9 (Ph), 125.6 (Ph), 123.4 (Ar Cmeta), 123.2 (Ph), 58.4 (CH2CMe3, 1JCH = 125 Hz), 51.4 (CHCMe2Ph), 33.7 (CH2C(CH3)3), 33.6 (CH2CMe3), 32.3, 31.4 (CHC(CH3)2Ph), 30.8 (CHMe3), 29.5 (CHMe3), 28.2 (CH2, c-C5H9), 27.8 (CH2, c-C5H9), 27.5 (CH2, c-C5H9), 27.4 (CH2, c-C5H9), 24.2 (CH(CH3)2), 23.7 (CH(CH3)2), 22.7 (CH, c-C5H9), 22.6 (CH, c-C5H9). 29Si{1H} NMR: δSi = −65.00 (T3), −65.20 (T3), −65.90 (T3), −102.2 (Q4). Anal. Calcd for C62H115NMoSi8O13: C, 53.49; H, 7.53; N, 1.01; Mo, 6.89. Found: C, 53.33; H, 6.85; N, 0.90; Mo, 7.66.
4.1.7 Monitoring by 1H NMR the formation of [(c-C5H9)7Si7O12Si–O–Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2m-Ph)
Solid [(c-C5H9)7Si7O12Si–OH] (14 mg, 19 μmol) was added to a frozen solution (−30 °C) of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) (8 mg, 19 μmol) in benzene-d6 (0.5 mL) in a J. Young NMR tube and the tube was sealed. The solution was kept in liquid nitrogen to the NMR probe, where it was brought to room temperature. Following the reaction by 1H NMR by monitoring the disappearance of alkylidene and methylene protons of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (2-Ph) and the apparition of the alkylidene and methylene protons of [(c-C5H9)7Si7O12Si–O–Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2m-Ph), and 2,2-dimethylpropane (0.90 ppm) shows that 1-Ph is transformed into (2m-Ph) quantitatively within 30 min. No signal for tertiobutylbenzene was observed.
4.1.8 Conversion [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) into [(tBuO)3Si–O–Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2n-Ph) monitored by 1H NMR
Solid [(tBuO)3Si–OH)] (8 mg, 33 μmol) was added to a frozen solution (−30 °C) of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) (18 mg, 33 μmol) in benzene-d6 (0.5 mL) in a J. Young NMR tube and the tube was sealed. The solution was kept in liquid nitrogen to the NMR probe, where it was brought to room temperature. Following the reaction by 1H NMR by monitoring the disappearance of alkylidene and methylene protons of [Mo(NAr)(CHCMe2Ph)(CH2tBu)2] (1-Ph) and the apparition of the alkylidene and methylene protons of [(tBuO)3Si–O–Mo(NAr)(CHCMe2Ph)(CH2tBu)] (2n-Ph), and 2,2-dimethylpropane (0.90 ppm) shows that 1-Ph is transformed into 2n-Ph quantitatively within 30 min. No signal for tertiobutylbenzene was observed. The following signals were: 1H NMR: δH = 12.46 (s, 1H, CHC(CH3)2Ph), 7.40 (d, 2H, CHCMe2Ph, 3JHH = 8 Hz), 7.18 (m, 3H, CHCMe2Ph), 7.05 (m, 3H, ArH), 3.92 (sept, 2H, CHMe2, 3JHH = 6 Hz), 2.69 (d, 1H, CHHCMe3, 2JHH = 13 Hz), 2.38 (d, 1H, CHHCMe3, 2JHH = 13 Hz), 1.67 (s, 1H, CHC(CH3)2Ph), 1.45, 1.39 (s, 27H, OSi(OC(CH3)3)3), 1.31 (d, 12H, CH(CH3)2, 3JHH = 6 Hz), 1.29 (s, 9H, CH2C(CH3)3), 0.90 (CMe4). 13C NMR: δC = 279.9 (CHC(CH3)2Ph, 1JCH = 116 Hz), 152.7 (Cipso), 149.9 (Ph), 145.6 (Cortho), 126.8 (Cpara), 126.2 (Ph), 126.0 (Ph), 123.4 (Cmeta), 72.7 (OSi(OC(CH3)3)3), 56.8 (CH2C(CH3)3, 1JCH = 118 Hz), 53.0 (CHC(CH3)2Ph), 34.0 (CH2C(CH3)3), 32.8 (CH2C(CH3)3), 32.3 (OSi(OC(CH3)3)3), 31.8 (CHC(CH3)2Ph), 29.0 (CH(CH3)2), 24.4 (CH(CH3)2), 24.1 (CH(CH3)2), 15.2 (CMe4).
4.1.9 Reaction with propene (500 equiv.)
The solid 2-R (R = Me or Ph) (20 μmol) was contacted with propene (500 Torr, 10 mmol, 500 equiv.) in a 370-mL reactor flask at 25 °C. Small aliquots were analysed by GC over time to follow the conversion and the formation of cross metathesis product. Analysis of the gas phase showed, for 2-Me, the formation of 0.65 equiv. of 3,3-dimethylbutene and 0.22 equiv. of 4,4-dimethyl-2-pentene, which are the cross-metathesis products between neopentylidene and propene in contrast to 2-Ph, where only 0.013 equiv. of 3,3-dimethylbutene and 0.003 equiv. of 4,4-dimethyl-2-pentene were obtained under identical reaction conditions. After 20 min, propene was equilibrated (34% conversion) into a 1:1 mixture of ethene and butenes (2.6:1 mixture of trans and cis isomers) in both cases.
4.1.10 Propene metathesis in a flow reactor
Representative procedure: The solid 2-Me (10.7 μmol) was loaded in a flow reactor in the glove box, the isolated reaction chamber was then connected to the propene line, the propene pressure was set to 1 bar, and the tubes were flushed with propene (purified on R-3/11 from BASF and MS 4 Å) for 2 h. Before opening the reaction, the flow rate was set to 320 mL/min (20.7 mol propene/(mol Mo.min), the temperature was set to 30 °C. The opening of the valve corresponds to the beginning of the catalysis and the reaction was monitored by GC using an auto-sampler. The same procedure was used for 2-Ph (12 μmol).
Acknowledgment
F.B. is grateful to the French Ministry of Education, Research and Technology (MENRT) for a graduate fellowship. A.S. thanks BASF AG for a graduate student fellowship. The authors thank N. Legagneux, A. Puissegur and L. Weinberg (CPE project students) for their participation in part of this work. We are indebted to the CNRS, ANR (ANR JC05_46372), ESCPE Lyon and the National Science Foundation for financial supports.
1 Because the catalyst deactivates, the number of active sites decreases, hence the contact time by active sites changes so that the selectivity changes and the observed butene ratio tends towards the kinetic ratio.