

1 Introduction
Paramagnetic polyoxometalates (POMs) with sandwich structure [MnM′m(H2O)n+m−2(P2W15O56)2](24−2n−2m)− with M, M′ = Mn, Co, Ni, … are characterized by the clustering of transition-metal (TM) cations, which make them interesting in different fields such as catalysis and electrocatalysis, magnetochemistry and medicine [1–4]. Indeed, the properties of these materials can be modulated by substitution of metallic elements. Therefore, precise knowledge of their structure is important to tune the POMs' properties. The structural characterization of these paramagnetic polyoxometalates is essentially made, in solution, by 31P NMR spectroscopy and, in the solid-state, by IR spectroscopy and possibly by single-crystal X-ray diffraction. To the best of our knowledge, no solid-state 31P NMR of this type of species has ever been reported, although solid-state NMR is in principle able to deliver more structural information, i.e. tensorial interactions, than solution NMR can do.
In the homometallic complexes [M4(H2O)2(P2W15O56)2]16− (M2+ = Mn2+, Co2+, Ni2+, Zn2+ …), the four transition metals (two internal and two external) are arranged in a sheet sandwiched between two [P2W15O56] (shortly {P2W15}) subunits (Fig. 1a) [5–8]. The whole anion is relatively symmetrical (C2h symmetry) with the two {P2W15} moieties being equivalent. So, the two phosphorus atoms close to the metallic centres, labelled P(1), are equivalent like those far from the metallic centres, labelled P(2). Consistently, only two signals are observed by 31P NMR in solution: for the paramagnetic species, e. g. [Co4(H2O)2(P2W15O56)2]16−, one resonance appears at high frequency (δ = +1483 ppm), attributed to the two equivalent P(1) nuclei, whereas the second one, attributed to the two equivalent P(2) nuclei, is observed at relatively low frequency (+9.9 ppm) [9]. On the other hand, in the case of mixed-metal sandwich complexes, i.e. when the metallic sheet contains two types of TM cations, or for “lacunary” sandwich complexes, i.e. when the metallic sheet contains less than four TM cations, the whole anion is generally less symmetrical (Cs symmetry) and the four P atoms may become non-equivalent (Fig. 1b). Hence, the 31P solution NMR spectra of mixed-metal cobalt complexes [MCo3(H2O)2(P2W15O56)2]17− (M2+ = Mn2+, Ni2+, Zn2+ and Cd2+) exhibit four NMR signals assigned to the four non-equivalent P(1) and P(2) atoms [10]. Similarly, the 31P solution NMR spectrum of the dissymmetrical tricobalt complex αβ-[Co3Na(H2O)2(P2W15O56)2]17− presents also four well-separated resonance signals covering a very large spectral range (nearly 2000 ppm) [11]. While solution NMR gives only isotropic chemical shifts, it can be interesting to obtain additional structural information from solid-state NMR of these paramagnetic compounds.
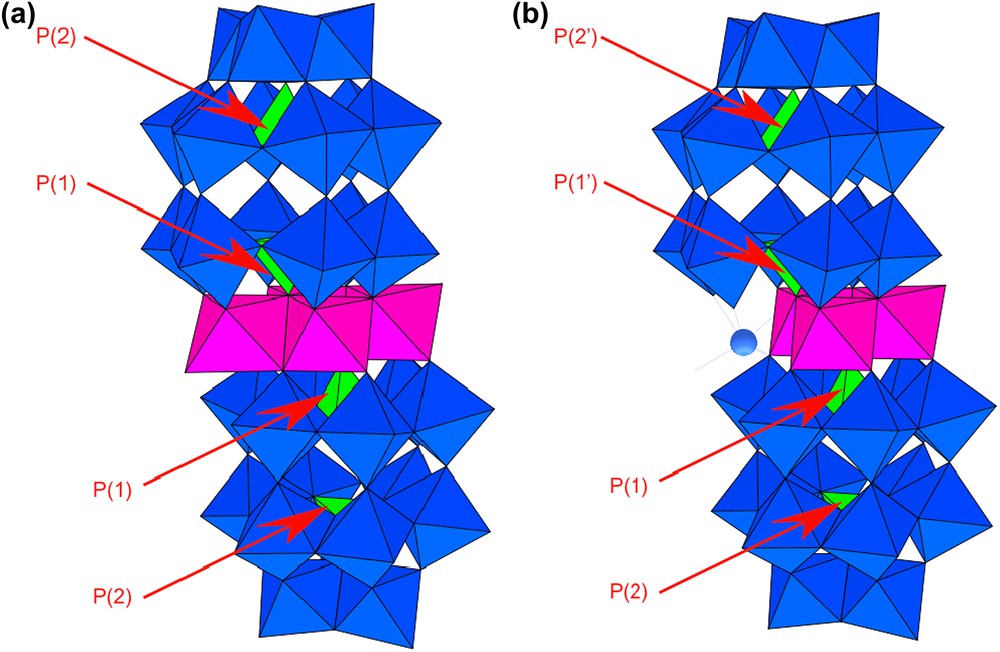
Polyhedral representation of (a) the saturated sandwich complex [Co4(H2O)2(P2W15O56)2]16− and (b) its lacunary derivative [Co3Na(H2O)2(P2W15O56)2]17−.
The aim of this paper is to show the complementarity of solution and solid-state 31P NMR techniques to analyze paramagnetic sandwich complexes; for this purpose we have chosen the “lacunary” cobalt complex αβ-[Co3Na(H2O)2(P2W15O56)2]17− because the presence of the paramagnetic Co2+ cation induces dramatic shifts of the 31P isotropic signals without strong incidence on the line width. In the following we present preliminary results obtained on this compound by MAS NMR and we discuss on the technical problems encountered with the high spinning speeds required. We present also variable temperature 31P NMR experiments, as illustrated with a simpler paramagnetic POM α2-[Ce(III)(P2W17O61)]7−.
2 Experimental
2.1 Preparation of αβ-[Co3Na(H2O)2(P2W15O56)2]17− and α2-[Ce(III)(P2W17O62)]10−
The dissymmetrical tricobalt complex αβ-[Co3Na(H2O)2(P2W15O56)2]17− and the lanthanide complex α2-[Ce(III)(P2W17O61)]7− were prepared according to the methods described in the literature [11,12].
2.2 NMR spectroscopy
Solution and solid-state 31P NMR spectra were recorded at room temperature and variable temperature on Bruker Avance 300 and Avance 400 spectrometers operating, respectively, at 7.1 and 9.4 T (Larmor frequencies 121.5 and 161.9 MHz, respectively). A 85% H3PO4 solution was used as a chemical shift reference.
The solution spectra were obtained from ca 0.02 mol/L solutions in unbuffered D2O/H2O (1:1). The one-pulse 31P 161.9 MHz MAS NMR spectra were recorded at spinning speeds of 12 and 34 kHz. A 0.5 μs pulse (π/9) was used with 10 ms recycling time.
The 31P echo–MAS NMR spectra recorded at 9.4 T were acquired at different spinning rates (from 30 to 35 kHz), with 30 ms repetition time. The first and second pulse durations were 2.6 (π/2) and 5.2 μs (π), respectively, and the delay between the pulses was calculated for synchronization with the rotor frequency (between 25 and 29 μs). For recording 7.1 T echo–MAS NMR spectra, the spinning speed was 29 kHz, the first and second pulses were 0.5 μs and 1 μs, respectively, and the recycling delay was 60 ms. In both cases, the recycling delay is sufficient to allow relaxation of the P(1) nuclei, while the P(2) resonances become more or less saturated.
3 Results and discussion
3.1 31P NMR of αβ-[Co3Na(H2O)2(P2W15O56)2]17−
The “lacunary” tricobalt complex αβ-[Co3Na(H2O)2(P2W15O56)2]17− (shortly {Co3P4W30}) derives formally from [Co4(H2O)2(P2W15O56)2]16− (shortly {Co4P4W30}) by removal of one external Co2+ cation from the tetrametallic sheet, the resulting lacuna being occupied by a Na+ cation. [13]. In this dissymmetrical species (Cs symmetry), the two {P2W15} moieties connected to the metallic sheet are non-equivalent and they present four types of non-equivalent phosphorus atoms subdivided into two groups: the P atoms in the PW6 subunits close to the paramagnetic centres, labelled P(1) and P(1′), and those in the PW9 subunits far from the paramagnetic centres, labelled P(2) and P(2′) (Fig. 1b).
The 31P solution spectrum of the tricobalt complex αβ-[Co3Na(H2O)2(P2W15O56)2]17− is shown in Fig. 2. The two relatively narrow resonances of equal intensity at −10 and +21 ppm (fwhm = 110 Hz, 0.7 ppm; Fig. 2b) are attributed to P(2) and P(2′) atoms far from the cobalt atoms, while the two broader resonances at +1136 (fwhm = 560 Hz, 3.5 ppm) and +1692 ppm (fwhm = 730 Hz, 4.5 ppm; Fig. 2c) are assigned to P(1) and P(1′) atoms, which are characteristic of phosphorus nuclei close to the cobalt nuclei. The positions of the signals as well as their line widths reflect the different influence of the paramagnetic Co2+ cations. Actually, for both P(2) and P(2′), their relative positions with respect to the trimetallic sheet are similar with a rather large closest P–Co distance (>7 Å) resulting in moderate paramagnetic frequency shifts and broadening. The situation is more contrasted for P(1) and P(1′): while P(1), in the PW6Co3 subunit, is nearly equally distant from the three cobalt centres (dP–Co ca 3.3 Å), P(1′), in the PW6Co2Na subunit, is far from the external Co atom (dP–Co ca 4.5 Å). Therefore, we may tentatively assign the broadest and the most shifted signal (+1692 ppm) to P(1) and the less shifted one (+1136 ppm) to P(1′).
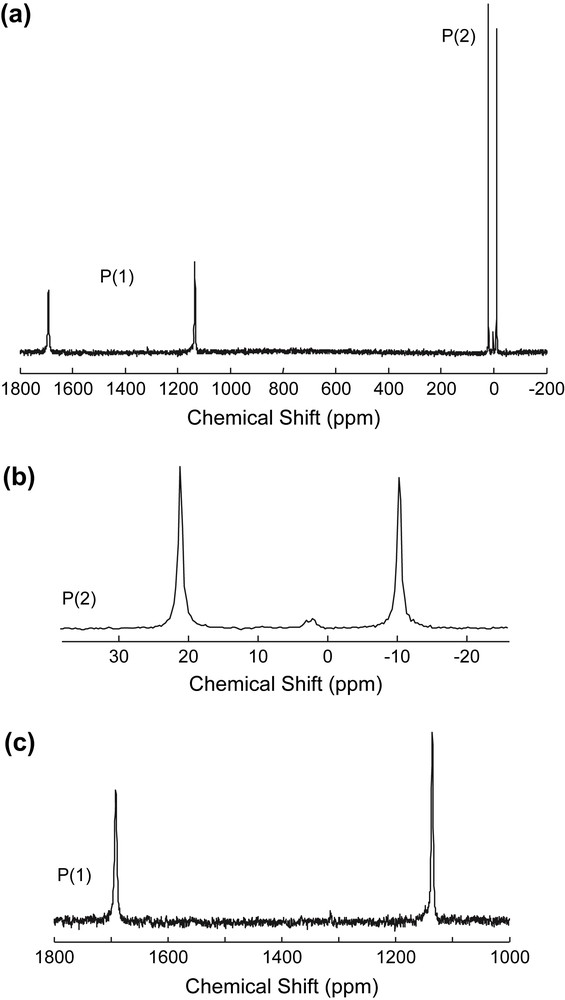
31P NMR spectrum (9.4 T) of a ca 0.02 mol/L solution of [Co3Na(H2O)2(P2W15O56)2]17− in unbuffered D2O/H2O (1:1): full spectrum (a) and abscissa expansion of P(2) region (b) and P(1) region (c).
In order to obtain supplementary data, in particular the chemical shift tensors, solid-state 31P NMR spectra of the tricobalt complex were recorded (Figs. 3–5). Fig. 3a shows the wide-range one-pulse 31P MAS NMR spectrum of this complex obtained at 34 kHz spinning rate. This spectrum presents clearly two overlapping subsets of spinning-side bands (SSB): only two SSB are observed around the two intense signals around 0 ppm, while the second subset of low-intensity SSB extends over ca 3000 ppm (nearly 500 kHz at 162 MHz). Because of phasing errors due to the large spectral width, the spectrum is of very poor quality, which does not allow us to extract useful parameters for the second SSB subset. Fig. 3b presents the one-pulse 31P MAS NMR spectrum acquired at lower spinning rate (12 kHz) with a reduced spectral width. The two signals of P(2) and P(2′) present well-separated SSB patterns from which we are able to determine both the isotropic chemical shifts and the chemical shift anisotropy: δiso = −30 ppm, CSA ∼ 500 ppm and δiso = −46 ppm, CSA ∼ 700 ppm. It should be noted that the chemical shift values differ notably from those obtained in solution (+21 and −10 ppm); this may be due to bulk magnetic susceptibility effects and/or to additional paramagnetic interactions between neighbouring complexes in the crystal.
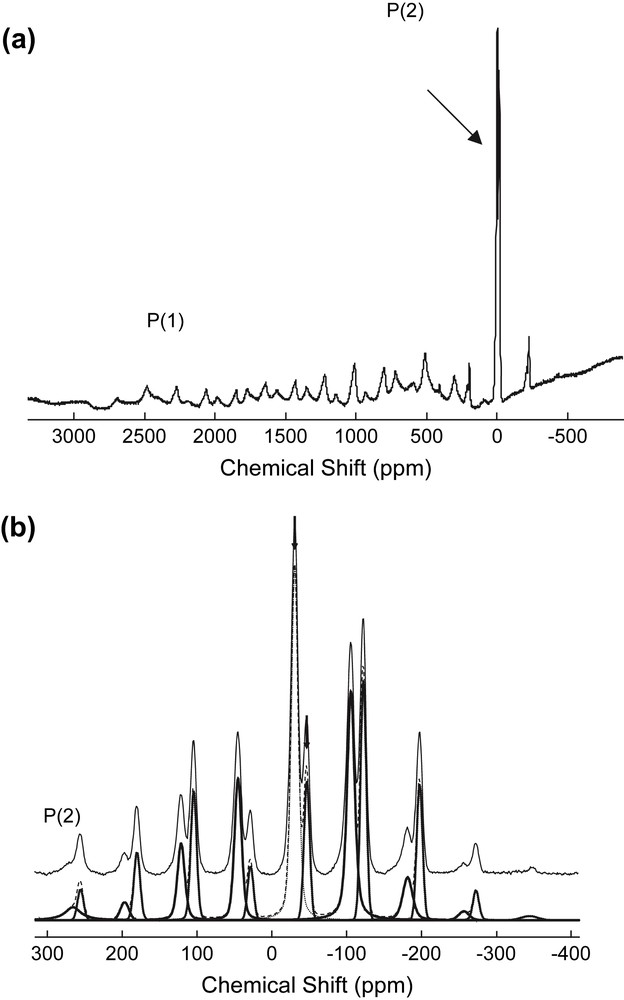
31P MAS NMR spectra (9.4 T) of [Co3Na(H2O)2(P2W15O56)2]17− recorded at a spinning speed of (a) 34 kHz and (b) 12 kHz (P(2) region only). Arrows indicate the position of the isotropic resonances. For the decomposition of the P(2) subspectrum, dotted lines and full line represent calculated and experimental spectra, respectively.
31P echo–MAS NMR spectra of αβ-[Co3Na(H2O)2(P2W15O56)2]17− recorded at (a) 9.4 T at a spinning speed of 34 kHz and (b) 7.1 T at a spinning speed of 29 kHz.
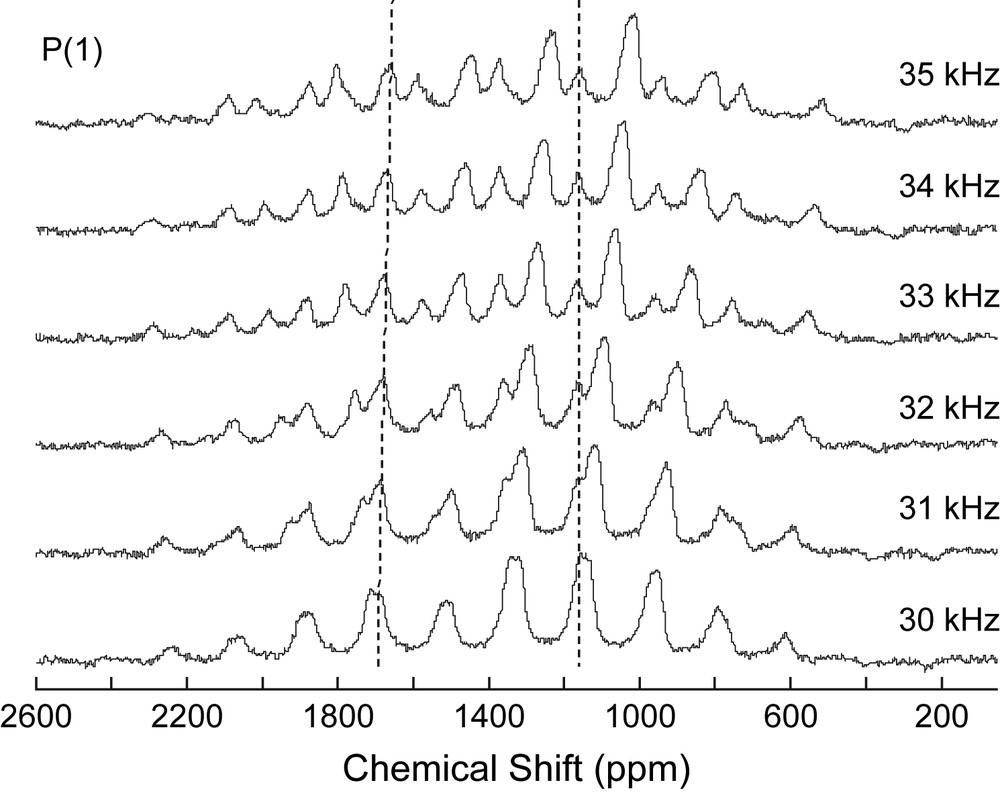
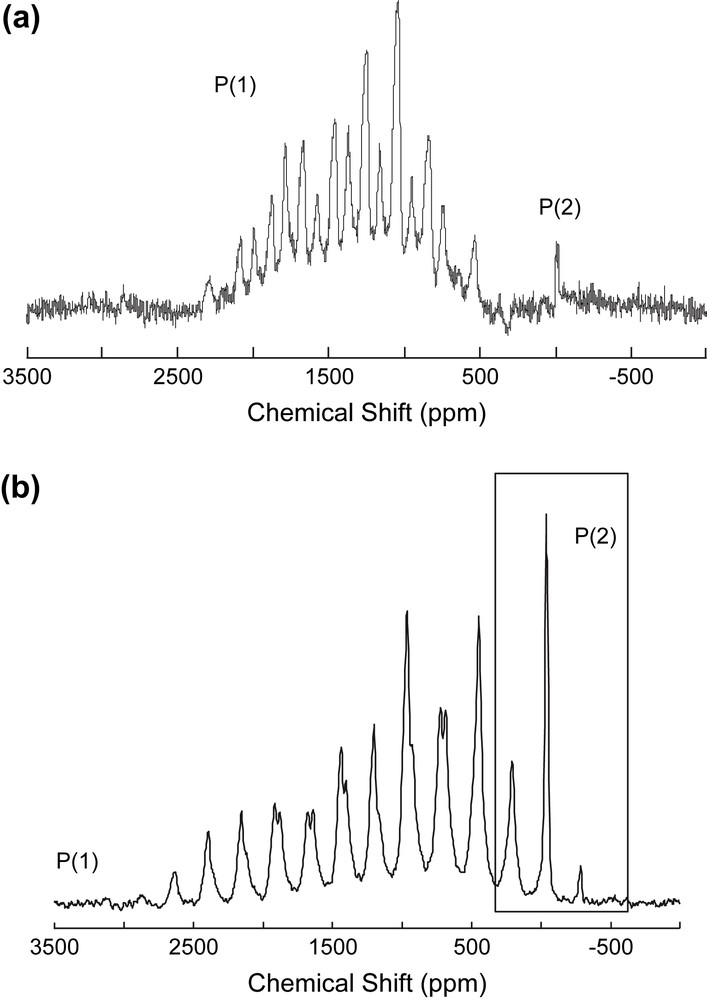
31P echo–MAS NMR spectra (9.4 T) recorded at different spinning rates (30–35 kHz) of αβ-[Co3Na(H2O)2(P2W15O56)2]17−. Dotted lines indicate the position of the isotropic resonances.
As expected, 31P NMR spectra recorded under echo–MAS conditions (Fig. 4) do not present the phasing errors of the single-pulse MAS spectrum. For the 9.4 T spectrum, the very short recycling delay (30 ms) leads to saturate the more slowly relaxing P(2) resonances (T1 ca 1 s), which become nearly extinguished. In principle the separation between the P(1) and P(1′) SSB patterns will be sufficient to determine at least coarse values of the CSA of both nuclei; however, spreading of the SSB is obviously far beyond the range anticipated from the single-pulse MAS spectra (Fig. 3a). Actually the relatively long π pulse (5.2 μs) does not allow complete excitation over the bandwidth covered by the SSB. This was also confirmed by changing the offset of the excitation pulse, which leads to different experimental spectra (not presented here). Therefore it was impossible to access directly the CSA parameters from these spectra.
The problem of defective excitation may be partially circumvented by working at lower field, which reduces the effective required spectral width by the Larmor frequencies ratio. The echo–MAS spectrum obtained at 7.1 T (125 MHz) and at a rotor speed of 29 kHz is presented in Fig. 4b. The SSB patterns of the P(1) and P(1′) resonances extend approximately over the same range as for the single-pulse MAS spectrum (Fig. 3a). Note also that, while still saturated, P(2) and P(2′) nuclei give rise to signals considerably more intense than that at 9.4 T. Better excitation at 7.1 T originates, not only from the narrower spectral width, but also from the shorter pulses allowed on this spectrometer (π pulse of 1 μs). Despite the better quality of the resulting spectra, they suffer from overlap of the SSB patterns: actually the resonances from both P(1) and P(1′) are hardly resolved for some bands only and, due to the more uniform excitation of the spectral width, the P(2) and P(1) subspectra are no longer fully separated. Nevertheless a rough value of ca 3300 ppm may be estimated for the chemical shift anisotropy of both P(1) and P(1′) nuclei. Concerning their isotropic chemical shifts, no direct measurement may be done by varying the spinning speed, because of temperature effects (see below); they can be evaluated by comparison with the 31P NMR solution data at δiso ∼ +1700 and ∼+1160 ppm for P(1) and P(1′), respectively.
Although the data obtained for the four non-equivalent P nuclei are not accurate, they represent the first complete set of parameters for such a type of paramagnetic species; in particular the very large CSAs of both P(1) and P(1′) and the moderate ones for both P(2) and P(2′) agree with the different relaxation rates observed in solution as well as in the solid state.
3.1.1 Temperature effects
It is well known that at high spinning speeds, the temperature of the external wall of the rotor increases and may reach up to 80 °C. In addition to this effect of mechanical nature, paramagnetic species may experience electromagnetic interactions capable also to modify the temperature of the sample. Moreover depending on the design of the probehead and on the thermal properties of the sample, more or less severe temperature gradients may occur inside the sample [14]. As NMR signals of nuclei in the vicinity of paramagnetic centers are very sensitive to the temperature, the spectra of paramagnetic species may be dramatically affected by the experimental conditions.
Solid-state 31P echo–MAS NMR spectra of αβ-[Co3Na(H2O)2(P2W15O56)2]17−) have been recorded at 9.4 T and at different spinning rates (30–35 kHz) in order to show the effect of temperature on the isotropic chemical shifts of the P nuclei close to the metallic centers (Fig. 5). This figure shows that the two subsets of spinning-side bands shift differentially: while these subsets are completely separated at 34 kHz, they are nearly overlapping at 30 kHz. Moreover, none of the signals remains strictly at the same chemical shift in all spectra; this precludes to locate the isotropic resonance of the P(1) and P(1′) nuclei. It is worth noting that, on the contrary, in the P(2) subset, the positions of the intense isotropic signals do not significantly vary by changing the spinning rate.
From these observations we can reach the following conclusions: (i) the shifts of P(1) and P(1′) resonances, likely due to a mean temperature increase of the sample by increasing spinning rate, agree with the close proximity of these nuclei to the paramagnetic centers; (ii) one of these two nuclei, namely that whose isotropic chemical shift was given the value of ∼+1700 ppm (see above) is more affected than the second one (δiso ∼ +1160 ppm), this may be consistent with the assignment of the +1700 ppm signal to P(1), which has three Co2+ cations as close neighbours.
3.2 31P NMR thermometer
In order to interpret the effects observed on the solid-state 31P NMR spectra recorded at different spinning speeds, it is essential to determine the mean temperature as well as the temperature gradient in the rotating sample. For the purpose of measuring directly the temperature of the NMR sample, a number of NMR thermometers have been proposed in the literature.
Lead nitrate is commonly used for solid-state NMR [14–17]; it is based upon the large temperature dependence of the chemical shift of the heavy 207Pb nucleus.
For solution NMR, methanol and ethylene glycol are considered as standards for use at low and high temperatures, respectively; in that case one makes use of the different temperature dependence of δ (1H) for the OH groups and the aliphatic protons [18]. Alternatively the great sensitivity of the cobalt-59 nucleus and the large temperature dependence of δ (59Co) (up to 3 ppm/K) has allowed one to propose two diamagnetic highly symmetrical cobalt(III) complexes, namely K3Co(CN)6 and Co(acac)3, as useful thermometers for multinuclear solution NMR spectroscopy [19]. For 13C NMR, in acetic acid–acetate buffer, the paramagnetic dysprosium nitrate induces paramagnetic shifts for the acetate resonances; the chemical shift difference is temperature dependent and for well-defined experimental conditions, the temperature sensitivity of Δδ has been reported to ca 4 Hz/°C (at 15 MHz), i.e. ca 0.3 ppm/°C [20].
In transition-metal substituted phosphorous-centered POMs, the paramagnetic shifts of the 31P resonances and also their sensitivity to temperature variations increase as the P–M distances decrease. Therefore, the chemical shift difference Δδ (31P) for two phosphorous nuclei not equidistant from the paramagnetic transition-metal cation should represent a measure of the sample temperature. As 31P NMR of POMs may be obtained easily in solution and the solid state, POMs can be considered as good candidates for 31P NMR thermometers in both states.
For these preliminary tests we have chosen a relatively simple monosubstituted cerium(III) POM complex, namely [Ce(III)(P2W17O61)]7− with only two non-equivalent phosphorous atoms (see Fig. 6) [12]. The choice of Ce(III) as substituting paramagnetic metal was directed by its moderate incidence on the 31P relaxation, leading to exceptionally narrow resonances as shown in Fig. 6.
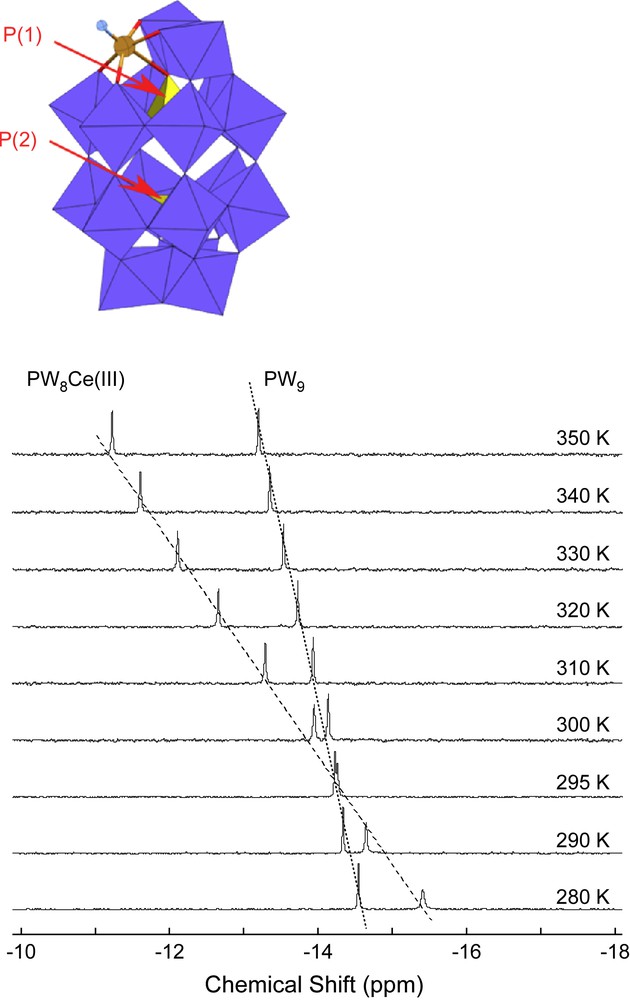
Schematic polyhedral representation of the paramagnetic POM α2-[Ce(III)(P2W17O61)]7− and variable temperature 125 MHz 31P NMR solution spectra of α2-[Ce(III)(P2W17O61)]7−.
31P spectra recorded in solution between 280 and 350 K exhibit two signals of same integrated intensity, as expected from the structure (Fig. 6). Whatever be the temperature, the two signals are, however, of unequal width, which indicates different relaxation rates. Increasing the temperature leads to a shift of both resonances to high frequency, with the highest shift observed for the broadest one. This is consistent with a closer proximity of the paramagnetic atom; this last resonance was therefore assigned to the phosphorous nucleus P(1), belonging to the PW8Ce(III) subunit [21]. Although the chemical shifts δP(1) and δP(2) and consequently Δδ (Δδ = δP(1) − δP(2)) do not vary strictly linearly with the temperature, their variations remain monotonous and Δδ, with a mean variation of ca 5 Hz/K, can be used for temperature measurement for 31P NMR spectroscopy. Further experiments are in progress to quantify more precisely the calibration curve.
While [Ce(III)(P2W17O61)]7− appears as a suitable NMR thermometer for 31P solution NMR, it is, however, not the case for solid-state experiments. Actually, the solid-state 31P spectra of the hydrated potassium salt of [Ce(III)(P2W17O61)]7−recorded between 300 and 350 K present two broad partially-overlapped main bands (Fig. 7). Contrary to what is observed in solution, the temperature variations are nearly the same for both signals and therefore their chemical shift differences do not vary sufficiently, as compared with their accuracy, to be a reliable temperature measurement. Moreover, some additional signals are present at higher temperature, likely from less hydrated samples.
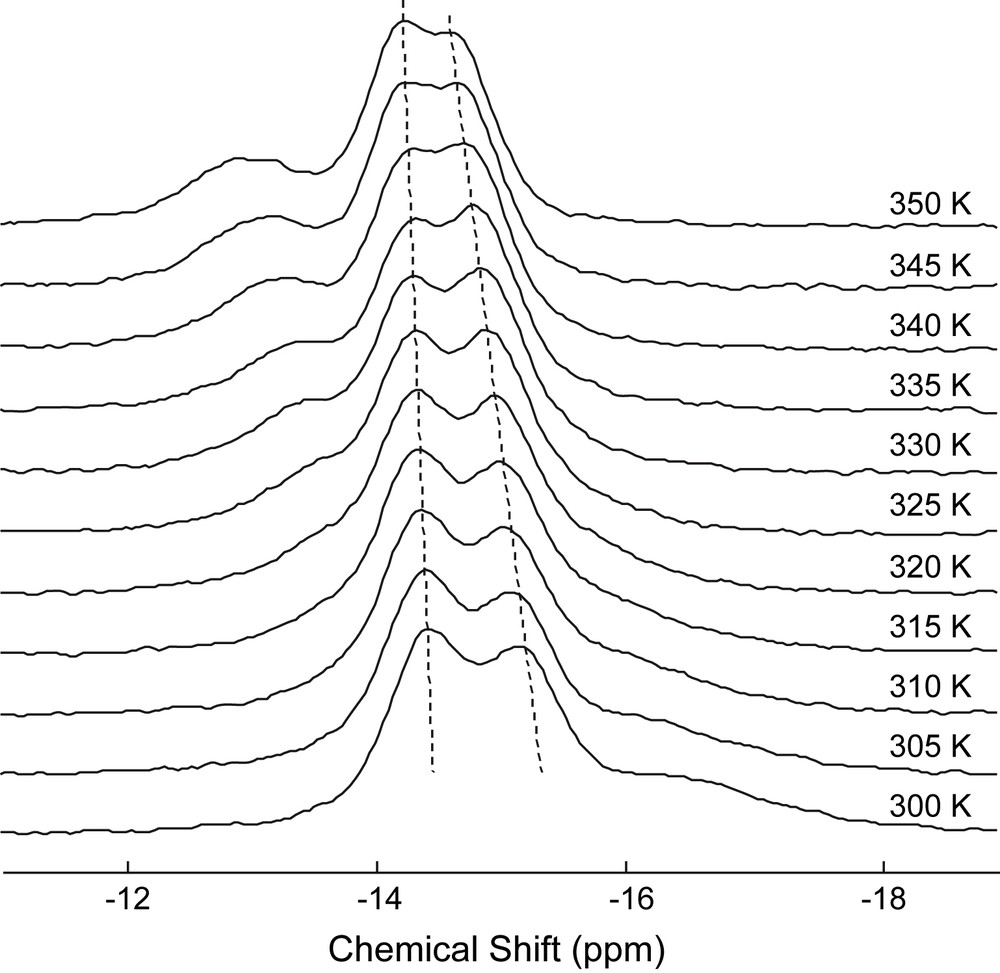
Variable temperature 31P MAS NMR spectra (9.4 T) of α2-[Ce(III)(P2W17O61)]7−.
We have, therefore, to search for other paramagnetic POMs which could present higher paramagnetic shifts and also a larger range of temperature stability; this work is currently under way.
4 Conclusion
Solid-state 31P NMR analyses on the cobalt complex αβ-[Co3Na(H2O)2(P2W15O56)2]17− are able to discriminate the phosphorus nuclei depending on their vicinity of the paramagnetic centres. Observation of the broad resonances of the closer 31P nuclei requires the use of echo–MAS techniques which allow one to obtain well phase-corrected spectra. Moreover, the presence of paramagnetic centres and high spinning speed leads to important temperature variations and temperature gradients inside the sample; this results in a shift of the isotropic signal shifts with increasing the spinning rate. At this time, this effect of temperature remains to be precisely analyzed. The monosubstituted paramagnetic derivative [Ce(III)(P2W17O61)]7− has been tested as 31P NMR thermometer; unfortunately this compound appears to be suitable for solution spectra only.
Acknowledgements
This work was supported by the CNRS and by the Université Pierre-et-Marie-Curie – Paris-6. The authors would like to thank M. Deschamps and D. Massiot (CRMHT – CNRS/Université d'Orléans) for recording 7.1 T NMR spectrum.