

1 Introduction
Depuis une trentaine d'années, de nombreux travaux ont montré que la mesure des variations locales du pH dans la cellule est un élément clé dans l'avancée des connaissances du métabolisme des organismes vivants [1–3]. La régulation des pH intracellulaire (pHi) et extracellulaire, ainsi que de celui des compartiments acides ou organelles insérés dans les membranes, est indispensable au fonctionnement optimal d'un grand nombre d'enzymes [1–3]. Des défauts dans l'homéostasie du pH apparaissent très rapidement dans les situations physiopathologiques, ainsi qu'en témoignent les études portant sur la mesure du pHi lors d'une ischémie [4], dans les tumeurs [5] ou les processus inflammatoires [6]. Des travaux récents ont montré l'importance de la régulation du pH au niveau subcellulaire, qui joue un rôle capital dans la signalisation cellulaire et l'apoptose [7,8]. Faisant suite aux travaux pionniers de Moon et Richards [9], qui, en 1973, ont utilisé le principe de la variation du déplacement chimique (δ) du phosphate inorganique principalement cytosolique (Pi) avec le pH, la RMN du 31P est intensivement utilisée comme technique de mesure non invasive du pHi [4,10]. Cependant, de nombreuses difficultés sont apparues, principalement liées à la trop faible variation de déplacement chimique (Δδab) entre les formes acides (δa) et basiques (δb) du Pi, dont le pKa de la seconde acidité est proche du pH physiologique. Ceci a pour conséquence de rendre peu sensible la mesure de faibles écarts de pH, tels que les gradients de pH intra- et extracellulaires [10]. De plus, la faible disponibilité du Pi dans le milieu extracellulaire tout comme dans certains compartiments et organelles ne permet pas localement d'accéder directement aux mesures de pH [11]. L'utilisation de marqueurs de pH synthétiques a donc été proposée, et tout particulièrement celle de dérivés alkylés de l'acide phosphonique, tels que le méthylphosphonate (MeP) ou le phénylphosphonate (PheP) ; mais elle n'apportait pas d'amélioration significative de la sensibilité (Δδab), ni de possibilité d'accès aux compartiments acides [10,12]. En revanche, l'utilisation de dérivés comportant une fonction amine, tels que l'aminométhylphosphonate ou le 2-aminoéthylphosphonate [13] a ouvert la voie vers l'étude des compartiments acides dans des préparations d'amibes Dictyostelium discoideum [14], puis de la mesure sélective des pH extracellulaire et cytosolique dans un modèle de foie isolé [15]. Le 3-aminopropylphosphonate, plus polaire, a été utilisé pour des déterminations de pH extracellulaire dans des cellules cancéreuses [16]. Dans la continuité de ces travaux, une nouvelle série de marqueurs de pH a été récemment développée afin (i) d'améliorer la pénétration intracellulaire et de permettre l'accès simultané aux compartiments intra- et extracellulaires ainsi qu'aux compartiments acides, (ii) d'accroître la sensibilité de la mesure du pH en augmentant la valeur Δδab, (iii) de disposer de molécules sensibles de différents pKa et de différentes lipophilies [17,18]. Ces aminophosphonates offrent, grâce à la présence de substituants, une gamme de pKa qui se situent entre 1,3 et 7 unités de pH, et présentent des valeurs Δδab de trois à quatre fois supérieures à celles des marqueurs existants [13,17,18]. Un de ces composés, le (2-méthylpyrrolidin-2-yl) phosphonate de diéthyle (2 ; pKa 6,99) a déjà fait l'objet d'une étude par RMN du 31P dans le cœur et le foie isolés au décours de l'ischémie et de la reperfusion [19]. Ces travaux ont décrit la mesure simultanée des pH cytosolique et extracellulaire grâce à la distinction des pics de résonance de 2, ainsi que, dans le cas du foie, la première exploration directe du pH des compartiments acides [19]. Néanmoins, les compartiments acides cellulaires fonctionnent à un pH inférieur à 5 [14,19], ce qui se situe dans la limite de précision que l'on peut obtenir avec le composé 2.
Le premier objectif de cette étude est donc de développer une nouvelle famille d'aminophosphonates, de pKa proches de la neutralité, mais inférieurs à celui de 2, tout en gardant des valeurs de Δδab avantageuses, ceci en faisant varier les substituants sur le groupement phosphoré. Il faut en outre souligner que les études par RMN du 31P, tout particulièrement lorsqu'elles sont réalisées simultanément dans plusieurs milieux biologiques (tampon de perfusion, milieu extracellulaire, cytosol ou organelles) ou à température variable, exigent la détermination préalable des paramètres de relaxation des composés phosphorés étudiés. Ceci conditionne l'acquisition des spectres et la quantification éventuelle des métabolites détectés [1–3]. De très nombreux travaux ont été réalisés dans le domaine de la relaxation des composés phosphorés endogènes [20–23] ou des sondes de pH exogènes [24], afin d'étudier, par exemple, la compartimentation cellulaire ou les flux de métabolites tels que le Pi, l'ATP ou la créatine phosphate, ainsi que les variations des volumes cellulaires et de gradients de protons.
Poursuivant l'objectif de développer de nouvelles sondes de pH dont les temps de relaxation sont de même ordre de grandeur que ceux des composés phosphorés endogènes [20–23], le présent travail se propose, dans une seconde partie, de déterminer les temps de relaxation longitudinale du phosphore 31, T1 (31P), pour une série de 27 aminophosphonates, incluant les nouvelles molécules, dans différentes conditions expérimentales, en faisant varier le milieu, le pH et la température. La vitesse de relaxation 1/T1 (31P), qui est elle-même dépendante du temps de corrélation de la molécule considérée τc, sera étudiée en fonction de la masse moléculaire des molécules (M). Pour mieux étudier ce phénomène, cette étude rassemble cinq familles d'aminophosphonates cycliques et linéaires, mono et diphosphorés, différemment substitués, dont les structures sont représentées sur la Fig. 1. Les valeurs de pKa et Δδab originales pour douze molécules sont présentées. La synthèse et la caractérisation physicochimique et toxicologique des trois composés 1, 3 et 9, jugés particulièrement représentatifs de la nouvelle série, sont décrites en détail.
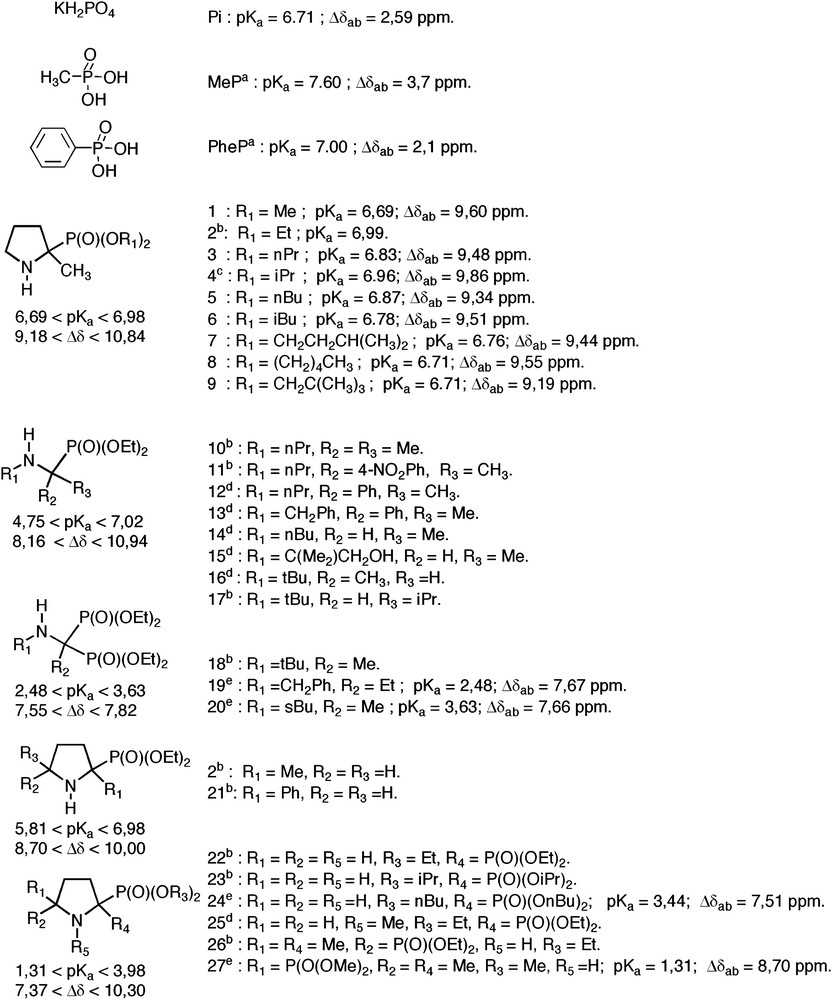
Structure des α-aminophosphonates, du Pi et des dérivés de l'acide phosphonique MeP, PheP. Me, CH3 ; Et, CH2CH3 ; nPr, CH2CH2CH3 ; iPr, CH(CH3)2 ; nBu, CH2CH2CH2CH3 ; iBu, CH2CH(CH3)2 ; sBu, CH(CH3)(CH2CH3) ; tBu, C(CH3)3 ; Ph, C6H5. (a) [14c] ; (b) [17] ; (c) [20] ; (d) [18] ; (e) [21].
2 Matériels et méthodes
2.1 Matériels
2.1.1 Réactifs et solvants
Les produits chimiques utilisés pour la synthèse, les titrages ou les milieux tampons ou de perfusion proviennent de Aldrich Chimie (Saint-Quentin-Fallavier, France) et sont utilisés sans purification préalable. De l'eau déionisée (ELGA, Purelab Option, France) est utilisée pour toutes les expériences de perfusion et de titrage. La synthèse et la caractérisation (RMN 1H, 13C et 31P) des aminophosphonates 2, 4, 10 à 27 ont été précédemment décrites [17,18,25,26], celle des molécules 5 à 8 sera décrite dans une publication ultérieure. La caractérisation par RMN des nouveaux aminophosphonates est obtenue sur un spectromètre Bruker Avance 300 MHz – 1H (300 MHz), 31P (121,49 MHz) et 13C (75,5 MHz) –, avec un découplage proton. Les valeurs des constantes de couplage J sont exprimées en Hz. Les déplacements chimiques (δ) sont exprimés en ppm par rapport au TMS (1H) et CDCl3 (13C) ou à une solution à 85% de H3PO4 dans l'eau (31P, référence externe).
2.1.2 Biologie
Les rats mâles de souche WISTAR proviennent de CERJ, France. Cette étude a été menée en conformité avec le Guide of Care and Use of Laboratory Animals, US Department of Health and Human Services, NIH Publication 85-23 (revised 1985). Les solutions tampons et les perfusats sont filtrés avant usage (Millipore, 0,2 μm).
2.1.3 Titrages et mesures du T1
Les titrages et les mesures de T1 sont faits sur un spectromètre Bruker Avance 400 MHz, équipé d'une sonde large bande (BBO) pour des tubes de 10 mm de diamètre : B0 = 9,4 T et νo = 161,98 Hz pour le 31P. La sonde est équipée d'un régulateur de température permettant de contrôler la température de l'échantillon tout au long des expériences. La référence externe est une solution de H3PO4 à 85% dans l'eau. Les pH sont mesurés avec un pH mètre Inolab 720 équipé d'une électrode Sentix 2I.
2.2 Expériences par RMN du 31P
2.2.1 Titrages
Les expériences de titrage ont été réalisées dans différents milieux : Krebs–Henseleit modifié (KHM, sans glucose ni CaCl2), milieu cytosolique cardiaque, solution de KCl (0,125 M) dont les procédés de préparation sont décrits ci-après.
Le pH des solutions d'aminophosphonates (5 mM), fraîchement préparées, est ajusté dans une gamme de valeur allant de 1 à 12 unités de pH (de 15 à 25 points par courbe de titrage) avec une solution de HCl 6 M ou de NaOH 6 M. Les solutions sont alors transférées dans un tube de 10 mm, dans lequel plonge un capillaire de D2O. Pour chaque pH testé, le spectre est enregistré par la séquence standard d'observation du 31P découplé proton, avec un temps d'acquisition de 2 s et 64 acquisitions.
Les caractéristiques de chaque composé (pKa, δa et δb) sont obtenues après tracé de la courbe expérimentale δ (31P) = f (pH), qui est ajustée sur l'expression de Henderson–Hasselbalch (Eq. (1)) par régression statistique non linéaire :
| (1) |
2.2.2 Mesure des T1 (31P)
Sur les mêmes solutions que celles décrites précédemment, les spectres pour la mesure du T1 sont enregistrés, à pH = pKa, avec une largeur spectrale de 100 ppm, en utilisant une impulsion P1 de 90° égale à 10 μs, pour une puissance de –3dB. Les T1 sont mesurés à l'aide de la séquence « inversion–récupération », l'acquisition du signal se faisant sur un cycle de huit acquisitions. Les 16 délais d'évolution, pour le retour de l'aimantation vers sa valeur d'équilibre, sont appliqués aléatoirement selon la série suivante (τ = délai d'évolution) :
τ = 0,01 s ; 35 s ; 4 s ; 15 s ; 0,1 s ; 25 s ; 2 s ; 20 s ; 8 s ; 6 s ; 12 s ; 30 s ; 0,5 s ; 10 s ; 1 s ; 35 s.
Un temps d'attente supérieur à 5 T1 (35 s pour les aminophosphonates) est utilisé entre deux mesures consécutives, pour laisser au système le temps de revenir à l'équilibre.
Le traitement des données se fait par ajustement non linéaire d'une fonction mono-exponentielle sur les intensités expérimentales I des signaux mesurés en fonction du délai τ, à l'aide de deux paramètres, suivant l'équation (2) :
| (2) |
Pour l'analyse de l'effet du pH sur le T1, les temps de relaxation longitudinale du Pi (1,2 mM) et du composé 2 (5 mM) ont été mesurés simultanément, dans un milieu KHM, dont le pH a été ajusté à des valeurs entre 3 et 10. Pour respecter les conditions de retour à l'équilibre, le délai entre les impulsions est de 55 s (soit 5 T1 du Pi).
2.3 Techniques de perfusion cardiaque : études de cytotoxicité
Les rats sont anesthésiés par une injection intrapéritonéale de pentobarbital sodique (50 mg/kg de poids corporel). La cavité thoracique est ouverte et le cœur rapidement prélevé et immergé dans du liquide de perfusion à 4 °C afin d'arrêter toute contraction, puis perfusé selon la technique de Langendorff par voie rétrograde aortique, dans laquelle le cœur est soumis à une pression constante appliquée en positionnant la colonne de perfusion à une hauteur de 90 cm [27]. Une petite incision dans l'artère pulmonaire permet l'écoulement et la mesure de l'effluent coronaire en fonction du temps (débit coronaire). Après ablation de l'oreillette gauche, un ballonnet en latex est inséré dans le ventricule gauche et rempli d'un volume d'eau distillée (50–60 μl) qui reste constant au cours du protocole expérimental. Ce ballonnet est relié à un capteur de pression (Gould) et à un enregistreur différentiateur permettant la mesure des paramètres hémodynamiques suivants : pressions diastolique et développée (Pdev) du ventricule gauche, la dérivée première de la pression développée dP/dt, la fréquence cardiaque (F). Le milieu de perfusion est une solution ionique de Krebs–Henseleit (KH) de pH 7,35, saturée avec un mélange gazeux (95% O2/5% CO2), contenant NaCl (119 mM), NaHCO3 (25 mM), KCl (5,9 mM), MgSO4 (1,2 mM), EDTA (0,5 mM), glucose (11 mM) et CaCl2 (2,5 mM). Les cœurs sont perfusés selon le protocole suivant : une période initiale d'équilibration de 30 min avec du Krebs–Henseleit, suivie de la perfusion de concentrations croissantes (10 min chacune) d'aminophosphonate (0,1 à 20 mM) ajouté dans le milieu de perfusion KH à partir d'une solution mère (50 mM) délivrée à l'entrée de l'aorte. Le débit coronaire (DC) ainsi que l'ensemble des paramètres hémodynamiques sont mesurés toutes les 5 min. Le travail cardiaque (RPP), représentatif de la fonction hémodynamique globale est calculé par le produit suivant : RPP = Pdev × F.
2.4 Préparation du milieu cytosolique cardiaque (MCytC)
Le milieu cytosolique riche en protéines hydrosolubles a été obtenu à partir du myocarde prélevé sur un rat, selon un procédé précédemment décrit [19,22]. L'homogénat de tissu cardiaque (1,25 g de tissu frais pour 3,25 ml de KCl 0,125 M) est préparé en hachant, puis mixant le tissu dans un blender (10 min). Le mélange est alors centrifugé à 3 °C pendant 20 min, et le surnageant, considéré comme représentatif du milieu cytosolique, est utilisé pour les expériences de titrage et de mesure des T1 [19,28]. Ce milieu contient déjà naturellement du Pi, mais l'addition de KH2PO4 (1,2 mM) permet d'améliorer les conditions d'observation du signal. L'aminophosphonate sélectionné est alors additionné à ce milieu, de manière à obtenir une concentration finale de 5 mM.
3 Résultats
3.1 Synthèse et caractérisation des aminophosphonates 1, 3 et 9
La voie de synthèse générale mise en œuvre pour préparer les aminophosphonates 1 à 9 est présentée sur la Fig. 2. La formation de l'α-aminophosphonate se fait en une ou deux étapes pour la série des amines différemment substituées sur le phosphore, selon la nature du phosphite de dialkyle utilisé (commercial ou non). L'α-aminophosphonate subit toujours une étape de purification sur colonne de silice pour obtenir le degré de pureté nécessaire pour une application en milieu biologique.
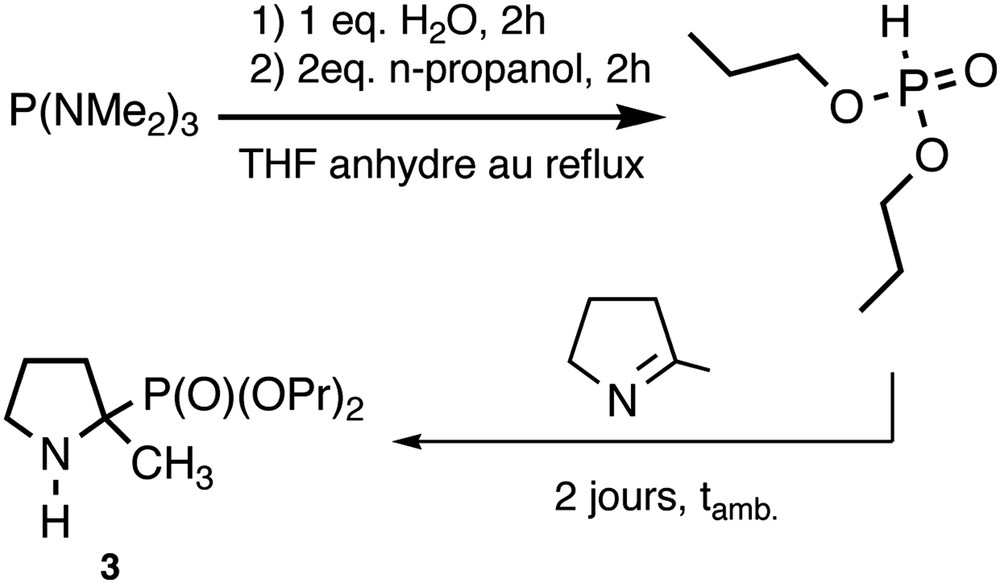
Synthèse des α-aminophosphonates 1, 3 et 9.
3.1.1 (2-Méthylpyrrolidin-2-yl) phosphonate de diméthyle (1)
On agite pendant 2 h le phosphite de diméthyle (10 g, 91 mmol) fraîchement distillé et la 2-méthyl-1-pyrroline (8,3 g, 100 mmol). De l'eau (50 ml) est ajoutée au mélange et le pH est porté à 2 par ajout d'HCl concentré (37%). La phase aqueuse est lavée trois fois par du terbutyl-méthyl éther (3 × 50 ml). Puis le milieu est basifié jusqu'à pH = 10 par ajout de pastilles de NaOH et extrait au dichlorométhane (4 × 50 ml). Les phases organiques sont rassemblées, séchées sur MgSO4. Les traces de solvant sont évaporées sous vide jusqu'à l'obtention d'une huile jaune visqueuse. Le résidu est chromatographié sur colonne de silice (CH2Cl2/EtOH : 86%/14%). On obtient 1 sous forme d'une huile jaune très visqueuse. (10,5 g, 60%). RMN 31P (CDCl3) : 33,27. RMN 1H (CDCl3) : 1,34 (d, J = 15 Hz, 3H, CH3) ; 1,55–1,90 (m, 4H, NH, CH2CH2N, CH2CN) ; 2,13–2,26 (m, 1H, CH2CN) ; 2,92–3,10 (m, 2H, CH2N) ; 3,77–3,78 (d, J = 3 Hz, OCH3) ; 3,81–3,82 (d, J = 3 Hz, OCH3). RMN 13C (CDCl3) : 24,18 (d, J = 6,8 Hz, CH3) ; 25,77 (d, J = 4,5 Hz, CH2CH2N) ; 34,67 (d, J = 3 Hz, CH2CN) ; 47,06 (d, J = 8,3 Hz, CH2N) ; 53,15 (d, J = 8,3 Hz, OCH3) ; 53,42 (d, J = 8,3 Hz, OCH3) ; 59,71 (d, J = 166 Hz, C). Analyse élémentaire de 1 (C7H16NO3P) : calculé : C 43,52%, H 8,35%, N 7,25% ; obtenu : C 41,77%, H 8,16%, N 7,23%.
3.1.2 (2-Méthylpyrrolidin-2-yl) phosphonate de dipropyle (3)
P(NMe2)3 (10 g, 61 mmol) est hydrolysé en 2 h au reflux du THF, sous atmosphère inerte, en acide bis(diméthylamino) phosphoreux, après l'ajout de 1 équiv d'eau (1,1 g, 61 mmol). À l'acide bis(diméthylamino) phosphoreux formé in situ, on ajoute du 1-propanol (2 équiv, 7,32 g, 122 mmol). Après 2 h au reflux et évaporation des solvants sous pression réduite, le phosphite de dipropyle est obtenu avec un rendement de 98%, sous forme d'une huile incolore. Un mélange de phosphite de dipropyle (9,9 g, 59 mmol) et de 2-méthyl-1-pyrroline (5,38 g, 65 mmol) est agité à température ambiante pendant 2 jours, l'évolution de la réaction pouvant être suivie par RMN du 31P. Le mélange subit le même traitement que précédemment. Le résidu est chromatographié sur colonne de silice (CH2Cl2/EtOH : 86%/14%). On obtient 3 sous forme d'une huile écrue très visqueuse (9,7 g, 64%). RMN 31P (CDCl3) : 30,65. RMN 1H (CDCl3) : 0,65 (t, J = 6 Hz, 6H, CH3–(CH2)2–O) ; 1,05 (d, J = 15 Hz, 3H,CH3) ; 1,26–1,60 (m, 8H, 2∗ CH2–CH3, HN, CH2CH2N, CH2CN) ; 1,80–2,00 (m, 1H, CH2CN) ; 2,66–2,80 (m, 2H, CH2N) ; 3,71–3,78 (m, 4H, CH2–O). RMN 13C (CDCl3) : 9,67 (s, CH3–(CH2)2–O) ; 23,59, 23,66 (CH2–CH3) ; 23,86 (d, J = 6 Hz, CH3) ; 25,33 (d, J = 4,5 Hz, CH2CH2N) ; 34,30 (d, J = 3 Hz, CH2CN) ; 46,70 (d, J = 7,5 Hz, CH2N) ; 59,20 (d, J = 164 Hz, C) ; 67,20 (d, J = 8,3 Hz, CH2–O) ; 67,45 (d, J = 8,3 Hz, CH2–O). Analyse élémentaire de 3 (C11H24NO3P) : calculé : C 53,00%, H 9,70%, N 5,62% ; obtenu : C 51,44%, H 8,88%, N 4,99%.
3.1.3 (2-Méthylpyrrolidin-2-yl) phosphonate de dinéopentyle (9)
P(NMe2)3 (10 g, 61 mmol) est hydrolysé en 2 h au reflux du THF, sous atmosphère inerte, en acide bis(diméthylamino) phosphoreux, après l'ajout de 1 équiv d'eau (1,1 g, 61 mmol). À l'acide bis(diméthylamino) phosphoreux formé in situ, on ajoute de l'alcool néopentylique : 1 équiv, 5,38 g, 61 mmol. Après 3 h de reflux, le deuxième équivalent d'alcool est rajouté au milieu, qui est laissé au reflux encore pendant 3 h. Les solvants sont évaporés sous pression réduite : le phosphite de dinéopentyle est obtenu avec un rendement de 80% sous forme d'une huile incolore. Un mélange de phosphite de dinéopentyle (10,9 g, 49 mmol) et de 2-méthyl-1-pyrroline (4,39 g, 53 mmol) est agité à température ambiante pendant 2 jours, l'évolution de la réaction pouvant être suivie par RMN du 31P. Le mélange subit le même traitement que celui décrit pour 1. Le résidu est chromatographié sur colonne de silice (CH2Cl2/EtOH : 86%/14%). On obtient 9 sous la forme d'une huile écrue très visqueuse (9,7 g, 64%). RMN 31P (CDCl3) : 30,35. RMN 1H (CDCl3) : 0,88 (s, 9H, (CH3)3–C–CH2–O) ; 0,93 (s, 9H, (CH3)3–C–CH2–O) ; 1,35 (d, J = 15 Hz, 3H, CH3) ; 1,50–1,86 (m, 4H, HN, CH2CH2N, CH2CN) ; 2,10–2,40 (m, 1H, CH2CN) ; 2,96–3,10 (m, 2H, CH2N) ; 3,70–3,78 (m, 4H, CH2–O). RMN 13C (CDCl3) : 24,60 (d, J = 6 Hz, CH3) ; 25,69 (d, J = 4,5 Hz, CH2CH2N) ; 26,13 ((CH3)3) ; 32,23 (C(CH3)3) ; (34,84 (d, J = 3 Hz, CH2CN) ; 47,15 (d, J = 7,5 Hz, CH2N) ; 59,83 (d, J = 164 Hz, C) ; 75,46 (d, J = 8,3 Hz, CH2–O) ; 75,76 (d, J = 8,3 Hz, CH2–O). Analyse élémentaire de 9 (C15H32NO3P) : calculé : C 58,99%, H 10,56%, N 4,59% ; obtenu : C 58,74%, H 10,83%, N 4,70%.
3.2 Cytotoxicité des aminophosphonates 1 à 9
Des paramètres hémodynamiques réguliers sont enregistrés au cours de la période d'équilibration sur l'ensemble des cœurs de rats perfusés (n = 22) dans les conditions normoxiques décrites précédemment : RPP (mm Hg × battements/min) = 24 220 ± 701 ; Pdias (mm Hg) = 12,7 ± 2,1 mm Hg ; DC (ml/min) = 16,2 ± 1,4. La perfusion des composés 1, 3 et 4 dans le myocarde de rat isolé perfusé à des concentrations croissantes dans des conditions normoxiques montre que ces composés ne perturbent pas ces index de la fonction cardiaque jusqu'à 10 mM pour 3, résultat voisin de celui obtenu pour le composé 2 [19,27], 5 mM pour 1 et 3 mM pour 4. Par exemple, on enregistre les paramètres hémodynamiques suivants dans le cas d'une perfusion de 4 à 3 mM pendant 20 min: RPP (mm Hg × battements/min) = 22770 ± 1502 ; Pdias (mm Hg) = 11,7 ± 2,4 mm Hg ; DC (ml/min) = 14,5 ± 1,7. La diminution du travail cardiaque de observée (−8%), bien que non significative (p = 0,3245, test t de Student) par rapport au groupe de cœurs recevant seulement du tampon KH, indique le seuil maximal d'utilisation de ce composé dans ce modèle. Ces résultats démontrent l'innocuité de ces composés dans une gamme de concentrations compatibles (de l'ordre du mM) avec les expériences de RMN en biologie [19]. Les molécules 5 à 9, qui possèdent des substituants plus encombrants sur le groupement phosphoré (Fig. 1), ce qui leur confère vraisemblablement une plus grande lipophilie, sont globalement plus cytotoxiques, et donc utilisables sur des modèles biologiques à des concentrations inférieures à 3 mM.
3.3 Titrages des nouveaux aminophosphonates 1, 3–9, 19, 20, 24 et 27
Les courbes de titrage obtenues dans KHM à 22 °C pour les molécules 1, 3, 4 et 9 sont présentées sur la Fig. 3 et sont comparées à celles du Pi. On obtient, pour trois de ces composés, des pKa inférieurs à celui de 2 (respectivement (1) 6,69 ; (3) 6,83 et (9) 6,71), jusqu'alors considéré comme une référence, car son pKa (6,99) est proche de la neutralité [17,19], ainsi qu'une différence de déplacement chimique Δδab comparable (respectivement Δδab (1) = 9,60 ; Δδab (3) = 9,48), sauf pour le composé 9, qui se situe un peu en retrait : Δδab (9) = 9,19. En revanche (Fig. 3), le composé 4 ne se démarque pas de 2 (pKa (4) 6,96 ; Δδab (4) 9,86). La plage de variation des valeurs de pKa obtenues pour les molécules de cette famille (1–9) est consignée sur la Fig. 1 et montre que les essais visant à modifier les substituants sur le phosphore, et non pas sur les groupements portés par le carbone en α (comme c'est le cas des composés 10 à 27), ne font varier le pKa que dans une gamme relativement réduite (6,7 < pKa < 7) et permettent de conserver d'excellentes valeurs de Δδab, supérieures à 9 ppm. Les valeurs individuelles des nouvelles molécules non décrites dans la Fig. 3 (pKa ; Δδab) sont respectivement : 5 (6,87 ; 9,60); 6 (6,78 ; 9,34); 7 (6,76 ; 9,44); 8 (6,71 ; 9,57).
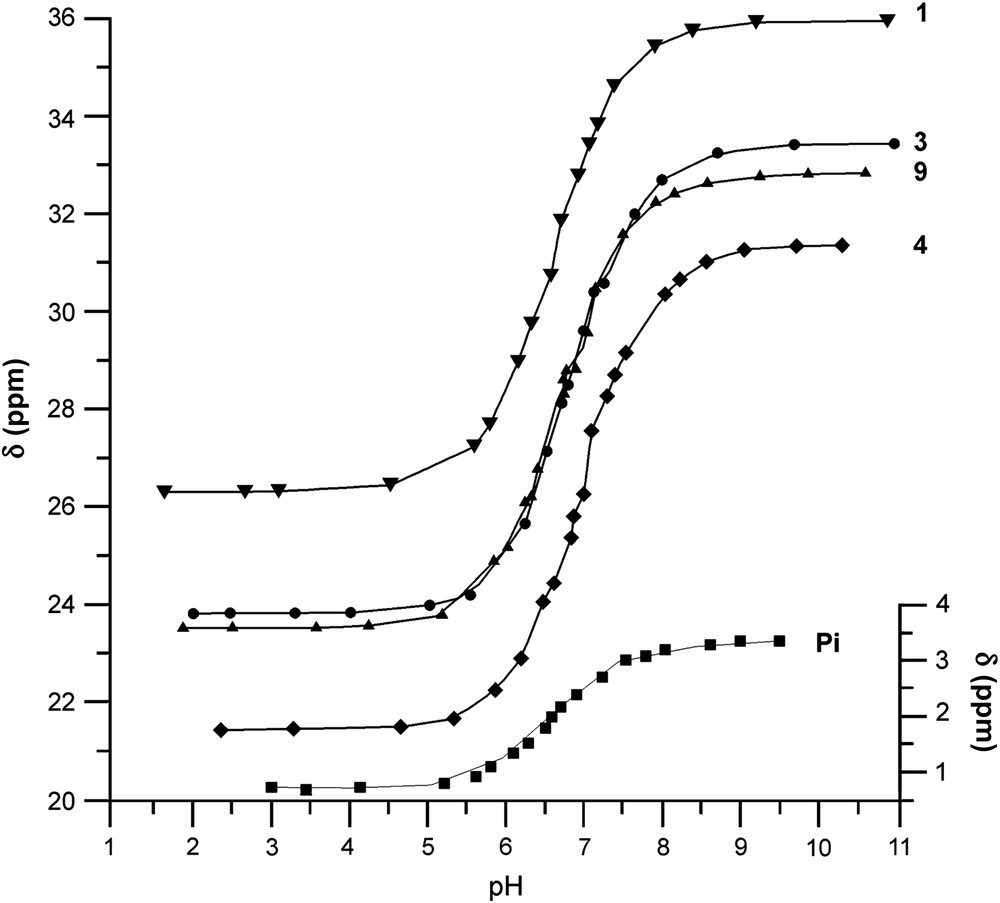
Variation du déplacement chimique du 31P du Pi et des aminophosphonates 1 (▾ ; pKa = 6,69), 3 (● ; pKa = 6,83), 4 (♦ ; pKa = 6,96) et 9 (▴ ; pKa = 6,71) en fonction du pH. Les expériences sont réalisées à 22 °C dans un milieu tampon de Krebs–Henseleit modifié.
Les courbes de titrage des molécules diphosphorées 19, 20, 24 et 27 ont été obtenues selon le même procédé, dans un tampon KHM à 22 °C. Les valeurs des (pKa ; Δδab) pour ces molécules sont respectivement : 19 (2,48 ; 7,67) ; 20 (3,63 ; 7,66) ; 24 (3,44 ; 7,51) ; 27 (1,31 ; 8,70). Ces valeurs sont homogènes (1,5 < pKa < 4) avec celles obtenues pour des molécules portant aussi deux groupements phosphorés (16, 22, 23 et 26 [17], et 25 [18]).
3.4 Effet du pH, du milieu et de la température sur les T1 (31P) des aminophosphonates 1, 2, 3, 10, 16, et du Pi
La mesure des T1 effectuée à différents pH dans le milieu tampon KHM modifié, pour le Pi et le composé 2, montre que la relaxation longitudinale de ces molécules n'a pas la même sensibilité vis-à-vis du pH (Fig. 4). Le T1 du Pi varie en fonction du pH et présente un minimum relativement marqué dans la gamme de pH proche de son pKa, alors que le T1 de 2 ne varie pas de manière significative dans la gamme de pH considérée.
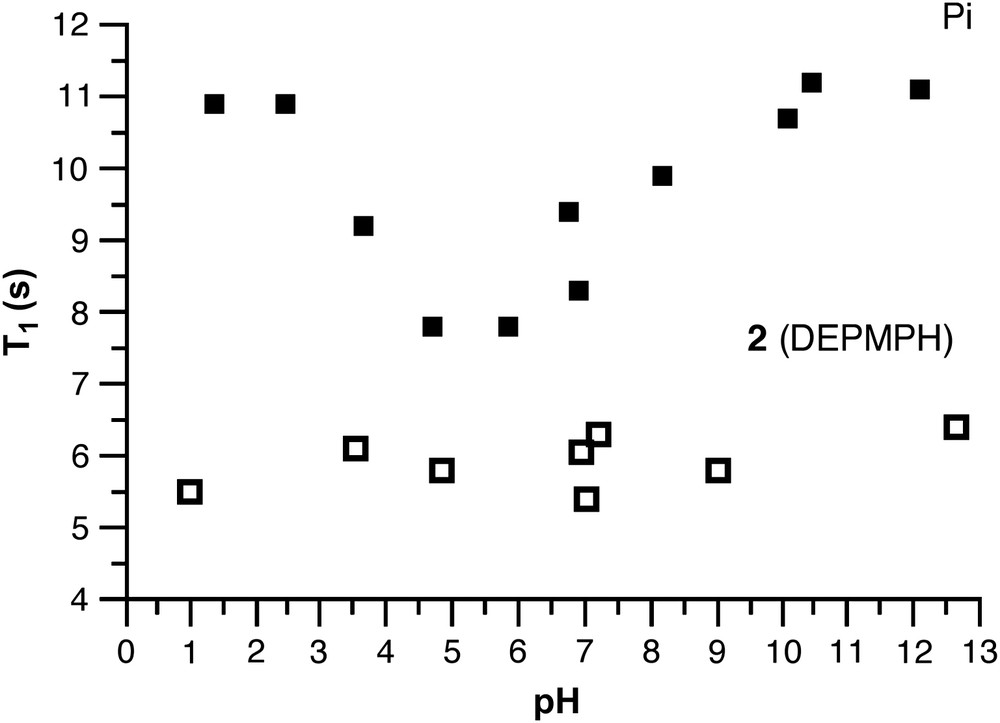
Effet du pH sur le T1 (31P) du Pi (■) et de l' α-aminophosphonate 2 (◘), mesurés à 22 °C dans un milieu tampon de Krebs–Henseleit modifié.
Les valeurs des T1 mesurés dans différents milieux pour une sélection de molécules représentatives des cinq familles étudiées (1, 2, 3, 10, 16) et du Pi sont consignées dans le Tableau 1. On observe que, dans les milieux ioniques usuels (KHM ou KCl), le T1 du Pi est plus élevé que celui de l'ensemble des aminophosphonates. En revanche, une très forte diminution du T1 est observée pour le Pi lorsque la mesure est faite dans une préparation de milieu cytosolique, alors que ce même milieu n'influence pas la relaxation des aminophosphonates de manière significative. Pour le composé 2, les expériences réalisées dans la présente étude (Tableau 1) ont été rapprochées de celles effectuées sur le modèle de cœur de rat perfusé [19,29], montrant que la valeur du T1 mesuré dans le cytosol n'est pas sensiblement modifiée au cours des expériences biologiques.
Valeurs du T1 (31P) de différents aminophosphonates et du Pi dans les milieux biologiques usuels
| Composés | Milieu | pKa | Δδab (ppm) | T1 (s) | 1/T1 (s−1) |
| Pi | KHM | 6,71 | 2,61 | 11,5 ± 0,5 | 0,087 |
| KHM∗ | 6,69 | 2,61 | 12,6 | 0,079 | |
| McytC | 6,78 | 2,60 | 3,8 | 0,263 | |
| cœura,b | 1,55 | 0,645 | |||
| KCl | 6,74 | 2,55 | 10,1 | 0,099 | |
| 1 | KHM | 6,69 | 9,60 | 7,90 | 0,126 |
| McytC | 6,77 | 9,65 | 4,50 | 0,222 | |
| 2 | KHMc | 6,98 | 965 | 6,3 ± 0,4 | 0,158 |
| KHM∗ | 6,81 | 9,58 | 8,2 | 0,122 | |
| McytCc | 7,09 | 9,69 | 5,4 ± 0,3 | 0,185 | |
| coeura,b | 4,8 | 0,208 | |||
| KCl | 6,95 | 9,69 | 5,4 | 0,185 | |
| 3 | KHM | 6,88 | 9,98 | 5,06 | 0,197 |
| KHM∗ | 6,50 | 9,59 | 6,03 | 0,166 | |
| MCytC | 6,88 | 9,55 | 5,64 | 0,177 | |
| 10 | KHM | 6,99 | 10,17 | 4,5 | 0,222 |
| KHM∗ | 6,77 | 10,20 | 6,1 | 0,164 | |
| MCytC | 7,09 | 10,20 | 4,2 | 0,238 | |
| KCl | 7,09 | 10,10 | 3,5 | 0,286 | |
| 12 | KHM | 5,89 | 8,19 | 2,8 | 0,357 |
| MCytC | 5,88 | 8,35 | 2,7 | 0,370 | |
| 14 | KHMc | 6,92 | 10,09 | 4,0 | 0,250 |
| MCytC | 7,05 | 10,16 | 3,7 | 0,270 | |
| KCl | 6,95 | 10,17 | 4,8 | 0,208 | |
| 16 | KHMc | 7,02 | 10,9 | 4,6 ± 0,2 | 0,217 |
| KHM∗ | 6,72 | 10,8 | 5,8 | 0,172 | |
| MCytC | 7,06 | 10,2 | 4,1 | 0,244 | |
| KCl | 7,03 | 10,26 | 8 | 0,125 | |
| 18 | KHM | 3,45 | 7,55 | 3,0 | 0,333 |
| KHM∗ | 2,90 | 7,18 | 5,3 | 0,189 | |
| 22 | KHMc | 3,63 | 7,61 | 3,3 ± 0,3 | 0,303 |
| KHM∗ | 3,47 | 7,65 | 4,5 | 0,222 | |
| MCytCc | 3,51 | 7,69 | 2,4 | 0,416 |
Enfin, une sélection d'aminophosphonates représentatifs des différentes familles de molécules, tels que 2, 3, 10, 16, 18, 22, ainsi que du Pi, ont été étudiés à deux températures (KHM, 22 °C et 37 °C). Dans tous les cas, on observe une augmentation du T1 lorsque la température augmente (Tableau 1).
3.5 Détermination des T1 (31P) dans KHM pour la série des 27 aminophospnonates
En complément des valeurs de T1 obtenues pour le Pi et les composés 1, 2, 3, 10, 16, 18, 22, consignées dans la Tableau 1, les valeurs de T1 (31P) des 20 autres aminophosphonates, obtenues dans les mêmes conditions (KHM ; 22 °C ; pH = pKa) sont présentées ci-après. Pour chaque composé, la valeur de T1 indiquée, en secondes, correspond à la moyenne de 2–3 mesures indépendantes. À partir de trois mesures indépendantes, la déviation standard (±SD) par rapport à la moyenne est présentée. Une variabilité d'environ 10% sur la mesure des T1 (31P) a été obtenue.
4 : 4,7 ± 0,2 ; 5 : 4,3 ; 6 : 4,2 ; 7, 3,6 ; 8 : 3,9 ; 9 : 3,9 ; 11 : 3,4 ; 12 : 2,8 ; 13 : 3,0 ; 14 : 4,1 ; 15 : 4,3 ; 17 : 4,9 ; 19 : 1,9 ; 20 : 2,4 ; 21 : 4,4 ; 23 : 2,8 ; 24 : 2,2 ; 25 : 3,1; 26 : 2,8 ± 0,3 ; 27 : 5,1 ± 0,4.
Les valeurs obtenues dans le KHM à pH = pKa, pour MeP et PheP, sont respectivement 8,3 ± 0,5 s et 6,3 ± 0,3 s et sont proches des valeurs trouvées dans la littérature pour PheP dans le milieu extracellulaire du foie isolé perfusé [12].
3.6 Étude des variations des 1/T1 (31P) des aminophosphonates avec la structure chimique et la masse moléculaire
Un des objectifs de cette étude est d'établir un modèle empirique qui permette de prévoir, avant synthèse, le T1 (31P) des aminophosphonates. Un examen rapide des T1 mesurés montre qu'ils sont corrélés à plusieurs grandeurs, les meilleures corrélations étant obtenues avec la masse moléculaire, le pKa et la constante de vitesse de protonation de l'amine (corrélations non montrées). Une difficulté est que ces grandeurs sont elles-mêmes corrélées entre elles dans la famille de molécules étudiées. La constante de vitesse de protonation kexch a une influence sur le temps de relaxation transversal T2 et peut, par un mécanisme différent, influer aussi sur T1. Mais les résultats obtenus pour l'ensemble des molécules de la littérature montrent que l'on doit s'attendre surtout à une forte dépendance de T1 vis-à-vis de la masse moléculaire [30–32]. Un résumé de la théorie rappelle que la relaxation du spin du 31P est due à plusieurs mécanismes indépendants, qui sont en particulier : les interactions magnétiques dipolaires intra- et intermoléculaires (DD), l'anisotropie du déplacement chimique (CSA), l'interaction spin–rotation (SR). La vitesse de relaxation longitudinale 1/T1 est la somme de leurs contributions :
| (3) |
La majorité des termes qui composent cette expression dépendent du temps de corrélation τc, qui décrit les fluctuations temporelles des champs magnétiques et dépend de la vitesse de réorientation de la molécule, elle-même fortement dépendante de la masse molaire M. Les interactions dipôle–dipôle s'écrivent de la manière suivante [31,32] :
| (4) |
| (5) |
Ces interactions dépendent de M par l'intermédiaire de τc, ou par le coefficient d'autodiffusion D. Il en est de même pour le terme de la relaxation CSA [31,33] :
| (6) |
La relaxation spin–rotation dépend de M à la fois par le moment d'inertie Ir et par τSR :
| (7) |
Dans ce contexte, il paraît justifié, pour l'ensemble des molécules étudiées (Fig. 1), de réaliser le tracé de 1/T1 en fonction de M (Fig. 5). Un développement polynomial du second degré (Eq. (8)), qui représente ces points de façon satisfaisante (écart-type σ1/T1 = 3,31 × 10−2 Hz), a été obtenu par régression :
| (8) |
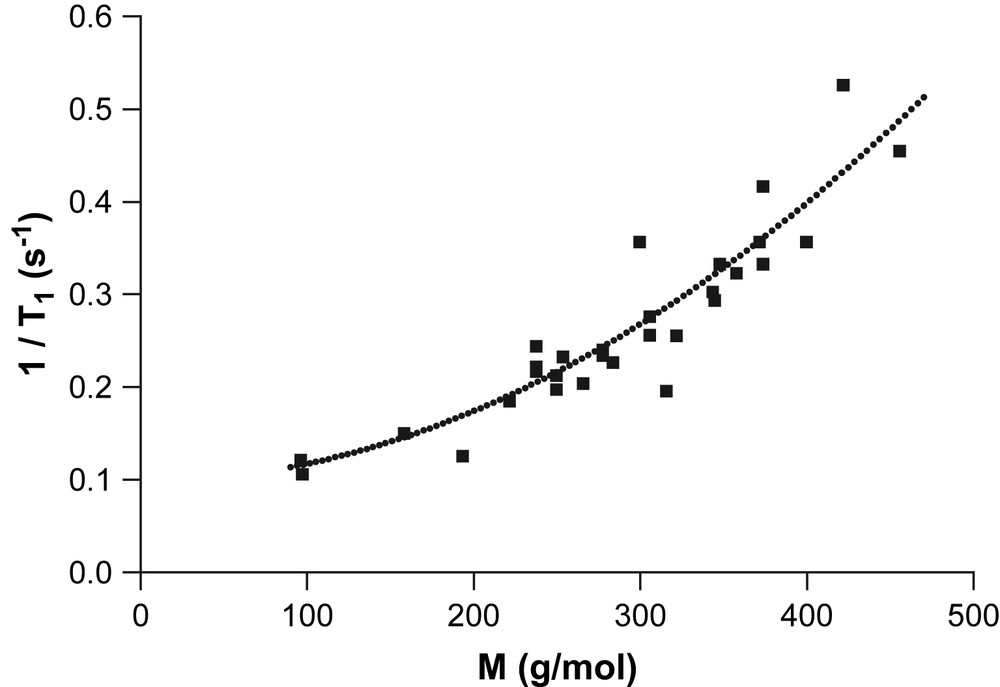
Dépendance de la vitesse de relaxation longitudinale 1/T1 (31P) vis-à-vis de la masse moléculaire M, pour les composés 2 à 26, obtenus par des modifications de substituants sur les atomes de phosphore, Pi, MeP et PheP.
On observe que les composés 1 et 27, qui ont des T1 élevés, n'entrent pas dans la relation précédente. Ces composés se trouvent être ceux dans lesquels le phosphore porte deux substituants méthoxy, particulièrement encombrants et qui peuvent par leur rotation entraîner des fluctuations de champ magnétique. En revanche, le Pi et les dérivés alkylés de l'acide phosphonique, MeP et PheP, s'y inscrivent parfaitement.
La corrélation statistique qu'on observe entre 1/T1 et le pKa (r2 = 0,68) n'a pas d'explication logique a priori. Un traitement statistique succinct va montrer qu'elle n'est qu'une redite de la corrélation entre 1/T1 et M. Compte tenu des arguments théoriques précédents en faveur d'une forte dépendance de 1/T1 vis-à-vis de M, il paraît logique de ne pas traiter le pKa sur un pied d'égalité avec M, mais comme un facteur secondaire. Ceci évite que le pKa, qui se trouve dans cette série de molécules être corrélé avec M (Fig. 6), puisqu'il dépend directement des substituants [19], ne s'approprie une partie de la corrélation de 1/T1 avec M. L'équation (8) a donc été conservée, en la multipliant par une fonction affine inconnue f(pKa) = a0 + a1 pKa. On obtient ainsi, par régression, l'expression (9) :
| (9) |
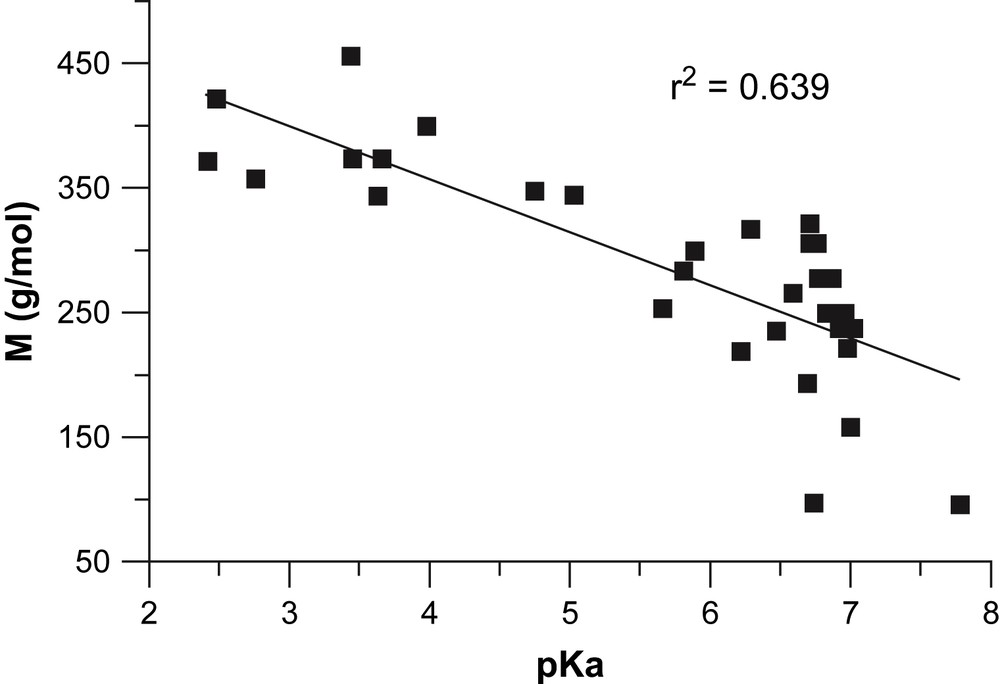
Corrélation entre le pKa et la masse moléculaire M pour les composés 1 à 26, Pi, MeP et PheP.
Le second facteur fluctue peu autour de sa moyenne, égale à 1 (écart-type = 1,5 × 10−2), ce qui prouve qu'une fois soustraite la corrélation de 1/T1 avec M, 1/T1 n'est plus corrélé au pKa. Ceci est corroboré par l'écart-type global en 1/T1 associé à l'Eq. (9) (σ1/T1 = 3,28 × 10−2 Hz), qui n'est pas meilleur que celui trouvé avec l'Eq. (8), que nous conserverons donc. Des considérations similaires conduisent à éliminer du modèle une autre grandeur, pourtant corrélée, qui est la constante de vitesse de protonation kexch, elle-même corrélée au pKa, donc à M. Par ailleurs, des relations liant 1/T1 au déplacement chimique δ = (δa + δb)/2 ou à (δb − δa), qui pourraient être partiellement corrélées à Δσ, ont été recherchées, sans résultat.
4 Discussion
Ce travail décrit, en premier lieu, une nouvelle famille d'α-aminophosphonates, différemment substitués sur le phosphore, et plus particulièrement la synthèse de trois nouveaux composés 1, 3 et 9, dont les valeurs de pKa sont inférieures (respectivement de 0,30, 0,15 et 0,28 unités de pH) à celle de 2, utilisé avec succès comme marqueur de pH en RMN du 31P sur des organes perfusés [19], ou pour ses propriétés protectrices sur la fonction et le métabolisme cardiaque [17,27,29]. La diminution du pKa, tout en restant proche du pH physiologique, peut dans certains cas offrir l'avantage d'une meilleure précision dans les bas pH, comme ceux des compartiments acides [14,19]. À ce titre, le composé 1 apparaît comme le plus sensible, puisqu'il associe une baisse de pKa à un maintien, voire à une amélioration de Δδab (Tableau 1; Fig. 3). Le composé 3, de plus faible cytotoxicité, peut aussi apparaître comme un meilleur compromis. Cette étude montre que, lorsqu'on fait varier les substituants sur le phosphore pour les composés 5 à 9, on obtient des courbes de titrage quasi superposables, qui se situent entre celles de 1 et 4, dont les valeurs de pKa sont les plus extrêmes (Fig. 3), suggérant que les modifications de structure autour du 31P (molécules 5 à 9) n'interviennent que faiblement dans l'acidité de l'amine. Enfin, l'ensemble de ces composés, quel que soit le pH étudié, résonnent (21,45 < δ < 35,89) loin des métabolites phosphorylés cellulaires, dont les pics de résonance détectés dans les modèles biologiques se situent entre − 15 et 5 ppm [18].
En comparant les quatre autres familles de molécules présentées dans ce travail, on observe que les substituants placés sur les carbones en α ou β du groupement phosphoré, influencent au contraire fortement le pKa avec une amplitude de variation de 6 unités de pH, tout en gardant des valeurs Δδab importantes. La contribution de chaque substituant à la modification du pKa de la fonction amine de la pyrrolidine, qui fait intervenir des effets inductifs et structuraux, a été étudiée en détail grâce à l'élaboration d'un modèle semi-empirique permettant leur incrémentation [17,18]. Ceci représente un avantage incontestable sur les dérivés de l'acide phosphonique, dont le pKa de la seconde acidité, qui seul présente un intérêt en biologie, ne varie significativement qu'en additionnant, par exemple, des substituants halogénés biologiquement peu compatibles [34]. Cette sensibilité moindre des dérivés de l'acide phosphonique pourrait provenir de la difficulté à faire varier la densité électronique autour de l'atome de phosphore, si les substituants lui sont directement liés [34]. Ce phénomène se trouve confirmé dans la présente étude, où de faibles variations de pKa sont observées dans la nouvelle famille de composés 1–9, portant différents substituants sur le groupement phosphoré.
La présente étude montre en parallèle que le T1 (31P) du Pi est très fortement sensible aux variations de milieu, alors que la relaxation des aminophosphonates n'est que très peu modifiée par leur environnement (ions, molécules biologiques hydrosolubles) et le pH, dans la limite des conditions expérimentales présentées. Le comportement du Pi est en accord avec différents travaux [11,21,35,36], qui montrent de même que d'autres molécules, telles que l'ATP, ou des dérivés alkylés de l'acide phosphonique, marqueurs de pH potentiels, réagissent avec le milieu environnant, tout particulièrement avec des ions métalliques [21]. Dans ce contexte, des mesures de T1 ont été réalisées dans le foie isolé lors de la perfusion de solutions contenant 10 mM de diméthyl méthylphosphonate ou de PheP, et des différences significatives ont été déterminées entre le milieu intracellulaire (valeurs de T1 de 2,4 s et 1,7 s, respectivement) et extracellulaire (valeurs de T1 de 8,2 s et 6,6 s, respectivement) [12]. D'autres composés portant des groupements phosphate, comme l'ATP, peuvent avoir des temps de relaxation très différents (de 0,4 à 14 s), selon qu'ils se trouvent dans des compartiments cellulaires distincts [36,37]. La connaissance des valeurs des T1 des aminophosphonates, comprises entre 1 et 10 s selon leur structure chimique, permettra de sélectionner une sonde qui sera proche des valeurs des composés endogènes à étudier. De plus, leur faible sensibilité à leur environnement peut être un atout certain pour des études nécessitant l'emploi de séquences d'acquisitions courtes, pour suivre par exemple l'évolution rapide de composés labiles.
Les résultats obtenus sont à rapprocher de ceux décrits dans la littérature pour des composés en solution, dans lesquels le T1 augmente avec la température pour les petites molécules en milieu peu visqueux [32]. En revanche, il a été également décrit que, pour les molécules de taille plus importante, le T1 diminue avec la température en milieu très visqueux [32]. Ceci illustre encore le fait que l'analyse de la relaxation des composés ne peut se faire qu'en prenant en compte un nombre très important de paramètres, et en premier lieu les conditions expérimentales. De manière intéressante, l'effet de l'accroissement de la température, qui, dans le cas de la présente étude, conduit à une augmentation du temps de relaxation longitudinale T1 du Pi en solution tampon, n'est pas directement transposable aux études dans les systèmes biologiques. En effet, lorsque le Pi se trouve dans des compartiments cellulaires, la situation peut être très différente. Par exemple, aucune variation significative du T1 (31P), si ce n'est une légère diminution, n'a été observée pour le Pi cytosolique et le Pi mitochondrial, dont les T1 (31P) ont été mesurés dans le foie isolé entre 4 et 37 °C [23].
En conclusion, outre une série de nouvelles molécules, ce travail apporte un ensemble exhaustif de valeurs du temps de relaxation T1 (31P) pour 27 α-aminophosphonates, et analyse les variations de ce paramètre dans différentes conditions expérimentales. Une tentative d'exploration de la contribution du temps de corrélation τc dans la relaxation du 31P a été réalisée en considérant l'impact de la masse moléculaire M de chaque composé sur la vitesse de relaxation, étant logiquement attendu que cette dernière augmente avec M. Dans le contexte d'études biologiques à venir, ces données sont indispensables. Il faut par exemple souligner que la connaissance des temps de relaxation T1 s'est avérée déterminante pour l'observation sélective du Pi extracellulaire (par saturation, T1 = 8–10 s) et du Pi intracellulaire relaxant 10 fois plus rapidement [20]. Dans le cas plus précis des marqueurs de pH, la connaissance des temps de relaxation des dérivés alkylés de l'acide phosphonique (tels que PheP) a permis une utilisation optimale de ces molécules réparties dans les différents compartiments cellulaires [12,37]. En prenant en compte les présents résultats, on peut s'attendre à ce que l'utilisation des aminophosphonates permette d'obtenir des résultats concomitants et plus directs.