

1 Introduction
Anodic oxide films formed on noble metals, especially Pt and Au, have formed an important part of electrochemical surface science owing to their electrocatalytic properties towards many organic electro-oxidation reactions. The development of thin and thick oxide films on these metal electrodes can be studied in detail by the application of electrochemical, optical and electron spectroscopy techniques such as cyclic voltammetry (CV) [1,2], linear sweep voltammetry [3,4], X-ray photoelectron spectroscopy (XPS) and Auger electron spectroscopy (AES) [5,6]. CV is of particular importance to studies of electrochemically formed oxide films on noble metal electrodes, since it allows in situ evaluation of the thickness of submonolayer, monolayer and multilayer oxide films. CV is also of great importance in the elucidation of the catalytic activity of the oxide films.
In aqueous H2SO4, which is of interest to the work reported here, the oxide formation at Pt starts at 0.12 V/SHE. The following oxide-formation mechanism at Pt electrodes was proposed [7,8]:
| (1) |
| (2) |
| (3) |
A large number of investigations have been reported on the electro-oxidation of aromatic compounds, particularly phenol, using metal oxide [9–13] in order to improve the catalytic activity of platinum. In general, the oxidation of phenolic compounds produces phenoxy radicals that can be further oxidized to quinones or can react to form a film of dimers and/or polymers [11,14–16]. This adherent film is insulate and thus passivates the surface of the working electrode [11,15,16]. Numerous treatment techniques have been developed to overcome the problems of surface fouling [17–20]. The electrochemical pre-treatment of electrode surface has been shown to reduce fouling [17]. For example, the use of higher applied potentials combined with a lower initial concentration of phenol in acidic medium (pH < 3) was shown to reduce electrode fouling and increase the amount of oxidized phenol [16,20]. However, the activity of noble metal electrodes is shown to be short lived. Platinum is one of the most widely used electrodes in electrochemical applications. It is generally recognized that oxidation of phenol and other phenolic compounds always produce a very rapid decrease in the first oxidation peak during cyclic voltammetry only after the first scan. This step is believed to generate a rapid passivation of the Pt electrode [11,15,20,21]. The mechanism of phenol oxidation and its derivatives at the platinum electrode was already described by Gattrell and Kirk [11] and Comninellis [12,13]. More recent data were reported by Andreescu et al. [22] and Arslan et al. [23]. This mechanism depends on electrode pre-treatments and experimental conditions. The electrode behaviour changes if the electrode was pre-oxidized, producing a platinum oxide coating which slightly inhibits the formation of the passivating layer [11,20].
In this paper bulk and pre-adsorbed phenol oxidation at pre-treated polycrystalline platinum electrodes were examined in acidic and alkaline solutions using CV technique to get further insights into the important electrocatalytic properties of these electrodes. The results obtained are compared with those determined for hydroquinone and p-benzoquinone on the same metal electrodes with a view to obtain more information concerning the influence of structural effects on the electrochemical reactivity of these compounds.
2 Experimental
2.1 Reagents
Phenol (Prolabo), catechol, hydroquinone (Aldrich) and p-benzoquinone (Across) were of analytical grade and were used as received. All the solutions were freshly prepared with bidistilled water, using 0.5 M H2SO4 (Prolabo) as supporting electrolyte.
2.2 Electrochemical measurements
Runs were made in a three-electrode compartment Pyrex glass cell (100 cm3) thermostated at 25 ± 0.5 °C. A platinum disk electrode (Tacussel) with a diameter of 0.2 cm was used, after its treatment, as working electrode. The counter electrode is a bar of graphite (∅ = 0.2 cm, L = 12 cm immersed). The working potential electrode was measured versus an Hg/Hg2SO4/K2SO4(sat) electrode (E = 0.61 V/SHE).
Polarisation measurements were carried out using a Tacussel potentiostat model PJT 35-2. The signal for triangular potential sweeps was produced by Tacussel scanner model GSTP 4. Voltammograms were recorded with the aid of a GSTP4 Tacussel X–Y recorder. The amount of electricity (Q) was measured by an IG 5-LN Tacussel-type integrator.
2.3 Pre-treatment of the platinum electrode
The working electrode was a polycrystalline platinum disk of ca. 0.0314 cm2 exposed geometric area. This electrode was mechanically polished with fine alumina powder and washed with bidistilled water before transferring to the cell. The electrode was then cycled between −0.61 and +0.89 V in 0.5 M H2SO4 at a scan rate of 5 V s−1. By subjecting the electrode to repeated cycles, there was an increase in the charge of the voltammogram, suggesting an increase in thickness of the oxide film adherent to Pt. An examination of the treated platinum surface by electronic scan microscopy suggested that the oxide layer deposited on Pt electrode is amorphous with sponge-like morphology (Fig. 1).

Scanning electron micrographs (1095×) of the surface of Pt electrode (a) before treatment and (b) after treatment.
2.4 Electrolyses
The electrolyses of the phenol aqueous solutions (H2SO4, pH 2) were carried out in a single-compartment Pyrex glass cell of 500 mL volume at 25 °C. The anode is a cylindrical polycrystalline platinum grid (∅ = 4.3 cm, L = 5 cm) covered with platinum oxide layer (Q = 480 mC) by CV under the conditions described in the previous paragraph. The counterelectrode was a rectangular platinum plate with a geometrical area of 15 cm2. The electrolysis was carried out by CV at 50 mV s−1.
2.5 Analyses
The analyses of phenol and its oxidation intermediate products were made by high-performance liquid chromatography using a Hewlett-Packard 1090 HPLC apparatus. The products formed are separated on a Hamilton PRP X300 column and then analyzed using a diode array detector measuring the optical density at 220 nm. The mobile phase was a mixture of methanol and 0.05 M H2SO4 with the percentage by volume of methanol varying linearly with time as follows: from 2 to 25% for the first 10 min then from 25 to 40% up to 20 min and finally from 40 to 60% up to 40 min. The mobile flow rate was fixed at 1.3 mL min−l.
3 Results and discussion
3.1 Bulk reactions
The electrochemical response of the treated electrode is presented in Fig. 2 (curve Pt). The cyclic voltammogram was characterized by a large peak beginning at +0.10 V and corresponding to the platinum electro-oxidation, while the reduction peak of the oxide was observed at +0.02 V. The charge Q considered as the main characteristic of the electro-active surface was measured for a single cathodic scan at 50 mV s−1 from +0.89 to −0.30 V. The treatment period varied from some minutes to 70 h to get surface states characterized by a charge between 0.3 and 4.8 mC. For Q values higher than 4.8 mC, the platinum oxide layer does not adhere to the Pt substrate.

Cyclic voltammograms of 0.1 M phenol + 0.5 M H2SO4; Q = 3.4 mC; v = 50 mV s−1.
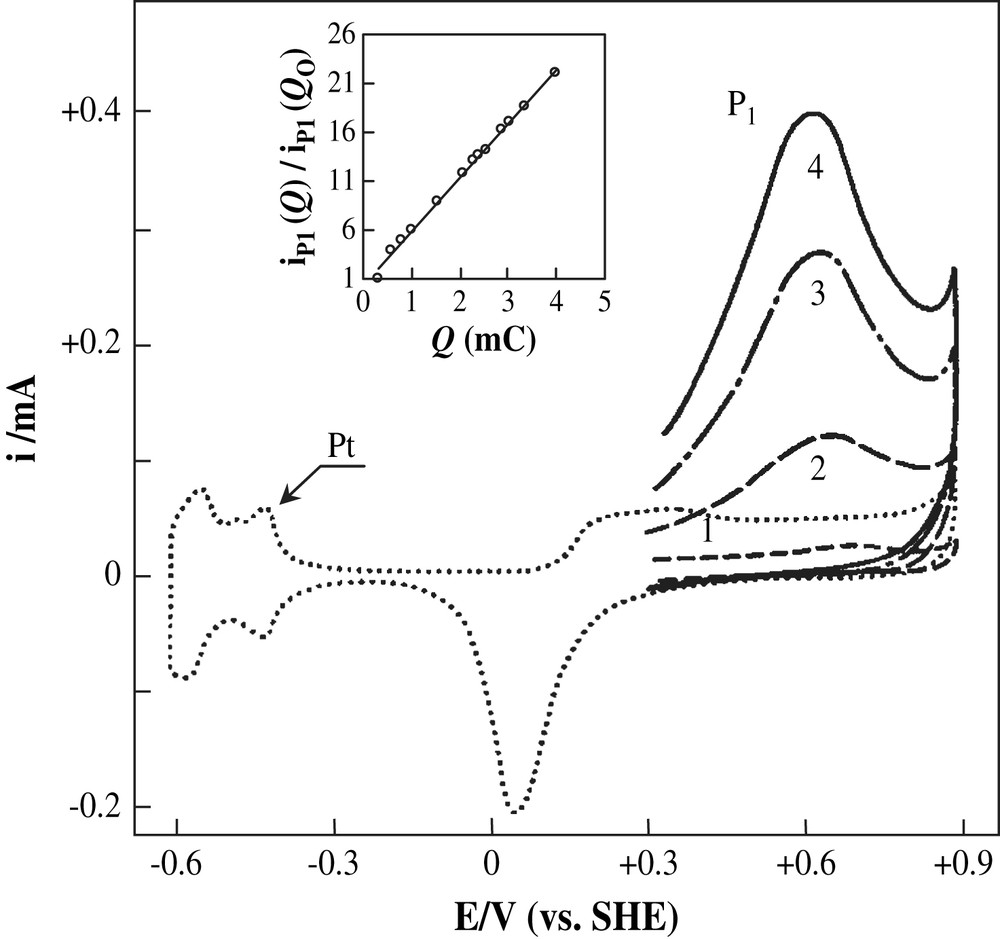
The cyclic voltammogram of 0.1 M phenol + 0.5 M H2SO4 solution at a pre-treated platinum electrode (Q = 3.4 mC) is shown in Fig. 2. During the first forward positive scan, phenol exhibits two anodic peaks P1 and P2 at approximately 0.50 and 0.78 V, respectively, within the region corresponding to the oxidation of the platinum surface. On the reverse scan, a broad reduction peak (P3), probably due to the reduction of the adsorbed oxidation product and of platinum oxide, appears near 0 V. Moreover, it can be observed that most of the adsorption sites of the platinum surface are blocked, as indicated by the low electric charge recorded in the potential region between −0.61 and +0.30 V. During the second and successive potential cycles, the cyclic voltammogram displays two features in the positive scan P4 and P5, located at 0.02 and 0.22 V, respectively. These peaks are, respectively, ascribed to the oxidation of hydroquinone and catechol by comparison with their authentic cyclic voltammograms recorded under identical experimental conditions. The current densities of peaks P1, P2 and P3 depend on experimental conditions, particularly the charge Q.
Fig. 3 shows the voltammograms obtained during the oxidation of 0.1 M phenol solution for Q values in the range 0.9–4.0 mC. The current intensity of the peak P1 increases linearly with the value of Q. It is multiplied by a factor 46 when Q increases from 0.3 to 4.0 mC (inset of Fig. 3). If the acidic medium was changed by an alkaline one using 0.5 M K2CO3 as electrolyte, all other experimental conditions were similar (Fig. 4), this current is multiplied by a factor 21 (inset of Fig. 4). These important factors could result from one or the two following causes:
- (1) an increase in the active surface of the electrode after pre-treatment;
- (2) an increase in the catalytic activity of the electrode surface.

Cyclic voltammograms of 0.1 M phenol (first cycle) + 0.5 M H2SO4; v = 50 mV s−1; Q: (1) 0.9; (2) 1.5; (3) 3.2; (4) 4.0 mC. Inset: multiplicative factor of P1 peak current as a function of Q values; Q0 = 0.3 mC.

Cyclic voltammograms of 0.1 M phenol + 0.5 M K2CO3; v = 50 mV s−1; Q = 3.5 mC. Inset: multiplicative factor of P1 peak current as a function of Q values; Q0 = 0.3 mC.
The increase in surface due to pre-treatment was evaluated by CV. The electrode was immersed in a 0.1 M ferrocyanide + 0.5 M K2CO3 solution and then cycled for different values of Q (inset of Fig. 5). It is shown that when Q increases from 0.3 to 4.0 mC, the surface increases only by a factor of about 1.9, so the amplification of the phenol current according to Q is not mainly due to an increase in the active electrode surface. It is therefore necessary to consider the increase of the electrocatalytic effect of the platinum surface.

Multiplicative factor of the platinum pre-treated surfaces as a function of Q values. Inset: cyclic voltammogram of 0.1 M ferrocyanide (first cycle) + 0.5 M K2CO3; v = 50 mV s−1; Q = 4 mC.
For comparison purposes, cyclic voltammograms were also recorded at the pre-treated Pt electrodes in p-benzoquinone and hydroquinone aqueous solutions.
Fig. 6 shows the first cyclic voltammograms for 10−2 M p-benzoquinone + 0.5 M H2SO4 solution at pre-treated platinum electrodes at different values of Q. These voltammograms were carried out between 0.3 and 0.9 V. In each voltammogram, only one oxidation peak at around 0.62 V was observed. The inset of Fig. 6 shows a linear dependence of the current of this peak on the Q values. It results in the amplification of about 21 when Q increases from 0.3 to 4.0 mC. Thus, as for phenol, one can admit that the catalytic activity of the platinum surface was increased by the electrochemical pre-treatment.
Cyclic voltammograms of 0.01 M p-benzoquinone (first cycle) + 0.5 M H2SO4; v = 50 mV s−1; Q: (1) 0.3; (2) 0.8; (3) 2.1; (4) 3.0 mC and Q0 = 0.3 mC. Inset: multiplicative factor of oxidation peak current as a function of Q values.
In similar experiments, the pre-treated electrode was immersed in 0.1 M hydroquinone + 0.5 M H2SO4 solution and then recorded between −0.61 and +0.89 V at various Q values. As can be seen from the cyclic voltammograms in Fig. 7, a quasi-reversible peak appear at approximately 0 V, which corresponds to the transformation of hydroquinone to p-benzoquinone and vice versa within a quasi-reversible two-electron process. The oxidation current densities slightly increase with the Q values. It results in an amplification of about 3 when Q increases from 0.3 to 4.0 mC (inset of Fig. 7). This value is much lower than that of both phenol and p-benzoquinone. One can see that this factor is close to that of the electrode surface and thus the electrocatalytic effect of the Pt surface is not important in this case.

Cyclic voltammograms of 0.1 M hydroquinone (first cycle) + 0.5 M H2SO4; v = 50 mV s−1; Q: (1) 0.3; (2) 1.1; (3) 2.6 mC. Inset: multiplicative factor of oxidation peak current as a function of Q values; Q0 = 0.3 mC.
It is generally believed that the degree of oxidation of aromatic compounds, irreversibly adsorbed from aqueous solutions onto platinum electrodes, was dependent on the chemical structure, the type of adsorption and the surface electrode composition [11,24–31].
With regard to the oxidation potential of phenol, p-benzoquinone and hydroquinone presented in Table 1, it should be noted that contrarily to both phenol and p-benzoquinone, the oxidation of hydroquinone begins at potentials lower than + 0.12 V where the Pt oxidation wave begins. So the oxidation of hydroquinone is carried out directly in the platinum double-layer region where the surface of the electrode is covered with a monolayer of adatoms Pt∗ (reduction product of Pt oxide). This oxidation involves a small number of electron transfers per mole while both phenol and p-benzoquinone are oxidized at higher potential values than that of the platinum. This oxidation is attributed to chemical reaction with reversibly deposited OH at open platinum metal sites as it has been explained by Conway [32].
Starting potentials and potentials values of oxidation peaks of phenol, p-benzoquinone, hydroquinone and their multiplicative factors with Q = 4 mC
| Estarting peak (V) | Epeak (V) | Multiplicative factors | |
| Phenol | +0.24 | +0.50 | 46.0 |
| p-Benzoquinone | +0.30 | +0.62 | 21.0 |
| Hydroquinone | −0.09 | +0.02 | 3.0 |
| Pt surface | +0.12 | – | 1.9 |
The importance of PtOH species on the catalytic surface is underlined by the data shown in Fig. 8. In this figure, curve (Pt) shows the platinum voltammogram in solution containing only 0.5 M H2SO4. During the drawing of this voltammogram, if scanning is stopped at −0.61 V, the electrode surface will be covered by a layer of adatoms Pt∗; on the other hand, if scanning is stopped at 0.89 V, the electrode will be covered by an oxide film essentially formed from PtO species following Eq. (3) [7,8]. Curves (1) and (2) show the first scans between 0.30 and 0.89 V in 0.5 M H2SO4 + 10−2 M p-benzoquinone solution, respectively, for these two surface qualities of platinum. It is clear that the p-benzoquinone oxidation peak on the PtO surface disappears (curve 2), whereas this oxidation (curve 1) takes place on the incipient hydrous oxide PtOH readily formed according to the following equation:
| (4) |

Cyclic voltammograms of 0.01 M p-benzoquinone (first cycle) + 0.5 M H2SO4; v = 50 mV s−1; Q = 0.8 mC; voltammograms (1) and (2) registered, respectively, on the pre-treated Pt electrodes where the scanning in potential on curve 0 was stopped before (−610 mV) and after (+890 mV) the Pt oxides reduction peak P3.
It appears from the preceding results that when Q increases by pre-treatment of platinum, the average coordination of the atoms on the surface of the electrode decreases and the energy level of these atoms increases. Consequently, the active sites with respect to the discharge of water (Eq. (4)) become much more numerous and thus the concentration of PtOH species on the surface of the electrode increases.
3.2 Pre-adsorbed phenol oxidation
The adsorption of organic substrates at a platinum electrode in aqueous medium was widely investigated [11,33–35]. In the present work, oxidation of the pre-adsorbed layer of phenol was studied as a function of charge Q. The pre-treated platinum electrode was immersed in a 4 × 10−2 mol L−1 phenol solution for 1 min, then rinsed with bidistilled water. After that, it was transferred to the working solution (0.5 M H2SO4), which was free of phenol, held for 30 s at the adsorbed potential and cycled between −0.61 and 0.89 V. The resulting voltammogram is shown in Fig. 9 along with the results for clean platinum. It can be observed that the hydrogen adsorption reaction between −0.61 and 0.3 V was partially blocked by the adsorbed species. During the first anodic sweep to 0.89 V, the oxidation of the adsorbed layer of phenol gives an oxidation wave in the potential zone where the formation of a surface oxide layer on the platinum electrode occurs.
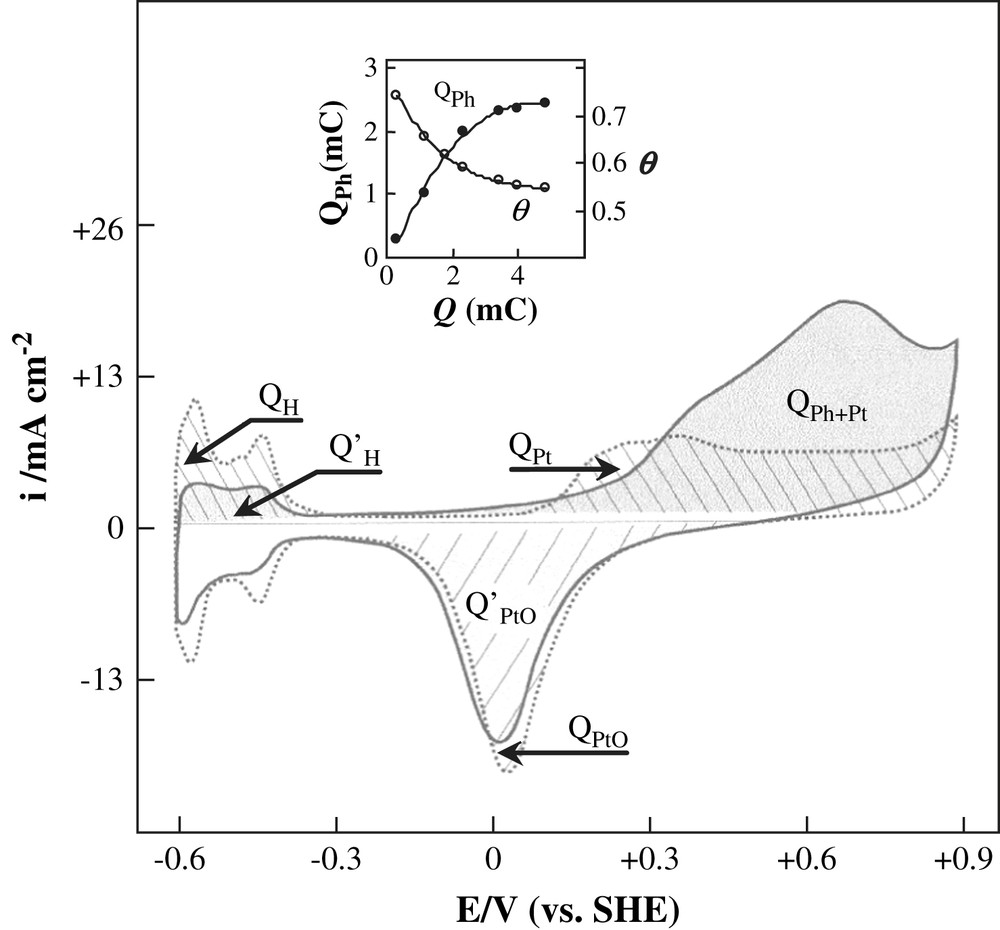
Cyclic voltammogram of pre-adsorbed phenol + 0.5 M H2SO4 on pre-treated Pt electrodes; v = 50 mV s−1; Q = 3.5 mC, QPt and QPh + Pt designed the charge of the anodic waves in the absence and in the presence of adsorbed phenol, respectively. QPtO and are those corresponding to the reduction peaks (P3) of the platinum oxide. corresponds to the platinum oxide formation in the presence of adsorbed phenol. QH and are the quantities of electricity associated with the desorption of hydrogen in the absence and presence of adsorbed phenol, respectively.
Integration of the voltammetric current for anodic and cathodic reactions yielded a coulometric charge Q. In Fig. 9, QPt and QPh + Pt designed the charge of the anodic waves in the absence and in the presence of adsorbed phenol, respectively. QPtO and are those corresponding to the reduction peaks (P3) of the platinum oxide. The charge which would correspond to the platinum oxide formation in the presence of adsorbed phenol could be deduced from the following equation:
| (5) |
The charge QPh corresponding only to the oxidation of the adsorbed phenol layer could be evaluated:
| (6) |
As can be seen in the inset of Fig. 9, QPh increased approximately up to 2.5 when Q increased from 0.3 to 4.0 mC and tended to stabilize for higher values of Q. This result suggests that the increase of QPh was essentially due to the increase of the active surface of the electrode which results from the formation of Pt adatoms (Pt∗). Thus, electro-oxidation is limited by the amount of the adsorbed layer of phenol.
On the other hand, the degree of coverage θ of the electrode surface by the adsorption of the phenol and its own oxidation products can be calculated by the following formula:
| (7) |
The degree of coverage θ decreased as a function of Q; it passed from 74 to 55% when Q increased from 0.3 to 4.0 mC (inset of Fig. 9). In this case, if one admits that the number of exchanged electrons remains constant, the observed decrease of θ is due to the increase of the specific surface area.
3.3 Degradation of phenol
At first, electrolyses of 0.1 M phenol solutions (V = 0.5 L) on platinum oxide electrodes were tested in potentiostatic mode between 400 and 900 mV. However, in all cases, after about 1 min, the reaction was inhibited by the formation of a passivating film which avoids further phenol oxidation and the formation of platinum surface oxide. To overcome this problem, electrolyses were carried out by CV under the same experimental conditions which were used in cyclic voltammograms of phenol. This technique has the advantage of allowing the reactivation of the electrode during the negative sweep [11,36,37]. Conversely, this technique presents a disadvantage: in the cathodic sweep, many phenol oxidation products can be reduced. It results in the reduction of phenol degradation rate and a useless consumption of the electric energy. Consequently, it will be advisable to optimize the relative values of the positive and negative scan rates.
Thus, during an electrolysis test, the phenol solution presents a yellowish color after about 30 min, and a brownish one after 4 h characteristic of the presence of quinonic structures. When the electrolysis was prolonged, these substances were dissolved and the brown color disappeared. Moreover, the oxidation process involves the formation of insoluble product which could be dimers and/or polymers. The electrolytic medium was regularly assayed by liquid chromatography. The analyses were conducted with reference to commercially available products used as standards. Thus, catechol, hydroquinone, p-benzoquinone, oxalic and maleic acids were detected.
4 Conclusion
This study showed that platinum electrodes treated by CV acquire important electrocatalytic properties towards many organic electro-oxidation reactions. The increase of the charge Q which characterizes the surface quality of the pre-treated platinum electrode entails an increase in active sites with respect to the discharge of water to form PtOH species which typically starts at ca. 0.12 V. Once such species are formed they react with both free and adsorbed phenol and p-benzoquinone. It results in the multiplication of the oxidation current by factors of 46 and 21 for phenol and p-benzoquinone, respectively, when Q increases from 0.3 to 4 mC. Because of its chemical structure, this factor, in the case of hydroquinone, is only about 3 for the same increase of Q. In fact, hydroquinone oxidation takes place at a potential lower than 0.12 V and so the oxidation mechanism is different.
The results show that phenol is quantitatively oxidized by CV on a pre-treated platinum electrode to produce, by the intermediary of catechol, hydroquinone, p-benzoquinone, oxalic and maleic acids, whose degradation leads to carbon dioxide.
Acknowledgements
The authors gratefully acknowledge the financial support of the, Comité mixte franco-tunisien pour la coopération universitaire, (Project 96/F 1113).