

1 Introduction
Helicenes have attracted great attention due to their distinct helical backbone [1–9], which incorporates nonplanar π-electrons and makes them highly stable in harsh experimental conditions [10–13]. These helically shaped molecules have shown exceptional chiroptical properties [14–21] and have been applied as suitable components for chiral discotic crystal materials [22,23], conjugated polymers [24–26], and as rotors [27,28]. They have also been used as organic materials for light emitting diodes [29–32], photovoltaic cells [33,34], and circularly polarized luminescence [35–43], thanks to their suitable HOMO and LUMO levels and their thermal and chemical high stability. Their high configurational stability allows appropriately functionalized heterohelicenes to be exploited as optically active or pure ligands and catalysts for enantioselective transformations [44–53].
In this article, we report the synthesis and structural characterization of a new S-containing helically chiral architecture substituted with nitrile groups at defined positions. Our synthetic approach highlights the use of 10-bromobenzo[b]naphtho[2,1-d]thiophene-7-carbonitrile as a new key teracyclic building block to achieve the target helically shaped compound. The nitrile function in this chiral helicene tends to enhance its solubility in organic solvents and may improve its photophysical properties. To the best of our knowledge, no positional isomers of cyano-5-thia[6]helicene have been reported before for making a comparison.
2 Results and discussion
The synthetic pathway to the helical compound began with the condensation of commercially available p-bromophenylacetonitrile (1) with benzo[b]thiophene-2-carboxaldehyde (2) in the presence of sodium methoxide in dry MeOH (Scheme 1). The reaction mixture was stirred for 6 h at room temperature to provide (Z)-3-(benzo[b]thiophen-2-yl)-2-(p-bromophenyl)acrylonitrile (3) in 90% yield. The resulting α,β-unsaturated nitrile was irradiated with a 500 W high-pressure mercury immersion lamp, on an 800 mg scale per 1.2 L of toluene, in the presence of stoichiometric amounts of iodine and a large excess propylene oxide [54], to produce the expected 10-bromobenzo[b]naphtho[2,1-d]thiophene-7-carbonitrile (4) in 95% yield, and overall 85% yield.

Synthetic approach for the benzo[c]phenanthrene-like system (4).
After getting the desired new [4]helicene 4, we have planned to complete the synthesis of the helical chiral compound 5. Thus, 4 has been coupled with 4-cyanostyrene (1.5 equiv) using 0.5 mol % of Hermann's catalyst and sodium acetate in N,N-dimethylacetamide (DMA) according to Scheme 2. After heating at 140 °C, for about 12 h, the coupling product 6 was obtained in 74% yield. Different fractions of the latter compound were subjected to photolysis in dilute solutions using a 500 W high-pressure mercury immersion lamp. The photolysis was carried out on a 200 mg of compound 6 per fraction in a reactor filled with 1.2 L of toluene in the presence of iodine and propylene oxide, leading to 7,14-dicyano-5-thiahexahelicene (5) in 60% yield, and overall 38% yield, after 2.5 h of irradiation.
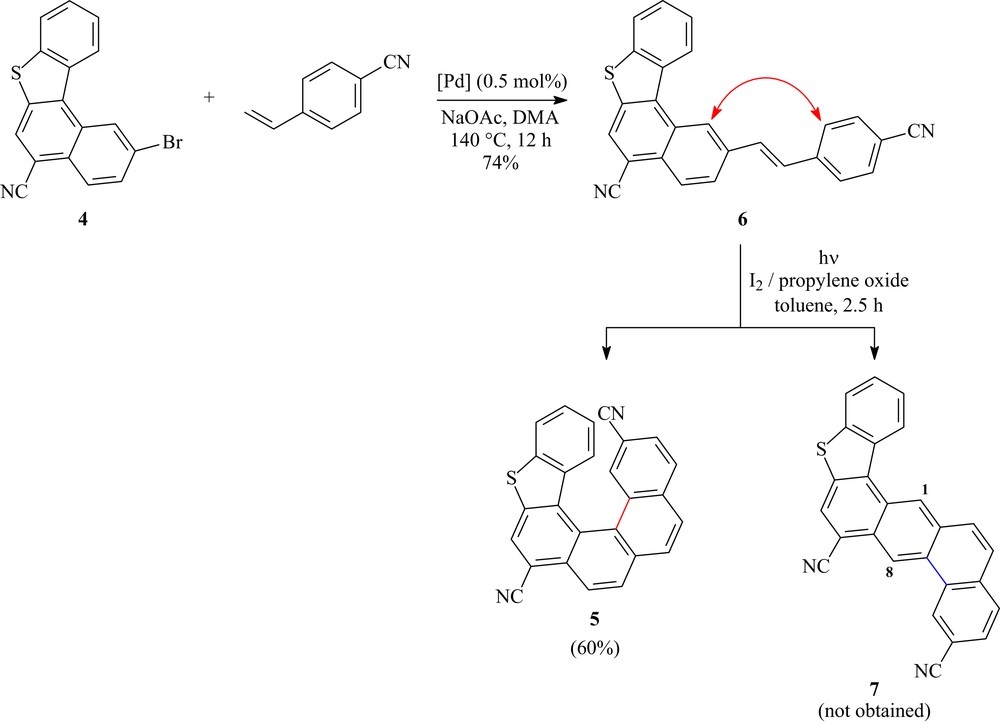
Synthetic pathway leading to racemic helical compound 5.
The photocyclization reaction did not lead to other regioisomers of helicene 5 indicating that ring-closure of alkene 6 had occurred in the periposition of the [4]helicene skeleton. In particular, the S-shaped compound 7 was not formed along this step, as these derivatives would be supposed to exhibit characteristic signals for protons H-1 and H-8 at low field in the proton nuclear magnetic resonance (1H NMR) spectrum [55–57].
Single crystals of 5 were collected as yellow plates by slow evaporation of a dichloromethane solution at room temperature. The product appears to be highly stable in air and to light. The X-ray analysis was carried out on a single crystal obtained from the racemic form of 5 as shown in Fig. 1. The torsion angles, at the inner helical rim, represented by C19C20C21C22 and C22C23C24C25 are unequal and were found to be shorter than C20C21C22C23 and C21C22C23C24 (Table 1).
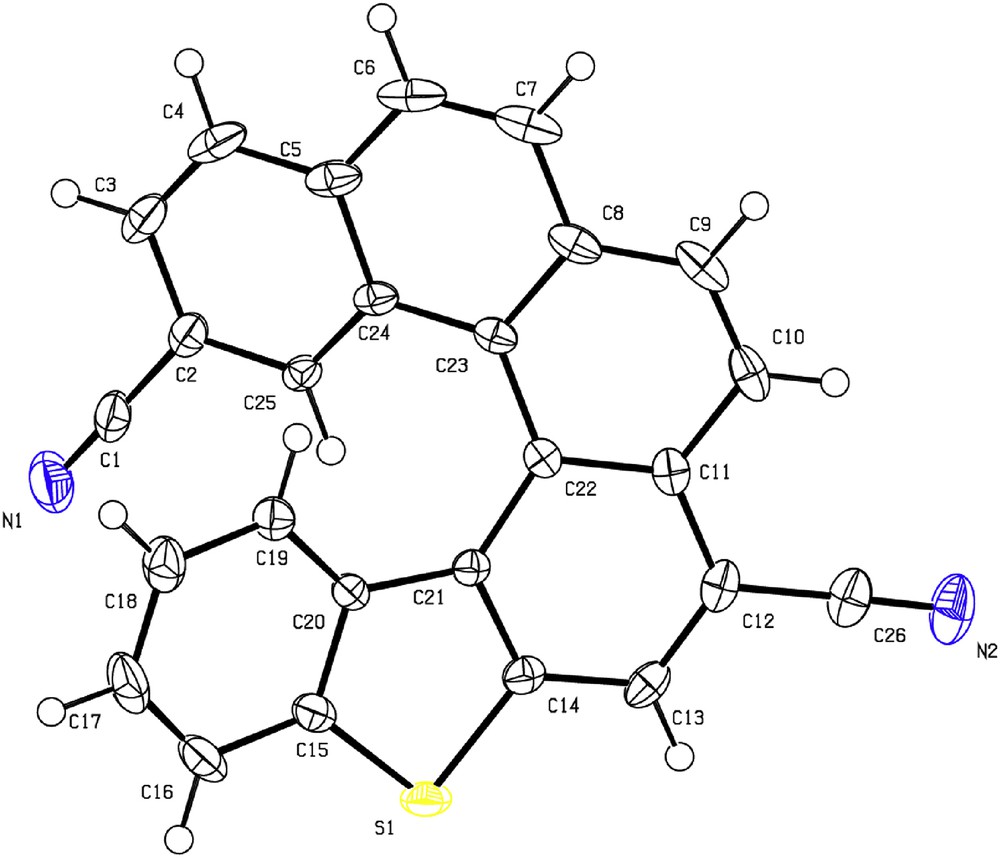
X-ray crystal structure of helicene 5: Oak Ridge Thermal Ellipsoid Plot (ORTEP) drawing.
Torsion angles of compound 5.
| Compound | Torsion anglea (°) | |||
| φ1 | φ2 | φ3 | φ4 | |
| Helicene 5 | 7.3 | 22.6 | 10.85 | 30.0 |
a φ1 = C19C20C21C22; φ2 = C20C21C22C23; φ3 = C22C23C24C25; and φ4 = C21C22C23C24.
The separation of the racemic helicene 5 into its enantiomers was accomplished on a preparative scale by HPLC using a Chiralpak IG column (250 × 10 mm) and n-hexane/2-propanol/dichloromethane (70:20:10) as the mobile phase. The sample was dissolved, then injected on the chiral column, and detected with a UV detector at 280 nm. Thus, a total of 48 mg of pure product was separated, starting from 50 mg of rac-5, equivalent to a yield of 96% (Scheme 3). The earlier eluting fractions contained the dextrorotatory enantiomer in 48% yield with 100% ee. Later eluting fractions gave the levorotatory enantiomer in 48% yield and 100% ee. The enantiomeric purity of both enantiomers was checked by chiral HPLC using the same stationary phase.
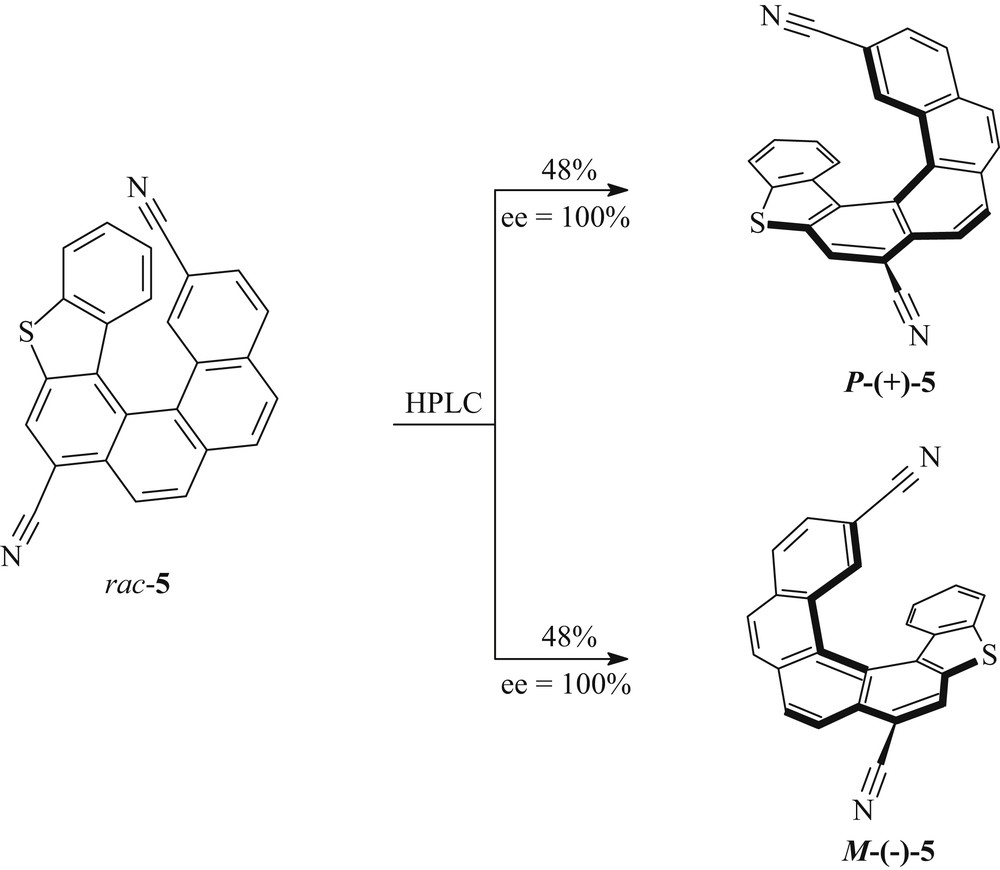
Enantiomeric resolution of the hexacyclic helicene 5.
Optical rotations were measured in dichloromethane using a Jasco P-2000 polarimeter with a sodium lamp (589 nm), a halogen lamp (578, 546, and 436 nm), in a 10-cm cell, thermostated at 25 °C with a Peltier controlled cell holder. The specific rotations obtained for both enantiomers of helicene 5 are mirror image and are summarized in Table 2.
Optical rotations of both enantiomers of helicene 5 in dichloromethane.
| λ (nm) | First eluted enantiomer (+)-5 | Second eluted enantiomer (−)-5 |
| [α]λ25 (c 0.15, CH2Cl2) | [α]λ25 (c 0.15, CH2Cl2) | |
| 589 | +2750 | −2750 |
| 578 | +2900 | −2900 |
| 546 | +3600 | −3600 |
| 436 | +8800 | −8870 |
The chiroptical properties of 7,14-dicyano-5-thihexahelicene (5) in the form of the circular dichroism (CD) spectrum were also measured in dichloromethane (Fig. 2), indicating that complete optical resolution had occurred, which makes assignment of the absolute configurations possible. The dextrorotatory enantiomer (+)-5 recorded a distinct positive maximum at 324 and two distinct negatives maximum at 230 and 352 nm, as depicted in Fig. 1. According to the case of (+)-P-helicene derivatives reported [58,59], the absolute configuration of (−)- and (+)-helicenes 5 could be attributed to M (left-handed helix) and P (right-handed helix), respectively.
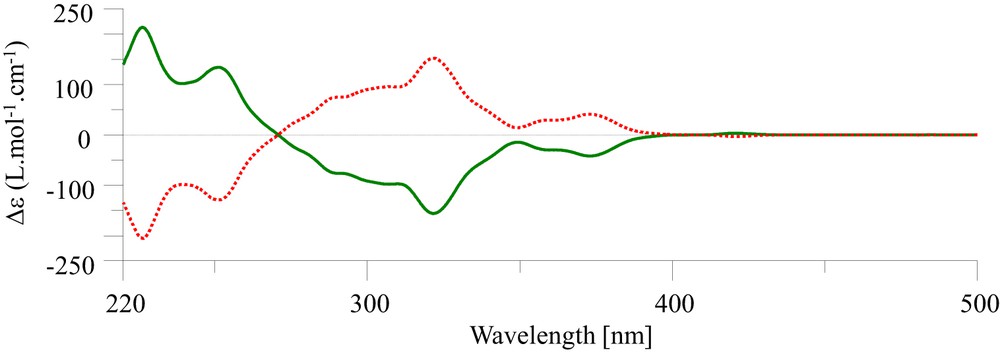
CD spectra of (P)-(+)-5 (red dotted line) and (M)-(−)-5 (green line) in dichloromethane (2.31 × 10−4 M).
The optical properties of helicene 5 have been evaluated in solutions, on the basis of UV–vis absorption and photoluminescence (PL) spectroscopies, at room temperature and the results are gathered in Table 3. The UV–vis absorption spectrum, recorded from a dilute chloroform solution (ca. 1 × 10−6 M) is characterized by several bands between 287 and 450 nm. Three typical strong absorption bands can be found around 314, 354, and 374 nm, additionally two lower-energy band shifts from 421 to 398 nm, with a shoulder peak at 291 nm, which could be assigned to π–π* and n–π* electronic transitions. Thanks to the UV–vis absorption spectroscopy, the optical band gap (Eg-op) was easily determined from the absorption band edge giving an approximately value of 2.87 eV.
Photophysical properties of helicene 5.
| Compound | Absorption | Photoluminescence | |||
| a (nm) | λonset (nm) | Eg-opb (eV) | λemsc (nm) | FWHMd (nm) | |
| Helicene 5 | 314 | 432 | 2.87 | 426 | 42 |
a Absorption maxima, measured in CHCl3 solution (1 × 10−6 M) at room temperature.
b The optical gap (Eg-op) was estimated from the onset point of the absorption spectrum: Eg-op = 1240/λonset.
c Emission maxima, measured in CHCl3 solution (1 × 10−6 M) at room temperature; λext = 375 nm.
d Spectrum full width at half maximum.
Fluorescence study of helicene 5 in a chloroform solution (ca. 1 × 10−6 M) was also conducted (Fig. 3). Irradiation of compound 5 at 375 nm produced emissions with maximum wavelengths (λmax) at 425, 446, and 451 nm with a shoulder peak at 479 nm where the first and the third wavelength are being the most prominent.
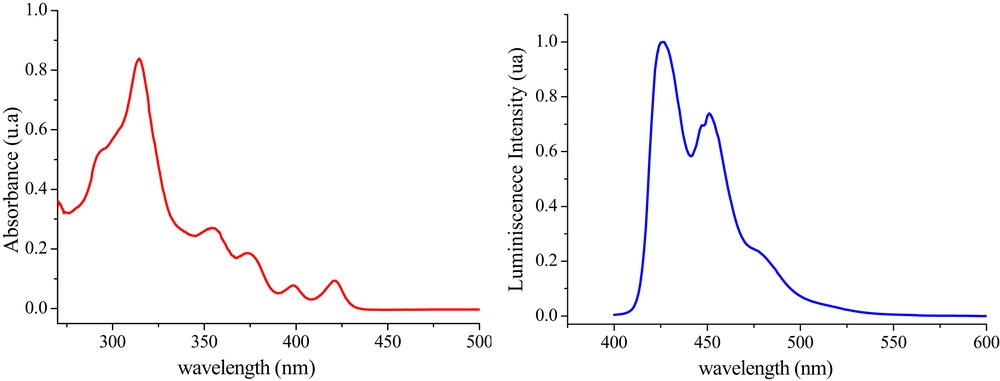
UV–vis absorption (red line) and PL (blue line) spectra of compound 5 in dilute chloroform solutions (ca. 1 × 10−6 M).
The electrochemical behavior of helicene 5 has been studied by cyclic voltammetry experiment. The CV measurement was performed at room temperature, under inert atmosphere, in a solution of n-Bu4NClO4, as the electrolyte, in CH3CN using a three-electrode cell at a scan rate of 50 mV/s. The resulting voltammogram showed irreversible anodic and reversible cathodic peaks (Fig. 4). As listed in Table 4, the onset of oxidation (Vonset-ox) was at 1.27 V, whereas the onset of reduction (Vonset-red) was found to be at −1.10 V, all measured versus (Ag/AgCl). From these latter potentials, the ionization potential and the electron affinity (Ea) have been estimated, and consequently the electrochemical band gap (Eg-el) could be simply calculated using the following empirical method [59,60]:
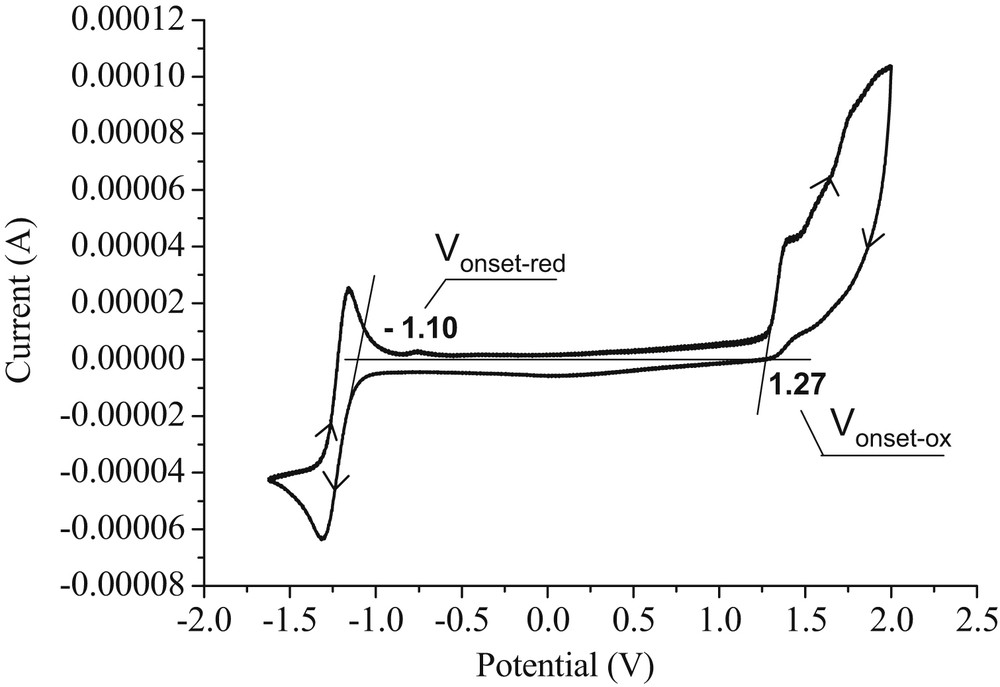
Cyclic voltammogram of helicene 5 in 0.1 M (n-Bu)4NBF4/acetonitrile at a scan rate of 50 mV/s.
Electrochemical properties of helicene 5 in acetonitrile.
| Compound | Vonset-ox (V) | Vonset-red (V) | EHOMO (eV) | ELUMO (eV) | Eg-el (eV) |
| Helicene 5 | 1.27 | −1.10 | −5.57 | −3.20 | 2.37 |
3 Conclusions
We have described a short procedure for the preparation of a new sulfur-containing hexacyclic helicene, using 4 as a suitable key teracyclic building block. We prepared the target racemic 5 in only four steps with a 38% overall yield, then we accomplished its enantiomeric resolution in high optical purity. This optical pure helicene may serve as a ligand in asymmetric synthesis or as a building block for supramolecular architectures.
4 Experimental section
4.1 General
Most experiments were performed under anhydrous conditions and an inert nitrogen atmosphere. Unless otherwise noted, starting materials and solvents used in this work were obtained from commercial suppliers. Column chromatography purifications were performed in the air over silica gel (40–63 nm; 230–240 mesh). The reactions were monitored by thin-layer chromatography (TLC) using commercial silica-gel plate 60 coated with a fluorescence indicator (Silicycle Chemical division, 0.25 mm, F254.). UV (254 nm) was used in the visualization of a TLC plate. Irradiation reactions were conducted using 500 mercury vapor lamp. 1H NMR and carbon nuclear magnetic resonance (13C NMR) spectra were acquired at room temperature in CDCl3 as a solvent using Bruker AM-300. Chemical shifts were reported in parts per million (ppm) relative to tetramethylsilane as an external standard. Signals due to the solvent served as the internal standard (CHCl3: δ 7.26 for 1H, δ 77.16 for 13C). The acquisition parameters are shown on all spectra. The terms s, d, t, m indicate, respectively, singlet, doublet, triplet, and multiplet; b is for broad; dd is a doublet of doublets; td is a triplet of doublets; the list of coupling constants (J) corresponds to the order of the multiplicity assignment. Melting points were measured using a Bibby Scientific Stuart Digital. UV–vis spectroscopy was conducted using a spectrophotometer UV-1600PC. Cyclic voltammetric experiments were performed using three-electrode cell, consisting of a glassy carbon working electrode, a platinum counter electrode, and an Ag/Ag+ 0.1 M AgNO3 in acetonitrile nonaqueous reference electrode. Investigations have been carried out in millimolar anhydrous acetonitrile solutions containing 0.1 M of n-tetrabutylammonium perchlorate (n-Bu4NClO4) as a supporting electrolyte. The reference electrode Ag/Ag+ was calibrated with respect to the formal potential of the ferrocenium/ferrocene (Fc+/Fc) couple in MeCN. All measurements were scanned in negative and positive directions at a scan rate of 50 mV/s, which are controlled by a PGSTAT30 Autolab potentiostat. Optical rotations were measured in a 10 cm thermostated quartz cell using a Perkin–Elmer-241 polarimeter. CD spectra were measured using a JASCO J-815 spectrometer equipped with a JASCO Peltier cell holder PTC-423 to maintain the temperature at 25.0 ± 0.2 °C. A CD quartz cell of 1 mm of optical path length was used. The CD spectrometer was purged with nitrogen before recording each spectrum, which was the baseline subtracted. The baseline was always measured for the same solvent and in the same cell as the samples.
4.2 Experimental procedure and spectroscopic data for (Z)-3-(benzo[b]thiophen-2-yl)-2-(p-bromophenyl)acrylonitrile (3)
4-Bromophenylacetonitrile (1 g, 5.1 mmol) and benzo[b]thiophene-2-carboxaldehyde (825 mg, 5.1 mmol) are dissolved in 30 mL of dry MeOH and stirred at 0 °C for 10 min. Sodium methoxide (380 mg, 7.1 mmol) was added in small portions, and the solution was stirred for 15 min at 0 °C and then for 6 h at room temperature. The precipitate was collected by filtration, washed with 10 mL of MeOH, then with water, and dried at 50 °C to give the α,β-unsaturated nitrile 3 as a yellow powder (1.55 g, 90%); Rf = 0.36 (cyclohexane/EtOAc, 90:10); mp = 160–162 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.37–7.45 (m, 2H), 7.52–7.60 (m, 4H), 7.69 (s, 1H, Hvinyl), 7.82–7.87 (m, 3H); 13C NMR (75 MHz, CDCl3): δ (ppm): 109.68 (C), 117.35 (CN), 122.43 (CH), 123.58 (C), 124.65 (CH), 125.12 (CH), 126.67 (CH), 127.33 (2CH), 130.01 (CH), 132.31 (2CH) 132.86 (C), 134.86 (CH), 137.37 (C), 138.69 (C), 141.28 (C); HRMS (MALDI-TOF): [M]+ calcd for (C17H10BrNS), 338.9717; found, 338.9711.
4.3 Experimental procedure for the photocyclization reaction
Alkene 3 (800 mg, 2.3 mmol) or 6 (200 mg, 0.51 mmol) was dissolved in 1.2 L of toluene and a stoichiometric amount of iodine and an excess of propylene oxide (50 equiv) are then added. The resulting solution was subjected to irradiation under a 500 W high-pressure Hg-vapor lamp. The reaction evolution was monitored by TLC. After completion, toluene was removed under reduced pressure and the crude residue was purified by flash column chromatography [SiO2, cyclohexane/EtOAc (80:20)].
4.3.1 10-Bromobenzo[b]naphtho[2,1-d]thiophene-7-carbonitrile (4)
Starting from 800 mg of alkene 3, we obtained 755 mg (95% yield) of 4 as a pale yellow solid; Rf = 0.39 (cyclohexane/EtOAc, 90:10); mp = 208–210 °C; 1H NMR (CDCl3, 300 MHz): δ (ppm): 7.57–7.67 (m, 2H), 7.77 (d, J = 8.7 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 9 Hz, 1H), 8.25 (s, 1H), 8.65 (d, J = 7.8 Hz, 1H), 9.06 (s, 1H); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 108.57 (C), 116.64 (CN), 122.78 (C), 122.96 (CH), 124.59 (CH), 125.24 (CH), 125.77 (CH), 126.69 (CH), 127.56 (CH), 127.63 (CH), 128.11 (C), 129.75 (CH), 130.41 (C), 131.15 (C), 134.50 (C), 137.15 (C), 140.69 (C); HRMS (MALDI-TOF): [M]+ calcd for (C17H8BrNS), 336.9560; found, 336.9554.
4.3.2 7,14-Dicyano-5-thiahexahelicene (5)
Starting from 200 mg of alkene 6, we obtained 119 mg (60% yield) of 5 as a yellow solid; Rf = 0.38 (cyclohexane/EtOAc, 80:20); mp > 300 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 6.80–6.88 (m, 2H); 7.40 (td, J1 = 8.1 Hz, J2 = 1.5 Hz, 1H); 7.60 (dd, J1 = 8.1 Hz, J2 = 1.2 Hz, 1H); 7.96 (d, J = 8.1 Hz, 1H); 8.08–8.16 (m, 5H); 8.49 (d, J = 8.7 Hz, 1H); 8.56 (s, 1H); 13C NMR (75 MHz, CDCl3): δ (ppm): 108.70 (C); 109.24 (C); 117.63 (CN); 118.49 (CN); 123.17 (CH); 123.34 (CH); 124.26 (C); 124.84 (C); 125.48 (CH); 126.13 (CH); 127.38 (CH); 127.53 (CH); 127.95 (CH); 128.23 (CH); 128.58 (CH); 129.11 (C); 119.13 (CH); 119.28 (CH); 130.95 (C); 133.08 (C); 133.39 (C); 133.78 (CH); 134.32 (C); 134.39 (C); 138.72 (C); 140.33 (C); HRMS (MALDI-TOF): [M+Na]+ calcd for (C26H12N2NaS), 407.0613; found, 407.0611.
Crystal data for compound 5 (C26H12N2S) were recorded using a D8 VENTURE Bruker AXS diffractometer, M = 384.44, monoclinic, space group P 21/n. a = 6.8110(7) Å, b = 12.5553(11) Å, c = 21.831 Å, V = 1858.3(3) Å3, Z = 4, ρcalcd = 1.374 g cm−3, X-ray source Mo Kα, λ = 0.71073 Å, T = 150(2) K; observed reflections 3648; refinement method full-matrix least-squares on F2; parameters refined 262; R(F) = 0.0369, ωR(F2) = 0.0904. Crystallographic data for the structure in this article have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1539337. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 (0) 1223 336033; e-mail: deposit@ccdc.cam.ac.uk or via www.ccdc.cam.ac.uk/data_request/cif.
4.4 Experimental procedure and spectroscopic data for the Heck coupling
Compound 4 (500 mg, 1.4 mmol) and NaOAc (133 mg, 1.6 mmol) are placed in a double-necked flask in the presence of 2 mL of N,N-dimethylacetamide and repeatedly degassed and purged with argon several times. 4-Cyanostyrene (1.6 equiv) was added and the mixture was heated to 100 °C. Next, a 7 mg solution of Hermann's catalyst (0.5 mol %) in 2 mL of N,N-dimethylacetamide was added and the mixture was heated to 140 °C for 12 h. After cooling the mixture to room temperature, 30 mL of distilled water was added and the organic phase was extracted with dichloromethane (3 × 30 mL). The combined organic phases were dried over MgSO4, the solvent was removed, and the residue was chromatographed through a flash silica gel column using cyclohexane/EtOAc (90:10), as the mobile phase, to give 422 mg of (E)-10-(p-cyanostyryl)benzo[b]naphtho[2,1-d]thiophene-7-carbonitrile (6): 74% yield; yellow solid; Rf = 0.28 (cyclohexane/EtOAc, 80:20); mp = 217–219 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.34 (d, J = 16.2 Hz, 1H, Hvinyl); 7.52 (d, J = 16.2 Hz, 1H, Hvinyl); 7.61 (d, J = 7.8 Hz, 1H); 7.66 (d, J = 7.8 Hz, 1H); 7.67–7.75 (m, 4H); 8.01 (d, J = 8.7 Hz, 1H); 8.06 (d, J = 7.5 Hz, 1H); 8.35 (s, 1H); 8.43 (d, J = 8.7 Hz, 1H); 8.92 (d, J = 8.4 Hz, 1H); 9.07 (s, 1H); 13C NMR (75 MHz, CDCl3): δ (ppm): 109.08 (C), 111.34 (C), 117.63 (CN), 118.82 (CN), 123.50 (CH), 123.63 (CH), 124.20 (CH), 125.43 (CH), 125.63 (CH), 127.13 (CH), 127.20 (2CH), 127.33 (CH), 128.12 (CH), 129.21 (CH), 130.21 (C), 130.58 (C), 131.82 (CH), 132.47 (C), 132.63 (2CH), 135.50 (C), 136.57 (C), 137.45 (C), 141.28 (C), 141.41 (C); HRMS (MALDI-TOF): [M]+ calcd for (C26H14N2S), 386.0877; found, 386.0876.
4.5 Enantiomeric resolution of racemic helicene (5)
The optical resolution of rac-5 was performed by HPLC according to the following conditions: column, Chiralpak IG (250 × 10 mm); mobile phase, n-hexane/2-propanol/dichloromethane (70:20:10); flow rate, 5.0 mL min−1; temperature, 298 K; detection, UV; k = 280 nm. The levorotatory enantiomer was firstly eluted in 48% yield, followed by the dextrorotatory enantiomer, which is obtained in 48% yield; retention times of the levorotatory and dextrorotatory enantiomers are 8.40 and 10.32 min, respectively.
4.5.1 Selected spectral data for (P)-(+)-7,14-dicyano-5-thiahexahelicene (5)
100% ee; yellow solid; mp > 300 °C; +2750 (c 0.15, CH2Cl2); HRMS (MALDI-TOF): [M + Na]+ calcd for (C26H12N2NaS), 407.0618; found, 407.0614.
4.5.2 Selected spectral data for (M)-(−)-7,14-dicyano-5-thiahexahelicene (5)
100% ee; yellow solid; mp > 300 °C; −2750 (c 0.15, CH2Cl2); ESI-MS: m/z = 384.07 [M+].
Acknowledgments
The authors are grateful to the DGRS (“Direction générale de la recherche scientifique”) of the Tunisian Ministry of Higher Education and Scientific Research for financial support. The authors thank Dr. Nicolas Vanthuyne (Aix-Marseille Université, Service 432-Plateforme de chromatographie chirale ISM2-UMR 7313) for the HPLC analysis.