

1 Introduction
π-Conjugated oligomers and polymers have emerged as promising materials for application in flexible, lightweight and low-cost electronic devices such as organic light-emitting diodes (OLEDs), field-effect transistors (FETs), plastic lasers and photovoltaic cells [1]. The optical and electronic properties of these derivatives depend significantly on their HOMO–LUMO gap and the electron density of the carbon backbone. In order to prepare novel conjugated frameworks with enhanced performances, several strategies have been developed to vary these parameters. A successful strategy in order to obtain such frameworks involves the incorporation of aromatic heterocycles with different aromatic character and electronic nature in the backbone of π-conjugated systems [1d,2]. Indeed, low aromaticity favours electron delocalisation along the main chain, while maximum conjugation is obtained in copolymers with alternating electron-rich and electron-deficient subunits due to intramolecular charge transfer (ICT) [2a–2d]. Heterocyclopentadienes have been extensively used for such purposes, since their electronic properties depend on the nature of the heteroatoms [2f,2j–2l,12]. In this context, the phosphole ring is an attractive synthon since it displays electronic properties that are markedly different from those of the widely used highly aromatic thiophene and pyrrole rings [3,4]. The phosphorus atom does not readily form s–p hybrids and mainly employs p-electrons for bonding. This “inert-pair effect” implies a pyramidal geometry of the tricoordinate phosphorus atom with the lone pair having a pronounced s-character [3,5]. These geometric and electronic features prevent efficient interaction of the phosphorus lone pair with the endocyclic dienic system [5a,5b]. In fact, the delocalisation within the phosphole ring arises from hyperconjugation involving the exocyclic P–R σ-bond and the π-system of the dienic moiety [6]. Thus, phospholes possess (i) a weak aromatic character, which should favour delocalisation of the π-system, and (ii) a reactive heteroatom, which potentially offers the possibility of tuning the HOMO and the LUMO levels via chemical modification such as coordination to metal ions. These unique properties, that set phosphole apart from thiophene and pyrrole, prompted us to prepare and study the physical properties of π-conjugated materials containing phosphole rings. Moreover, the presence of this reactive P atom allows the use of the tool of coordination chemistry for the 3D organization of π-conjugated chromophores. This organization in order to control and understand the electronic interactions between individual chromophoric subunits is of fundamental importance for the engineering of organic materials relevant to electronic or optoelectronic applications. New supramolecular assemblies have thus been characterized, whose physical properties depend on the intrinsic properties of the phosphole-based ligand as well as on their controlled specific organisation.
This account will summarise our results on the development of π-conjugated materials incorporating phosphorus moieties and metallic ions. The synthesis, the applications of organophosphorus complexes and their supramolecular arrangement will be detailed.
2 π-Conjugated derivatives incorporating phosphole ring
2.1 Synthesis and physical properties
The synthetic route to organophosphorus derivatives 3 employs the one-pot “Fagan–Nugent method” [7] involving the oxidative coupling of functionalized octa-1,7-diynes 1 with zirconocene followed by treatment with a dihalogeneted phosphine (Scheme 1) [8]. This synthetic pathway is rather simple and efficient, allowing gram-scale preparations, since the functionalized octa-1,7-diynes are obtained in good yields by Sonogashira couplings [8]. 1-Phenylphospholes 3a–g (Scheme 1) can be isolated as air-stable solids following flash column chromatography on basic alumina. They are readily soluble in common organic solvents such as CH2Cl2, THF or CH3CN. They exhibit classical NMR data and their structures were supported by high-resolution mass spectrometry and elemental analysis.
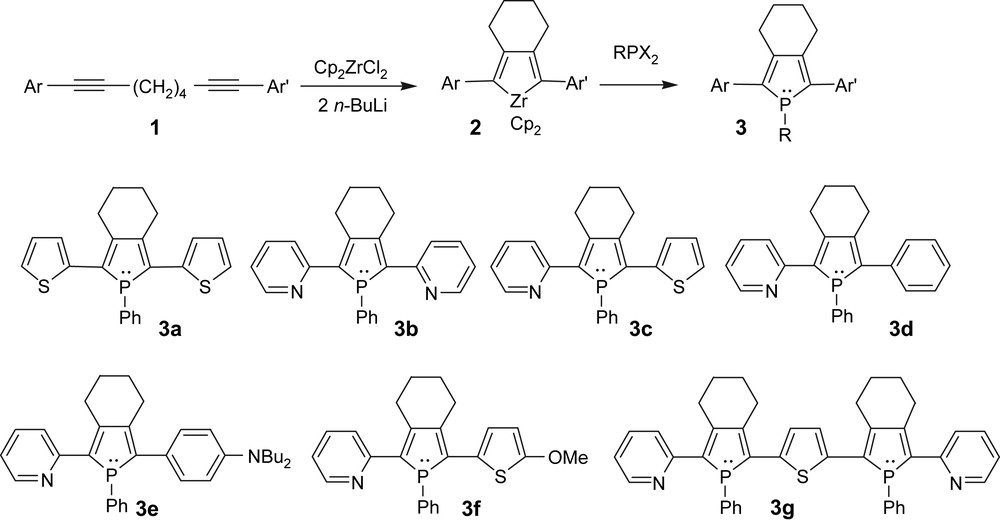
General synthetic route to 2,5-diarylphospholes.
It is noteworthy that this synthetic route allows the preparation of symmetrical (Ar = Ar′) and unsymmetrical (Ar ≠ Ar′) 2,5-disubstituted 1-phenylphospholes. This dissymmetry is easily introduced, at the level of the functionalized 1,7-diynes 1 through successive Sonogashira coupling reactions [9]. A large variety of functional groups can be introduced, leading to numerous 1-phenylphosphole derivatives 3a–f with markedly different physical properties [9]. Moreover oligo(heterocycle-phosphole) derivative 3g with precise length has also been obtained according to this synthetic procedure [10].
The X-ray diffraction study performed on the model compounds 3a [8b,d] and 3b [8c,d] (Scheme 1, Fig. 1) shows that the three heterocycles are almost coplanar, with the phosphorus atom being strongly pyramidalised. The lengths of the C–C linkages between the rings are in the range expected for Csp2–Csp2 bonds. These solid-state data indicate that an efficient delocalisation of the π-system takes place over the three heterocycles in oligomers 3a,b, as supported by theoretical studies [8d].
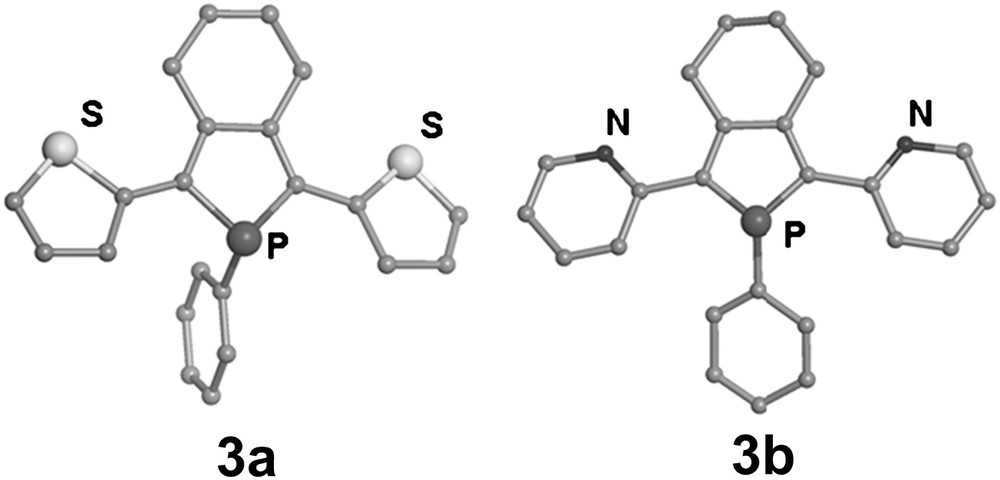
Perspective views of the crystallographic structures of the phosphole based ligands 3a and 3b. Hydrogen atoms have been omitted for clarity.
The presence of an extended π-conjugated system in phospholes 3a,b was also confirmed by the observation of a broad absorption in the visible region of their UV–vis spectrum. The energy of these absorptions, attributed to π–π∗ transitions, depends dramatically on the nature of the 2,5-substituents of the phosphole ring [8d]. The values of λmax and the optical end absorption λonset (the solution optical “HOMO–LUMO” gap) [8d], become red-shifted on replacing the 2-pyridyl groups (λmax = 390 nm) by 2-thienyl rings (λmax = 412 nm). Theoretical studies showed that this bathochromic shift is due to a better interaction between the HOMO of phosphole with the HOMO of thiophene, compared to that of pyridine [8d,11]. It is noteworthy that the λmax value recorded for 3a (412 nm) [8d] is considerably more red-shifted than that of the related tert-thiophene (355 nm) [12]. This observation is in agreement with theoretical studies predicting that heterocyclopentadienes with a low aromatic character are optimal building blocks for the synthesis of extended π-conjugated systems with low HOMO–LUMO gap [1d,8a].
Phospholes 3a,b are also fluorophores and their λem depend on the nature of the 2,5-substituents. A blue emission is observed for 1-phenyl-2,5-di(2-pyridyl)phosphole 3b (λem = 463 nm), whereas the emission of 1-phenyl-2,5-di(2-thienyl)phosphole 3a is red-shifted (Δλem = 35 nm). Cyclic voltammetry (CV) performed at 200 mV s−1 revealed that the redox processes observed for all the σ3-phospholes 3 are irreversible and that their redox properties are related to the electronic properties of the phosphole substituents [8d]. For example, derivative 3a featuring electron-rich thienyl substituents is more easily oxidised than compound 3b, which possesses electron-deficient pyridyl substituents [8d].
2.2 Fine tuning of the physical properties via chemical modifications of the phosphole ring
One of the appealing properties of phosphole rings is the versatile reactivity of the endocyclic heteroatom that allows creating structural diversity in these types of systems. This feature offers a direct access to a broad range of new π-conjugated systems from single P-containing chromophores, without the need for additional multi-step syntheses, as illustrated with dithienyl-phospholes 3a (Scheme 2). The chemical modifications of the nucleophilic P centre (Scheme 2) have a profound impact on the optical and electrochemical properties of the phosphole oligomers as a whole. For example, a chemical modification of the σ3-phosphole 3a leading to the neutral σ4-derivatives 4–6 (Scheme 2) induces a red shift in their emission spectra and an increase and decrease of their oxidation and reduction potentials, respectively. It is also noteworthy that the quantum yields of the gold complex 5 (Φ = 14%) is much higher than those of the corresponding phosphole 3a (Φ = 5%) or its thioxo-derivative 4 (Φ = 4.6%), illustrating that the nature of the P-modification has a considerable impact on the optical properties of these fluorophores [8d,13].
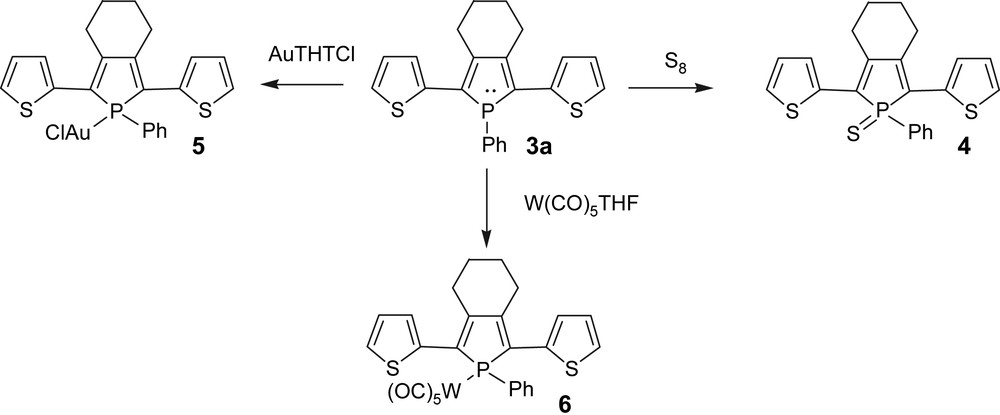
Chemical modifications on the nucleophilic P centre.
The comparison of the solution and thin film optical properties of the thioxophosphole 4 shows that this compound does not form aggregates in the solid state. This property, which is supported by an X-ray diffraction study, results in an enhancement of the fluorescence quantum yields in the solid state. In contrast, the solution and thin film emission spectra of the gold complex 5 are different. Two broad emission bands are observed for the thin films, one at a wavelength similar to that of the solution spectrum (550 nm), and a second which is considerably red-shifted (690 nm). The excitation spectra of 5 in thin film, recorded for emission at 550 or 700 nm, are similar to its absorption spectrum, clearly indicating that both emissions are due to the (phosphole)gold complex 5. The low-energy luminescence band obtained in thin film with phosphole–Au(I) complex 5 very probably arises from the formation of aggregates.
The uncommon emission properties of these organophosphorus derivatives prompt us to use them as emissive layer in single- and multilayer OLEDs. In particular, it has been shown that the emission color of the devices and their performances vary with the nature of the P material. Upon sublimation, phosphole 3a decomposed, while the more thermally stable thioxo-derivative 4 formed homogeneous thin films on an indium–tin-oxide (ITO) semi-transparent anode, allowing a simple layer OLED to be prepared [13]. This device exhibits yellow emission for a relatively low turn-on voltage of 2 V. The comparatively low maximum brightness (3613 cd m−2) and electroluminescence (EL) quantum yield (0.16%) can be increased by nearly one order of magnitude using a more advanced device, in which the organic layer consisting of 4 was sandwiched between hole- and electron-transporting layers (α-NPD and Alq3, respectively). The Au(I) complex 5 (Scheme 2) is thermally stable enough to give homogeneous thin films upon sublimation in high vacuum. The corresponding single layer device exhibited an EL emission covering the 480–800-nm domain [13], a result which is encouraging for the development of white-emitting OLEDs based on phosphole–Au complexes. Note that the low-energy emissions are very probably due to the formation of aggregates in the solid state [13]. These results constitute the first application in optoelectronics of π-organophosphorus materials.
In conclusion, co-oligomers with a phosphole core exhibit very interesting properties. They are air-stable, provided that the P-substituent is a phenyl group, they exhibit low optical gap and their electronic properties can be tuned by varying the nature of the 2,5-substituents (2-thienyl or 2-pyridyl) or chemical modification of the phosphole ring. Exploitation of this method of tailoring π-conjugated systems, which is not possible with pyrrole and thiophene units, has led to the optimization of the properties of thiophene–phosphole co-oligomers for their use as materials suitable for OLED fabrication [13].
3 Coordination chemistry of 2-(2-pyridyl)phosphole derivatives
Compared to organic molecules, metal complexes offer a larger variety of molecular structures and a diversity of electronic properties [14]. Since it is well-known that phospholes and pyridines behave as classical two-electron donor toward transition metals [14,15], we have explored the coordination behaviour of 2-pyridyl-substituted phospholes with the aim to obtain complexes exhibiting interesting photophysical properties and original structures.
3.1 Square-planar complexes exhibiting a NLO activity
The pyridylphosphole ligands possess two coordination centres with different stereo-electronic properties, which, in accordance with the Pearson's anti-symbiotic effect [16,17], allows the control of the orientation of a second chelating ligand in the coordination sphere of a square-planar d8-metal centre [18]. This property has been exploited in order to control the in-plane parallel arrangement of 1D-dipolar chromophores. Phospholes 3e and 3f (Scheme 1), have a typical “Donor–(π-bridge)–Acceptor” dipolar topology incorporating a 1,4-P,N chelate. They exhibit moderate NLO activities (β1, 9 μm, ca. 30 × 10−30 e.s.u.) compared to classical chromophores such as Dispersed Red 1 (DR1) (β, 50 × 10−30 e.s.u.) [9a]. These low values are consistent with the weak acceptor character of the pyridine group. However, their potential in NLO is considerably increased by their coordination behaviour toward Pd(II) centres. The reaction of two equivalents of 2-(2-pyridyl)phospholes 3e,f with [Pd(CH3CN)4][BF4]2 afforded complexes 7e,f as single diastereoisomers (Scheme 3) [9a], with the P and N donor in a mutual trans-arrangement. Clearly, the trans-influence can overcome the natural tendency of 1D-dipolar chromophores to adopt an anti-parallel alignment as the square-planar metal centre acts as a template imposing a non-centrosymmetric assembly of two identical 1D-chromophores 3e,f. Furthermore, the metal centre plays a puzzling role, since a considerable enhancement of the NLO activities is observed upon complexation. For example, complex 7e exhibits fairly high non-linear optical activities with β value of 180 × 10−30 e.s.u, a value which is much higher than the sum over the contribution of two sub-chromophores 3e. In a first approach, this effect could be related to an increase of the acceptor character of the pyridine groups [19]. However, it is very likely that the origin of this large β enhancement is due to metal-to-ligand (MLCT) or ligand-to-metal-to-ligand charge transfers (LMLCT) evidenced by UV–vis spectroscopy [9a] that contribute coherently to the second harmonic generation. In conclusion, the coordination chemistry offers a simple synthetic methodology for controlling the in-plane parallel arrangement of P,N-dipoles in a molecular assembly.
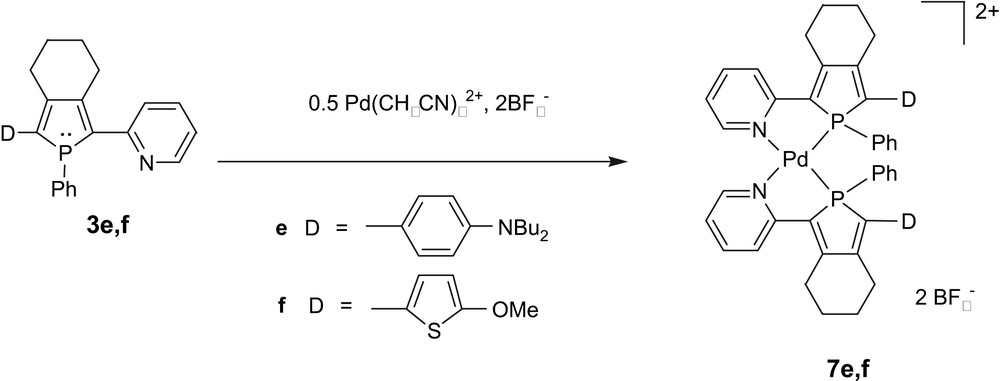
Synthesis of complexes exhibiting high NLO activities.
3.2 Ruthenium complexes
A great attention has been paid to the synthesis and photophysical properties of dinuclear ruthenium complexes containing bridging π-conjugated ligands [20]. The design of the bridging ligands allows tuning of the electronic properties of these dyads and has led to important progress in the field of photoinduced electron or energy transfer processes. These properties prompted us to investigate a new family of derivatives possessing two terminal (2-pyridyl)phosphole moieties joined by a 2,5-thienyl spacer as potential bridging ligands to build bimetallic ruthenium complexes.
The target complex 8 was obtained by addition of [(p-cymene)RuCl2]2 and two equivalents of KPF6 to the ligand 3g (Scheme 4) [10]. This coordination is diastereoselective, each diastereoisomer of 3g giving one diastereoisomer of 8. The UV–vis spectrum of 3g exhibits an absorption maximum in the visible region at 482 nm attributed to π–π∗ transitions of the π-conjugated system [10]. This value is considerably red-shifted (Δλmax = 84 nm) compared to that of 3c (Scheme 1). The coordination of the 2-pyridylphosphole moieties of 3g has almost no influence on the λmax observed for complex 8. This is probably due to the symmetrical structure of complex 8, which prevents efficient ILCT. It is noteworthy that this broad ligand-centred transition overlaps the possible MLCT transition arising from Ru(II) to π∗-orbital of the coordinated 2-pyridylphosphole moiety.

Synthesis of ruthenium complex 8.
4 Coordination chemistry of 2,5-(2-pyridyl)phosphole derivative: complexes bearing bridging phosphane ligands and coordination-driven supramolecular organization of π-conjugated chromophores
4.1 Bimetallic coordination complexes bearing a bridging phosphane ligand
Toward Pd(I) and Cu(I) metal centres, 2,5-bis(2-pyridyl)-phosphole ligand 3b revealed a very original coordination chemistry in comparison to the other 2-(2-pyridyl)-phosphole P,N ligands 3c–f. A family of coordination complexes exhibiting a bridging phosphane coordination mode was thus characterized, a very rare coordination mode for such a commonly used family of ligand.
4.1.1 Pd(I) bimetallic complexes
Ligand 3b was reacted with [Pd2(CH3CN)6][BF4]2 and afforded complex 9 in a 85% yield (Scheme 5) [9b,21]. The X-ray crystal structure of complex 9 (Fig. 2) evidences a dimer of square-planar Pd(I) centres gathered by two 2,5-bis(2-pyridyl)phospholes 3b acting as 6-electron μ-1κN:1,2κP:2κN donors.
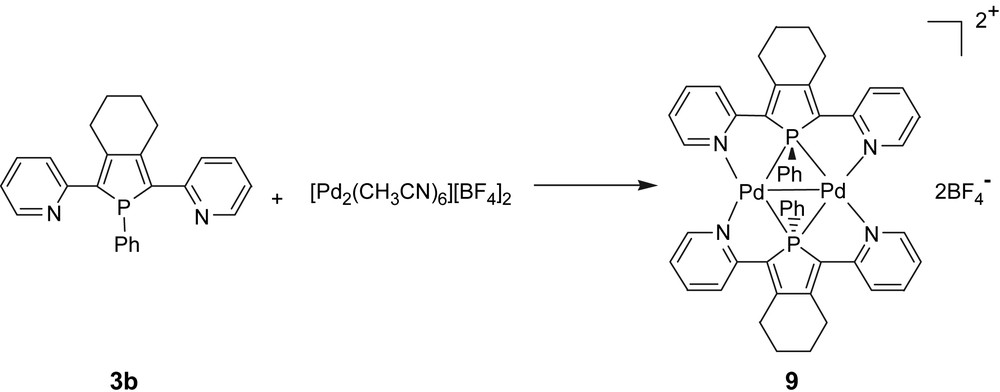
Synthesis of Pd(I) dimer 9.
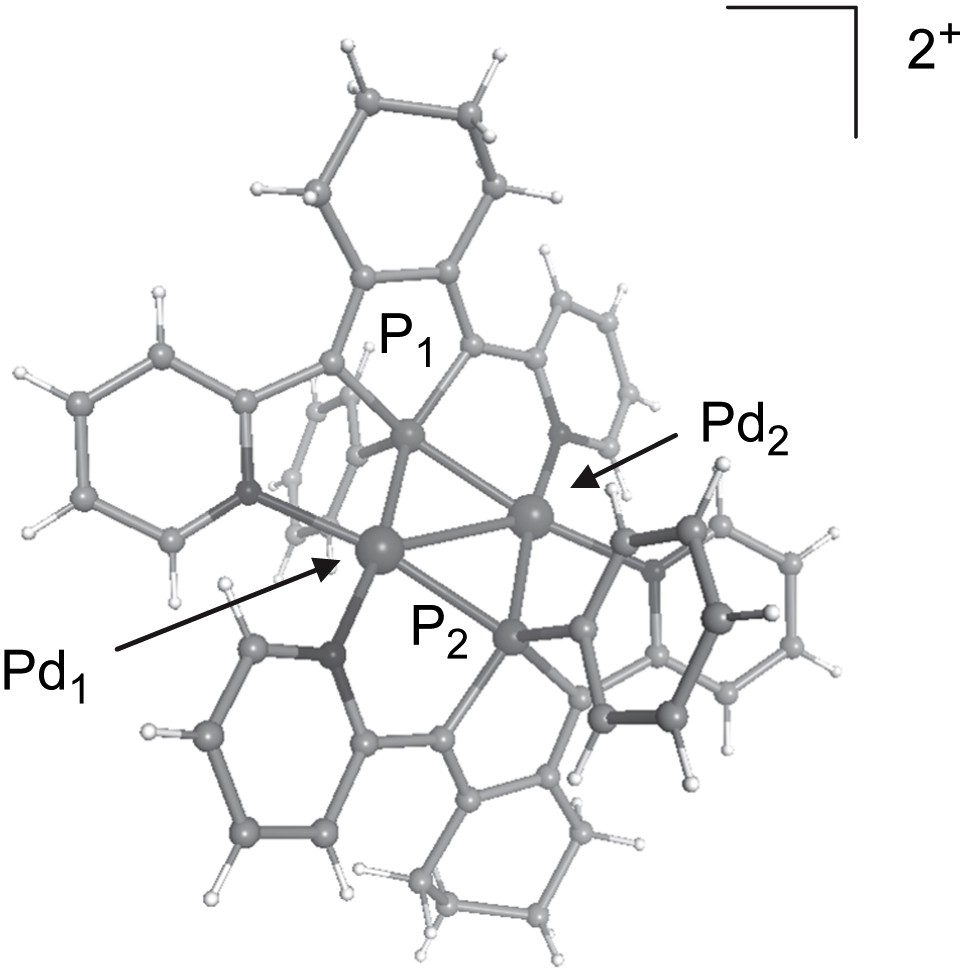
View of the crystallographic structure of the dicationic complex 9.
In this structure, the derivative 3b bridges the two Pd(I) centres via a symmetrically bridging phosphane coordination mode [Δ(Pd − P) = 0.01 Å]. This coordination mode was previously only observed in the case of a family of Rh(I) complexes [22] and constitute a breakthrough in coordination chemistry [23]. The phosphanes acting as binucleating ligands allow the synthesis of di- and polynuclear complexes that are of great interest in manifold fields such as catalysis, bio-inorganic and materials sciences [24].
4.1.2 Cu(I) bimetallic complexes
The coordination chemistry of ligand 3b with Cu(I) salts was also investigated in order to generalize this original bridging phosphane coordination mode to other metal ions [25]. Thus, reaction of di(2-pyridyl)phosphole 3b with Cu(CH3CN)4PF6 in a 1:2 ratio led to the quantitative isolation of the dimetallated complex 10 (Scheme 6).
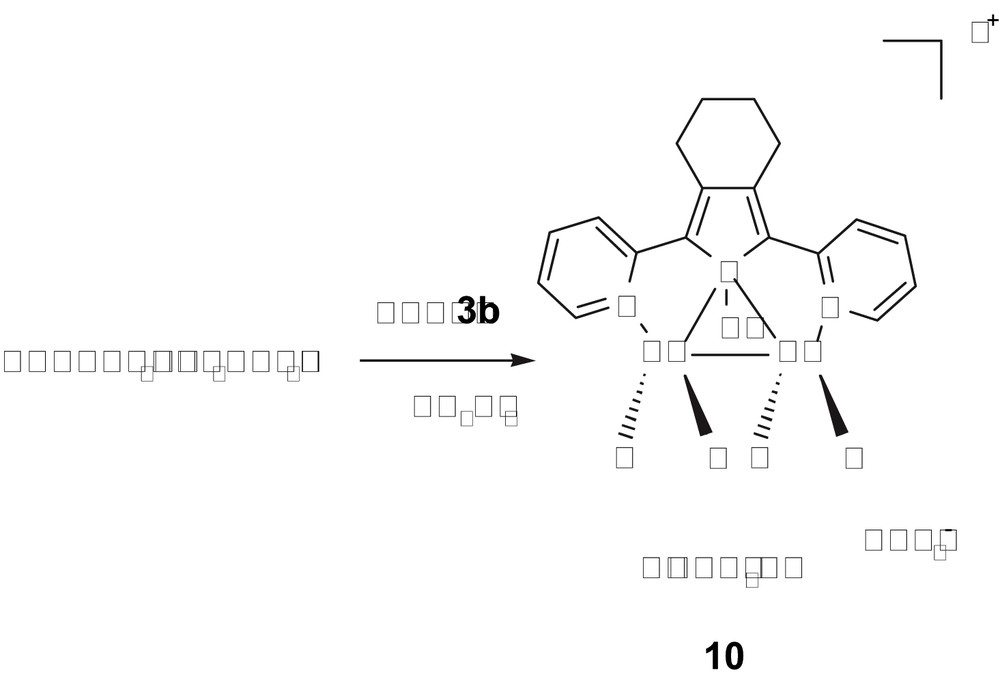
Synthesis of Cu(I) bimetallic complex 10.
Its X-ray diffraction study revealed that compound 10 is a [Cu2(3B)(CH3CN)4][PF6]2 complex (Fig. 3) in which the two Cu(I) centres are capped by a 2,5-bis(2-pyridyl)phosphole ligand 3b acting as a 6-electron μ-1κN:1,2κP:2κN donor.
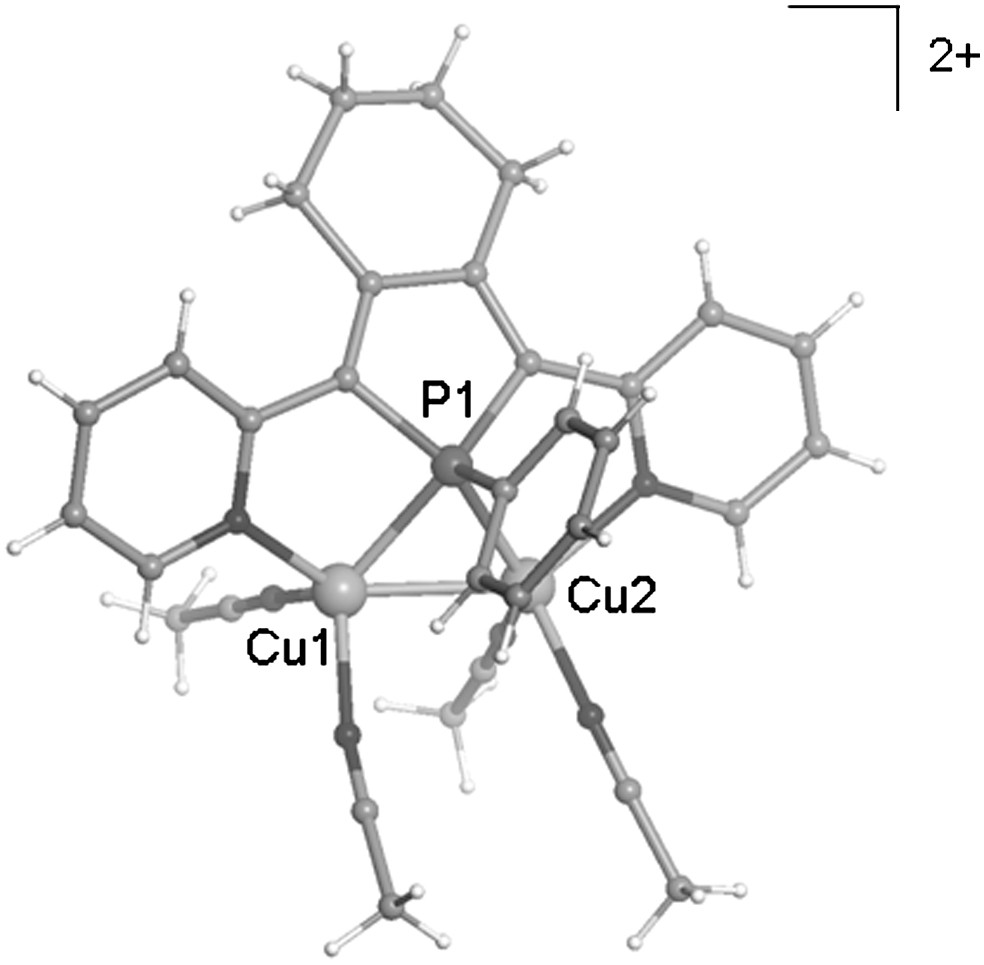
View of the crystallographic structure of the dicationic complex 10.
In this complex as observed in the derivative 9, the P atom presents an almost symmetrical coordination mode [Δ(μP − Cu) = 0.010(2) Å] and short intermetallic distance [2.568(10) Å]. In addition, derivative 10 is the first complex in which: (i) two d10-metal centres are bridged by a phosphane donor, and (ii) two metals are held together by one bridging phosphane and no other supporting ligands. Such results suggest that bridging phosphane ligands are able to stabilise a large variety of bimetallic complexes and confirm that “there is no inherent thermodynamic instability associated with this bonding situation” [23a].
Complex 10 can be reacted with one equivalent of phosphole 3b [25] or one equivalent of diphenylphosphinomethane ligand (dppm) [26] affording in very good yields the air-stable complexes 11a and 11b (Scheme 7). In addition, derivative 11a can be reacted with a third equivalent of phosphole 3b to give the air-stable bimetallic complex 12 [25].
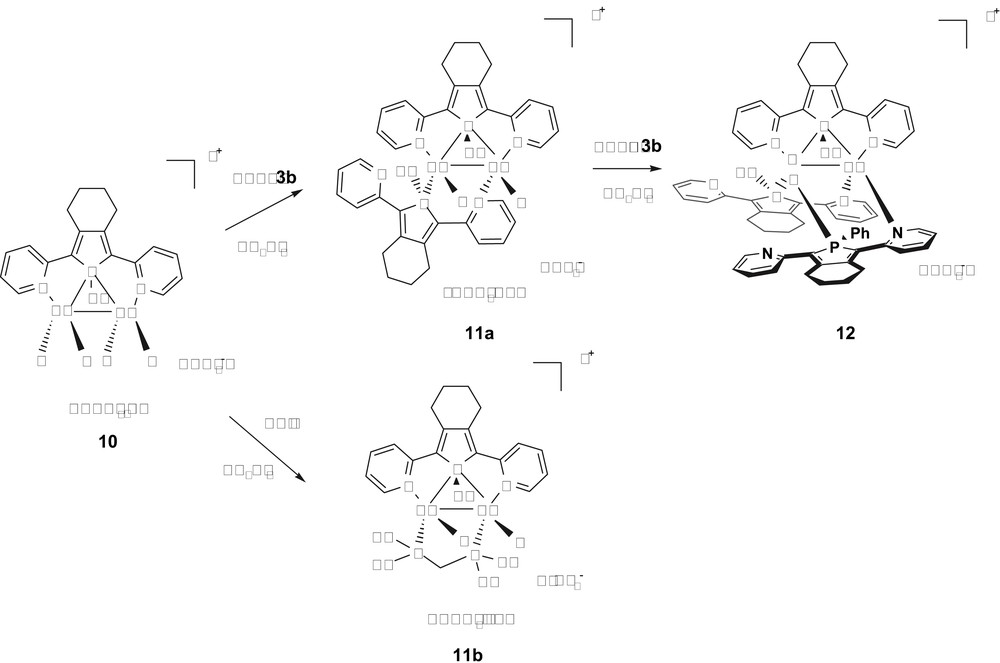
Synthesis of complexes 11a,b and 12.
X-ray diffraction studies (Fig. 4) showed that these three dicationic Cu(I) dimers possess one 2,5-bis(2-pyridyl)phosphole ligand 3b acting still as a μ-1κN:1,2κP:2κN donor. The fact that the μ-bridging coordination mode of the P centre is retained in this series highlights the robustness of bridging phosphanes as binucleating ligands. The tetrahedral geometry of the Cu(I) atoms in complex 10 prevents the formation of a doubly bridged structure, as observed in the Pd(I) dimers 9 (Fig. 2), which requires square-planar metal centres.
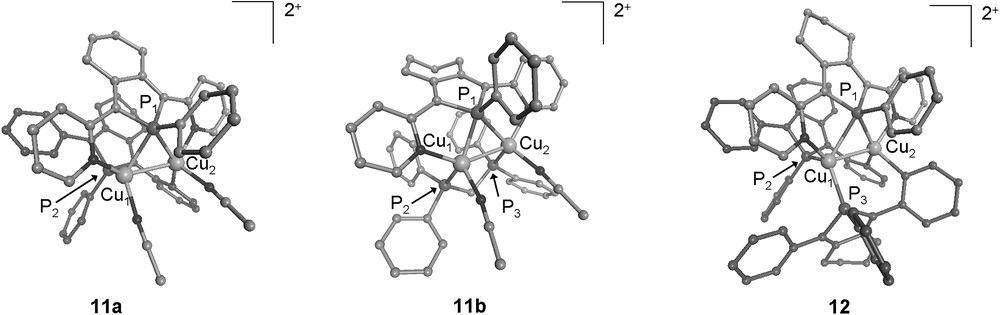
Views of the crystallographic structures of the dicationic complexes 11a, 11b and 12. Hydrogen atoms have been omitted for clarity.
These results demonstrate that chelates exhibiting bridging phosphanes are versatile binucleating ligands and moreover that phosphanes have to be considered as potentially classical bridging ligands.
4.2 Supramolecular organization of π-conjugated chromophores via coordination chemistry: synthesis of analogs of [2,2]-paracyclophanes
Due to their specific topological properties, bimetallic Cu(I) complexes 11a and 11b have been efficiently used as molecular clips [27] in order to organize various π-conjugated systems into rectangular supramolecular assemblies. The synthesis of π-stacked molecular assemblies is indeed of great importance in the understanding the electronic interactions between individual chromophores and thus affords potential new material in the field of molecular devices for optoelectronics. One fruitful approach to probe co-facial π–π interactions involves the assembly of chromophore pairs into well-defined [2,2]paracyclophanes [28], as these molecules provide incisive insights into bulk properties of conjugated systems and are suitable π-dimer models. However, straightforward routes to these assemblies as well as for tailoring their structure are still a challenge to chemical synthesis.
We have investigated a novel synthetic route to analogs of [2,2]paracyclophanes using supramolecular coordination-driven chemistry. Following the concepts of the “directional-bonding approach” [29], the design of metalloparacyclophanes with π-stacked walls requires a bimetallic clip possessing two cis coordinatively labile sites that are closely aligned (Scheme 8).
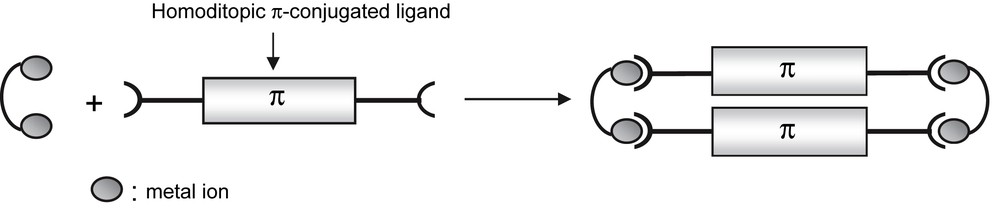
Principle of supramolecular rectangles' synthesis.
The two complexes 11a [25] and 11b [26] possess two acetonitrile ligands that have a cisoid arrangement. Furthermore, the bridging coordination mode of the phosphole ligand 3b imposes a short intermetallic distance [11a, 2.555(1) Å; 11b, 2.667(1) Å] and thus a close proximity of the two kinetically labile acetonitrile ligands (N–N distances, ca. 3.2 Å). Such geometrical characteristics are pre-required in order to allow the organization of homoditopic ligands coordinated on 11a,b in new supramolecular assemblies in which electronic interactions could be forced between these homoditopic ligands (see Scheme 9).

Synthesis of supramolecular assemblies 17a–20b.
Thus, bimetallic derivatives 11a,b have been reacted with linear homoditopic ligands 13–16 incorporating π-conjugated systems affording the new supramolecular rectangles 17a–20b (Scheme 9) [26]. X-ray diffraction studies of these supramolecular assemblies (Fig. 5) evidenced that the metric data of the dimetallic clips 11a,b do not change significantly upon their incorporation into the self-assembled structures. This result demonstrates the conformation rigidity of the Cu(I) based subunits. In all cases, the four Cu atoms are located in the same plane defining a rectangle and the aromatic moieties of the chromophores are parallel as result of a hindered rotation. Moreover, as a consequence of the short Cu(I)–Cu(I) intermetallic distance imposed by the bridging phosphane coordination mode, these aromatic moieties participate in face-to-face π interactions (phenyl centroid–centroid distance: 3.4–3.5 Å) with small lateral offsets. The dimensions of these nano-sized rectangles are fixed by the size of the ditopic ligands and reach 18.1 Å for 19a,b and 25.2 Å for 20a (Cu–Cu distances), with an overall dimension of 26a of about 39.0 Å in length [26].
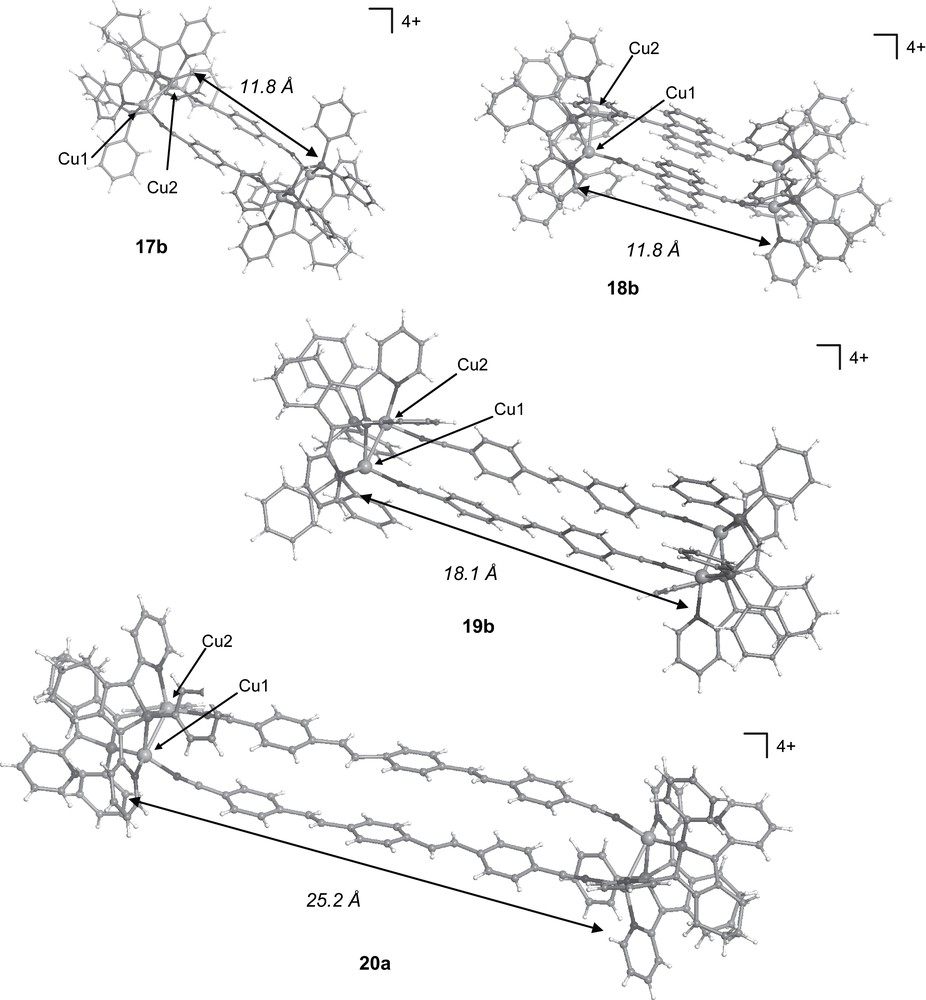
Views of the crystallographic structures of the tetracationic supramolecular rectangles 17b, 18b, 19b and 20a.
These results demonstrate that, despite the repulsive interactions between the closed-shell π-clouds of the homoditopic ligands, the molecular clips 11a,b can force face-to-face π-stacking of aromatic derivatives upon coordination into well-defined supramolecular metalloparacyclophanes. This proves that, due to their rigidity and unique topology, complexes 11a,b are unique building blocks for the synthesis of π-stacked molecular assemblies exhibiting a [2,2]paracyclophane-like topology. This result opens appealing perspective for the design of multifunctional molecular material using this supramolecular assembling approach.
Moreover, the stacking pattern of the metalloparacyclophanes 19a,b and 20a is remarkable. In the solid state, these supramolecular rectangles organize into infinite columns with short inter-molecular distances (ca. 3.6 Å). Within these columns, the cationic parts of the rectangles have a parallel-displaced arrangement along the a-axes. Unique infinite columnar stack resulting from intra- and inter-molecular π–π interactions of (para-phenylenevinylene)-based chromophores 15 and 16 are thus formed at the macroscopic scale with these novel metalloparacyclophanes (Fig. 6) [26]. Organization of the π-conjugated chromophores is thus successfully performed at two hieratical levels, the first within the metalloparacyclophanes and the second in the infinite 1D stack of these metalloparacyclophanes. Complexes 11a,b are thus versatile molecular clips that allow one to control the organization of π-conjugated systems in supramolecular assemblies having a [2,2]paracyclophane-topology. Elucidation of the electronic properties of these novel π-stacked molecular assemblies as well as the use of other ditopic conjugated systems to construct novel metallo[2,2]paracyclophane are under active investigations.
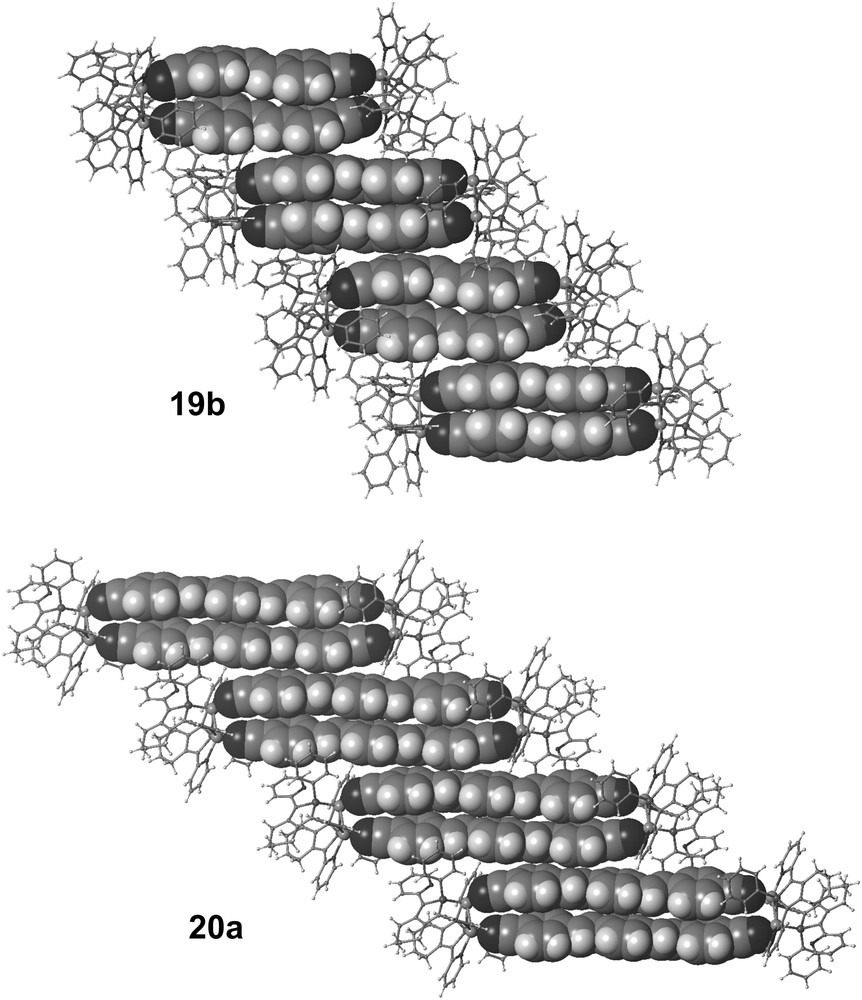
Views of the packing of 19b and 20a along the a-axes (H atoms, counteranions and solvent molecules have been omitted for clarity).
5 Conclusions
Oligomers based on phosphole rings revealed to be valuable building blocks for the preparation of π-conjugated systems. The optical and electrochemical properties of 2,5-diheteroarylphospholes can be easily tuned over a wide range by tailoring the 2,5-substituents or by performing simple chemical modifications involving the nucleophilic P atom. The good stability of the phosphole ring, along with its specific electronic properties, opens new perspectives in the chemistry of π-conjugated systems incorporating heavy heteroatoms. In addition, the association of the phosphole ring with the pyridine rings has allowed the synthesis of new P,N and N,P,N ligands. We have evidenced the original coordination chemistry of these derivatives and characterized new coordination complexes exhibiting appealing potential for the development of molecular materials for optoelectronics. Moreover, the discovery of the bridging phosphane coordination mode opens new perspectives, as it gives access to unprecedented dinuclear complexes. Finally, in order to achieve operative molecular devices with specific properties, an important step lies in the control of the organization of the chromophores in the solid state. We have shown that simple reactions of organic and coordination chemistry allow the access to a variety of molecular material where specific local and/or global organization of π-conjugated systems are induced. Coordination of P,N chelates integrated in phosphole based π-conjugated systems afford monometallic complexes where NLO activities are generated. Moreover, starting from original coordination complexes bearing bridging phosphole ligands, a variety of organic chromophores can be organized in nanometric supramolecular assemblies where π-conjugated system are in interaction at the level of the bulk crystalline solid.
Acknowledgements
We thank all researchers, including collaborators, colleagues and coworkers, whose work has been presented in this chapter. We thank the “Ministère de l'Éducation nationale, de la Recherche et de la Technologie”, the “Centre national de la recherche scientifique”, the “Conseil régional de Bretagne”, and the “Institut universitaire de France”.