

1 Introduction
Current approaches to the reconstruction of bone tissue in orthopedic surgery, stomatology and dental applications more and more rely on calcium phosphate (CaP) ceramics. CaP biomaterials are used in bone repair or substitution, as osteoconductive fillers to achieve bone coalescence. CaPs are the principal raw materials used for the elaboration of granules or blocks in bone substitutes, for which one of the most important criteria is that the scaffold consists of a highly interconnected porous network with pore size large enough for cell migration, fluid exchange, tissue ingrowth and vascularization.
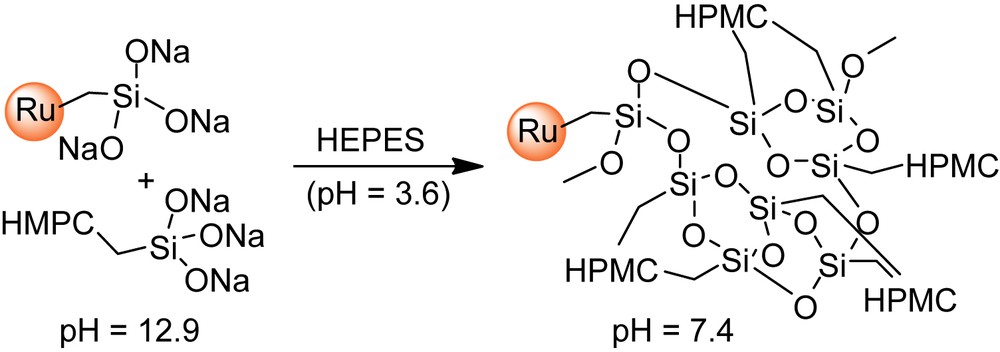
Water-swollen polymeric networks such as natural hydrogels are also potential materials for tissue engineering [1,2], since they can mimic the extracellular matrix. The design of injectable biomaterials was thus investigated using such hydrogels combined with calcium phosphates, in a manner that enables implantation with minimally invasive surgical techniques [3]. In this context, we have previously developed an Injectable Bone Substitute (IBS) [4] consisting of CaP ceramic granules suspended in a biocompatible cellulose-based hydrogel [hydroxypropylmethylcellulose (HPMC), denoted as E4M®], for the design of a ready to use material for the surgeon. The vascularization and bone ingrowth in this gel is very fast and CaP granules in the polymer carrier phase create the so-called “interconnecting macroporosity” [5]. The next step was to develop a second-generation material with self-hardening properties, in order to minimize possible dispersion of the bone substitute away from the implantation site. Therefore, polymerizable trialkoxysilyl groups were introduced onto the cellulose ether polymer, by reaction with 3-glycidoxypropyltrimethoxysilane [6–8]. Subsequent dissolution of the resulting composition in strong basic medium generates silanolate groups (–Si(O−Na+)3), which can then undergo polymerization that irreversibly forms gels, upon raising the pH close to neutral (pH = 7.4). This approach offers the advantage that the setting of the biomaterial is simply obtained by addition of a non-toxic buffer [HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)], prior to implantation. Gelation times along with mechanical properties of the corresponding gels, denoted as Si–HPMC, can be tuned through the amount of silanolate groups introduced onto the polymer and the amount of calcium phosphate loaded in the gel.
For its use as a scaffold for bone tissue engineering, an important question is to know whether the cellulose-based hydrogel component remains intact in the implantation site or is depredated by cells and then fully eliminated. The challenge was thus to find a labeling method that could deliver attractive information on that point, at acceptable constraints with regard to the flexibility required by in vivo biological evaluation. A wide variety of labels have been reported, among which are radioactive labels that can be detected at very low concentration. However, their use requires sophisticated equipment and the resulting limitation for safety reasons are thus of poor practical use for medium-term in vivo animal experiments such as that considered in the present work. Of particular value are non-radioactive molecules observable by emission, absorption and fluorescence spectrometry, which would potentially allow quantification of the label within slices of bone tissue taken from the implantation sites and the regions nearby. Examples of such compounds include organic chromophores or fluorophores used for labeling oligosaccharides (i.e. rhodamine B, fluorescein-5-isothiocyanate [FITC], 8-amino-1,3,-pyrene trisulfonic acid [APTS]…) [9–12]. On the other hand, organometallic-based labels offer a detection method which specifically assays the metal component of the organometallic compound. This can be advantageous, given that many metal elements are absent from biological systems. In this category, Ru-containing compounds have been extensively reported, including [Ru(2,2′-bipyridine)2L]2+ complexes where L is often a bidentate nitrogen-containing ligand, such as a bipyridine unit derivatized for the covalent attachment of the label to various biological molecules [13], including DNA [14,15]. In this context, tris(bipyridyl)ruthenium(II) complexes seemed suitable for our study because (i) of their high stability at acid pH, suggesting that no degradation will occur in the vicinity of bone tissues, thus avoiding any non-specific dissemination; (ii) as ruthenium is absent from biological fluids, this element should be easy to quantify on thin bone tissue samples, using X-ray microfluorescence maps recorded at a synchrotron light source [Ru Kα (19.15 keV)].
The selected strategy described in the present paper was thus to prepare Ru(bpy)3-based labels containing trialkoxysilane groups to be dispersed in the cellulose hydrogel, prior to condensation of the polysilsesquioxane network, to ensure the most homogeneous distribution of the label within the final gel. It enables the label to attach to the polymer through Si–O–Si bonds formed by cross-reticulation of the SiO3 blocks present on both the hydrogel and the label (Scheme 1). The number of alkoxysilane groups and their location on the ruthenium complex backbone was varied, and the influence of these parameters on the labeling yield and rheological properties of the hydrogel were investigated.
2 Materials and methods
2.1 Materials
Hydroxypropylmethylcellulose (HPMC) E4M® was purchased from Colorcon-Dow chemical (Bougival, France). 3-Glycidoxypropyltrimethoxysilane (GPTMS) was obtained from Acros (Geel, Belgium). The Si–HPMC hydrogels were prepared as previously reported [7], and the Si content of the two batches was determined by elemental analysis performed by the CNRS Analysis Laboratory (Vernaison). HEPES was purchased from Sigma–Aldrich (St. Louis, MO, USA). All other chemicals were purchased from standard laboratory suppliers and were of the highest purity available.
2.2 Preparation of bipyridine 1–4
5-Trimethylsilylmethyl-5′-methyl-2,2′-bipyridine was obtained according to Schubert et al. [16]. 5-Chloromethyl-5′-methyl-2,2′-bipyridine 1, 5-acetoxymethyl-5′-methyl-2,2′-bipyridine and 5-hydroxymethyl-5′-methyl-2,2′-bipyridine 2 were synthesized following similar methods used for the preparation of their analogues [17–19].
2.2.1 5-Trimethylsilylmethyl-5′-methyl-2,2′-bipyridine
Yield: 72%. 1H NMR (300 MHz, CDCl3): 0.00 (9H, s, CH3(Si)); 2.08 (2H, s, CH2(bpy)); 2.35 (3H, s, CH3(bpy)); 7.40 (1H, dd, 3J = 8.1 Hz and 4J = 2.1 Hz, H4); 7.56 (1H, dd, 3J = 8.1 Hz and 4J = 2.1 Hz, H4′); 8.17 (1H, d, 3J = 8.1 Hz, H3); 8.19 (1H, d, 3J = 8.1 Hz, H3′); 8.31 (1H, d, 4J = 2.1 Hz, H6); 8.44 (1H, d, 4J = 2.1 Hz, H6′). 13C NMR (75 MHz, CDCl3): −1.9 (CH3(Si)); 18.4 (CH3(bpy)); 24.1 (CH2(bpy)); 120.2 and 120.4 (C3 and C3′); 132.9 (C5′); 136.2 (C4); 136.4 (C5); 137.5 (C4′); 148.6 (C6); 149.6 (C6′); 152.5 and 154.0 (C2 and C2′). MS (EI; m/z, I): 256 (84, M+); 241 (27); 184 (29); 73 (100).
2.2.2 5-Chloromethyl-5′-methyl-2,2′-bipyridine 1
Yield: 90%. 1H NMR (300 MHz, CDCl3): 2.38 (3H, s, CH3(bpy)); 4.63 (2H, s, CH2(bpy)); 7.60 (1H, dd 3J = 8.1 Hz and 4J = 2.4 Hz, H4′); 7.82 (1H, dd, 3J = 8.1 Hz and 4J = 2.4 Hz, H4); 8.27 (1H, d, 3J = 8.1 Hz, H3′); 8.39 (1H, d, 3J = 8.1 Hz, H3); 8.49 (1H, d, 4J = 2.4 Hz, H6′); 8.64 (1H, d, 4J = 2.4 Hz, H6). 13C NMR (75 MHz, CDCl3): 18.5 (CH3(bpy)); 43.3 (CH2(bpy)); 120.8 (C3 and C3′); 132.9 (C5); 133.8 (C5′); 137.2 (C4); 137.6 (C4′); 149.1 (C6); 149.8 (C6′); 153.2 (C2′); 156.5 (C2). MS (EI m/z, I): 218 (34, M+); 183 (100).
2.2.3 5-Acetoxymethyl-5′-methyl-2,2′-bipyridine
Yield: 85%. 1H NMR (300 MHz, CDCl3): 2.12 (3H, s, CH3(CO)); 2.40 (3H, s, CH3(bpy)); 5.17 (2H, s, CH2(bpy)); 7.63 (1H, dd, 3J = 8.1 Hz and 4J = 2.1 Hz, H4′); 7.80 (1H, dd, 3J = 8.1 Hz and 4J = 2.1 Hz, H4); 8.29 (1H, d, 3J = 8.1 Hz, H3′); 8.37 (1H, d, 3J = 8.1 Hz, H3); 8.65 (1H, d, 4J = 2.1 Hz, H6′); 8.66 (1H, d, 4J = 2.1 Hz, H6). 13C NMR (75 MHz, CDCl3): 18.5 (CH3(bpy)); 21.0 (CH3(CO)); 63.81 (CH2(bpy)); 120.7 (C3); 120.8 (C3′); 131.3 (C5); 133.8 (C5′); 137.1 (C4); 137.6 (C4′); 149.2 (C6); 149.8 (C6′); 153.4 (C2); 156.4 (C2′); 171.0 (CO). MS (EI; m/z, I): 242 (100, M+); 199 (31); 182 (63).
2.2.4 5-Hydroxymethyl-5′-methyl-2,2′-bipyridine 2
Yield: 85%. 1H NMR (300 MHz, DMSO): 2.35 (3H, s, CH3(bpy)); 4.59 (2H, d 3J = 5.7 Hz, CH2(bpy)); 5.39 (1H, t 3J = 5.7 Hz, OH); 7.73 (1H, dd 3J = 8.1 Hz and 4J = 2.1 Hz, H4′); 7.85 (1H, dd 3J = 8.1 Hz and 4J = 2.1 Hz, H4); 8.27 (1H, d 3J = 8.1 Hz, H3′); 8.31 (1H, d 3J = 8.1 Hz, H3); 8.50 (1H, d 4J = 2.1 Hz, H6′); 8.59 (1H, d 4J = 2.1 Hz, H6). 13C NMR (75 MHz, DMSO): 17.8 (CH3(bpy)); 60.5 (CH2(bpy)); 119.6; 119.8 (C3 and C3′); 133.4 (C5); 135.5 (C4); 137.5 (C4′); 137.7 (C5′); 147.6 (C6); 149.5 (C6′); 152.8 (C2′); 154.1 (C2). MS (EI; m/z, I): 200 (100, M+); 183 (21); 171 (92).
4-Hydroxymethyl-4′-methyl-2,2′-bipyridine 3 was prepared as reported by Berg et al.[20]. 5,5′-Bis(hydroxymethyl)-2,2′-bipyridine 4 was obtained by reduction of dimethyl 2,2′-bipyridine-5,5′-dicarboxylate, as described by Telfer et al. [21] for the diethyl analog. The dimethyl dicarboxylate precursor was synthesized following an adaptation of a protocol reported by Will et al. [22] for the preparation of the 4,4′-isomer.
2.3 Preparation of complexes [Ru(bpy)2(L)](PF6)2
A mixture of bis(2,2′-bipyridine)-dichloro ruthenium dihydrate (ACROS – 1 g, 1.92 mmol) dissolved in 40 mL distilled ethanol and 1 equiv of the desired ligand L (2, 3 or 4) was refluxed under argon for 24 h. The reaction mixture was allowed to cool at room temperature, and then evaporated. Distilled water (25 mL) was added to the residue, and the aqueous phase was washed with hot ethyl acetate (3 × 25 mL), then concentrated under vacuum to ca. 15 mL volume. Then, ammonium hexafluorophosphate (0.32 g, 1.96 mmol) was added and the resulting precipitate was collected by filtration, rinsed with water (5 mL), diethyl ether (4 × 15 mL) and dried under vacuum.
2.3.1 [Ru(bpy)2(2)](PF6)2
Yield: 90%. 1H NMR (300 MHz, DMSO): 2.20 (3H, s, CH3(bpy)); 4.45 (2H, t, 3J = 5.4 Hz, CH2(bpy)); 5.38 (1H, t, 3J = 5.4 Hz, OH); 7.51–7.59 (6H, m, Har(bpy)); 7.70–7.73 (4H, m, Har(bpy)); 8.14 (2H, d, Har(bpy)); 8.19 (4H, t, Har(bpy)); 8.69–8.85 (6H, m, Har(bpy)). 13C NMR (75 MHz, DMSO): 18.0 (CH3(bpy)); 59.6 (CH2(bpy)); 123.4; 123.6; 124.4; 127.7; 135.6; 137.8; 138.0; 138.5; 141.9; 148.1; 150.6; 151.1; 153.9; 155.1; 156.5; 156.6 (Car(bpy)). HRMS (ESI +) calculated for C32H28N6O102Ru (M2+): m/z = 307.0684, found m/z = 307.0683. UV–visible (H2O – λ nm, ɛ): 453 (12420 M−1 cm−1).
2.3.2 [Ru(bpy)2(3)](PF6)2
Yield: 90%. 1H NMR (300 MHz, DMSO): 2.53 (s, 3H, CH3(bpy)); 4.73 (d, J = 5.4 Hz, 2H, CH2(bpy)); 5.73 (t, J = 5.4 Hz, 1H, OH); 7.36 (d, J = 5.5 Hz, 1H, Har); 7.46 (d, J = 5.7 Hz, 1H, Har); 7.52 (m, 5H, Har); 7.65 (d, J = 5.8 Hz, 1H, Har); 7.73 (m, 4H, Har); 8.16 (t, J = 7.8 Hz, 4H, Har); 8.72 (d, J = 8.2 Hz, 2H, Har); 8.83 (d, J = 8.1 Hz, 4H, Har); 13C NMR (75 MHz, DMSO): 20.7 (CH3(bpy)); 60.8 (CH2(bpy)); 117.8; 121.3; 121.4; 124.9; 147.9; 148.9; 149.0; 152.8; 155.2; 155.3 (Car). HRMS (ESI +) calculated for C32H28N6O102Ru (M2+): m/z = 307.0684, found m/z = 307.0690. UV–visible (H2O – λ nm, ɛ): 453 (12,420 M−1 cm−1).
2.3.3 [Ru(bpy)2(4)](PF6)2
Yield: 70%. 1H NMR (300 MHz, DMSO): 4.46 (d, J = 5.5 Hz, 4H, CH2(bpy)); 5.38 (t, J = 5.5 Hz, 2H, OH); 7.54 (dt, J = 6.8 Hz, J = 1.1 Hz, 4H, Har); 7.61 (s, 2H, Har); 7.72 (t, J = 5.1 Hz, 4H, Har); 8.03 (dd, J = 8.6 Hz, J = 1.1 Hz, 2H, Har); 8.18 (dt, J = 8.1 Hz, J = 1.1 Hz, 4H, Har); 8.76 (d, J = 8.4 Hz, 2H, Har); 8.84 (d, J = 8.2 Hz, 4H, Har); 13C NMR (75 MHz, DMSO): 59.9 (CH2(bpy)); 123.4; 123.3; 123.9; 127.2; 135.2; 137.4; 141.7; 148.7; 151.3; 156.7 (Car). Melting point: 230 °C. HRMS (ESI +) calculated for C32H28N6O2102Ru (M2+): m/z = 315.0659, found m/z = 315.0660. UV–visible (H2O – λ nm, ɛ): 453 (12,420 M−1 cm−1).
2.4 General procedure for the preparation of the ruthenium complexes I–III
The desired ruthenium complex ([Ru(bpy)2(2)](PF6)2, [Ru(bpy)2(3)](PF6)2 or [Ru(bpy)2(4)](PF6)2, 0.7 mmol) was dissolved in dry distilled acetonitrile (6.5 mL) under nitrogen, to which activated 4-Å molecular sieve was added. To this solution was added distilled triethylamine (2 equiv per OH group) followed by, after stirring for 20 min, addition of 3-(triethoxysilyl)propylisocyanate (2 equiv per OH group) in three portions, at 3-h intervals. After the last addition, the resulting mixture was stirred at room temperature for 36 h and then filtered and evaporated under reduced pressure. The residue was taken up in the minimum amount of dichloromethane and placed in a centrifugation tube. After addition of dry acetone, the desired product precipitated and the mixture was centrifuged (5000 rpm) for 10 min. The supernatant was removed and the above protocol was repeated on the oily residue until a solid red powder was obtained.
2.4.1 Ruthenium complex I
Yield: 85%. 1H NMR (300 MHz, CD3CN): 0.53 (m, 2H, CH2Si); 1.15 (9H, t, J = 7.0 Hz, CH3(CH2OSi)); 1.25 (t, J = 7.1 Hz, 2H, CH2); 2.18 (s, 3H, CH3(bpy)); 2.97 (dd, J = 13.1 Hz, J = 6.7 Hz, 2H, CH2(NH)); 3.75 (q, J = 7.0 Hz, 6H, CH2OSi); 4.89 (s, 2H, CH2(bpy)); 5.61 (t, J = 5.6 Hz, 1H, NH); 7.38 (dt, J = 6.7 Hz, J = 0.9 Hz, 4H, Har); 7.50 (d, J = 1.4 Hz, 1H, Har); 7.58 (d, J = 1.1 Hz, 1H, Har); 7.69 (m, 4H, Har); 7.85 (dd, J = 8.3 Hz, J = 1.1 Hz, 1H, Har); 7.95 (dd, J = 8.4 Hz, J = 1.7 Hz, 1H, Har); 8.03 (dt, J = 8.0 Hz, J = 3.6 Hz, 4H, Har); 8.35 (d, J = 8.4 Hz, 1H, Har); 8.40 (d, J = 8.4 Hz, 1H, Har); 8.48 (d, J = 8.1 Hz, 4H, Har). 13C NMR (75 MHz, CD3CN): 6.8 (CH2); 17.4 (CH3(bpy) and CH3(CH2OSi)); 22.8 (CH2); 43.0 (CH2); 57.8 (CH2(OSi)); 61.8 (CH2(bpy)); 123.1; 123.4; 124.0; 127.2; 136.1; 137.0; 138.1; 138.5; 137.4; 149.7; 151.3; 153.7; 155.3; 156.0; 156.7 (Car and CO). 29Si MAS NMR (reference: tetramethylsilane): (s) –45.1. Melting point: 90 °C. HRMS (ESI +) calculated for C42H49N7O5Si102Ru (M2+): m/z = 430.6304, found m/z = 430.6311. UV–visible (H2O – λ nm, ɛ): 453 (12,420 M−1 cm−1).
2.4.2 Ruthenium complex II
Yield: 63%. 1H NMR (300 MHz, CD3CN): 0.53 (m, 2H, CH2Si); 1.15 (9H, t, J = 7.0 Hz, CH3(CH2OSi)); 1.25 (t, J = 7.1 Hz, 2H, CH2); 2.18 (s, 3H, CH3(bpy)); 2.97 (dd, J = 13.1 Hz, J = 6.7 Hz, 2H, CH2(NH)); 3.75 (q, J = 7.0 Hz, 6H, CH2OSi); 4.89 (s, 2H, CH2(bpy)); 5.61 (t, J = 5.6 Hz, 1H, NH); 7.38 (dt, J = 6.7 Hz, J = 0.9 Hz, 4H, Har); 7.50 (d, J = 1.4 Hz, 1H, Har); 7.58 (d, J = 1.1 Hz, 1H, Har); 7.69 (m, 4H, Har); 7.85 (dd, J = 8.3 Hz, J = 1.1 Hz, 1H, Har); 7.95 (dd, J = 8.4 Hz, J = 1.7 Hz, 1H, Har); 8.03 (dt, J = 8.0 Hz, J = 3.6 Hz, 4H, Har); 8.35 (d, J = 8.4 Hz, 1H, Har); 8.40 (d, J = 8.4 Hz, 1H, Har); 8.48 (d, J = 8.1 Hz, 4H, Har). Melting point: 140 °C. HRMS (ESI +) calculated for C42H49N7O5Si102Ru (M2+): m/z = 430.6304, found m/z = 430.6308.
2.4.3 Ruthenium complex III
Yield: 70%. 1H NMR (300 MHz, CD3CN): 0.55 (m, 4H, CH2Si); 1.17 (t, J = 7.0 Hz, 18H, CH3(CH2OSi)); 1.48 (m, 4H, CH2); 3.00 (dd, J = 13.4 Hz, J = 6.8 Hz, 4H, CH2(NH)); 3.78 (q, J = 7.0 Hz, 12H, CH2OSi); 4.93 (s, 4H, CH2(bpy)); 5.60 (t, J = 5.6 Hz, 2H, NH); 7.40 (m, 4H, Har); 7.64 (s, 2H, Har); 7.71 (d, J = 4.4 Hz, 4H, Har); 7.99 (dd, J = 8.4 Hz, J = 1.4 Hz, 2H, Har); 8.07 (t, J = 7.9 Hz, 4H, Har); 8.45 (d, J = 8.4 Hz, 2H, Har); 8.50 (d, J = 8.1 Hz, 4H, Har). 13C NMR (75 MHz, CD3CN): 6.8 (CH2); 17.4 CH3(CH2OSi); 22.8 (CH2); 43.0 (CH2); 57.8 (CH2(OSi)); 61.8 (CH2(bpy)); 123.7; 124.0; 127.3; 136.1; 137.5; 149.9; 151.3; 155.3; 155.6; 156.7 (Car and CO). Melting point: 110 °C. HRMS (ESI +) calculated for C52H70N8O10Si2102Ru (M2+): m/z = 562.1899, found m/z = 562.1901.
2.5 General procedure for the labeling of the Si–HPMC
2.5.1 Preparation of Si–HPMC 3 wt% aqueous solutions A (pH = 12.9)
Si–HPMC (6 g; 0.52 or 0.81 wt% Si) was slowly introduced into a sodium hydroxide aqueous solution (0.2 mol L−1 – 194 mL). After homogenization under stirring into a closed system for 48 h, the polymer solution was transferred into dialysis tubes previously washed with a sodium hydroxide aqueous solution (0.09 mol L−1). The polymer solution was dialyzed twice against sodium hydroxide aqueous solutions (2 × 3.8 L; 0.09 mol L−1) for 16 and 2 h, respectively, and then stocked at 4 °C after steam sterilization in an autoclave at 121 °C for 30 min.
2.5.2 Preparation of HEPES aqueous solution B (pH = 3.6)
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (1.55 g, 6.5 mmol), sodium chloride (0.4 g, 6.8 mmol), distilled water (35 mL) and acid hydrochloride solution (0.1 mol L−1, 15 mL) were mixed and hand-shaked before steam sterilization in an autoclave at 121 °C for 30 min.
2.5.3 Labeling of Si–HPMC solutions
The desired ruthenium complex (I, II or III; 2.5–30 mg) and A (8 mL) were mixed at room temperature in a closed vessel and stirred for 3 h for homogenization. After addition of B (8 mL), the mixture was mechanically stirred for a few minutes and immediately transferred by 3-mL portions into dialysis tubes. The resulting tubes were placed under humidified atmosphere over 14 days to allow gel formation and to keep the dialysis tubes wet. The resulting labeled Si–HPMC gels were dialyzed several times against distilled water (30 mL) under mechanical transversal stirring (100 rpm) for 24–96 h for each cycle until colorless rinsing solutions were obtained.
2.6 Rheological measurements
Si–HPMC (8 mL, 0.52 wt% Si), 3 wt% aqueous solution A, and ruthenium complex I (7.5 or 15 mg) or III (7.5 or 15 mg), corresponding to a ∼0.1 or 0.2% label loading by weight with respect to the weight of gel, respectively, were stirred for 24 h at room temperature before addition of HEPES solution B (pH = 3.2).
Viscoelastic properties of the different crude hydrogels, as prepared above, were recorded at 25 °C. The storage (G′) and the loss (G″) moduli of the samples were measured as a function of time. Multiwave tests were realized in the linear domain at a total stress of 1 Pa and at three frequencies (1, 3.2 and 10 Hz). Dynamic measurements were performed on a controlled stress RheoStress 300 (ThermoHaake®, Germany) rheometer fitted with a cone-plate geometry (1° cone angle, 60 mm diameter, 53 μm gap). The Winter and Chambon method [23] was used to determine the gelation time of each sample, which corresponds to the point where the tan(δ) (i.e. G″/G′) factor is independent of the frequency [24].
3 Results and discussion
3.1 Preparation of the three Ru(bpy)3-based labels I–III
The Ru(bpy)3-based labels were prepared by coupling 3-(triethoxysilyl)propylisocyanate to ruthenium tris-bipyridine complexes, in which one of the 2,2′-bipyridine ligands was functionalized by one (in the 5 or 4 position [2 and 3, respectively]) or two (in the 5 and 5′ positions [4]) hydroxymethyl groups. The purpose was to investigate the amount of label incorporated along with the rheological properties of the resulting gels, as a function of the number of alkoxysilane groups, as well as the position on which they are located on the bipyridine backbone.
Reaction of 5,5′-dimethyl-2,2′-bipyridine with 2.2 equiv of LDA in THF at −78 °C, followed by addition of trimethylsilyl chloride and quenching after 1 min with ethanol yielded 5-trimethylsilylmethyl-5′-methyl-2,2′-bipyridine [16]. The latter compound was then reacted with hexachloroethane and cesium fluoride in acetonitrile to give 5-chloromethyl-5′-methyl-2,2′-bipyridine 1, by analogy with similar reactions used for the preparation of 5-bromomethyl or 4,4′-bischloromethyl analogues [17,18]. Conversion of 1 to 2 was performed by halogen displacement using sodium acetate in dry DMF and hydrolysis of the resulting acetate using aqueous Na2CO3 in THF [19] (Scheme 2). Ruthenium (5-hydroxymethyl-5′-methyl-2,2′-bipyridine)bis(2,2′-bipyridine)bis(hexafluorophosphate) was obtained by reaction of 2 and Ru(bpy)2Cl2·2H2O in refluxing ethanol and further precipitation using ammonium hexafluorophosphate according to a procedure adapted from Ghosh and Spiro [25]. This compound was then reacted with 3-(triethoxysilyl)propylisocyanate to yield complex I, via the formation of a carbamate linkage (Scheme 3). The 29Si MAS NMR analysis of complex I showed a single peak at −45.1 ppm, characteristic of a Q0 site, which means that no hydrolysis of the alkoxysilane groups (Si(OEt)3) occurred during the synthesis.
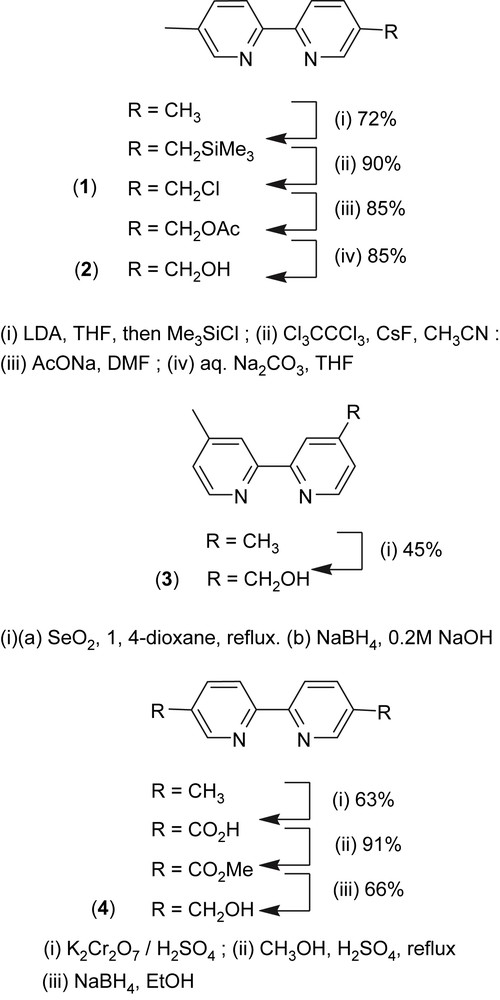
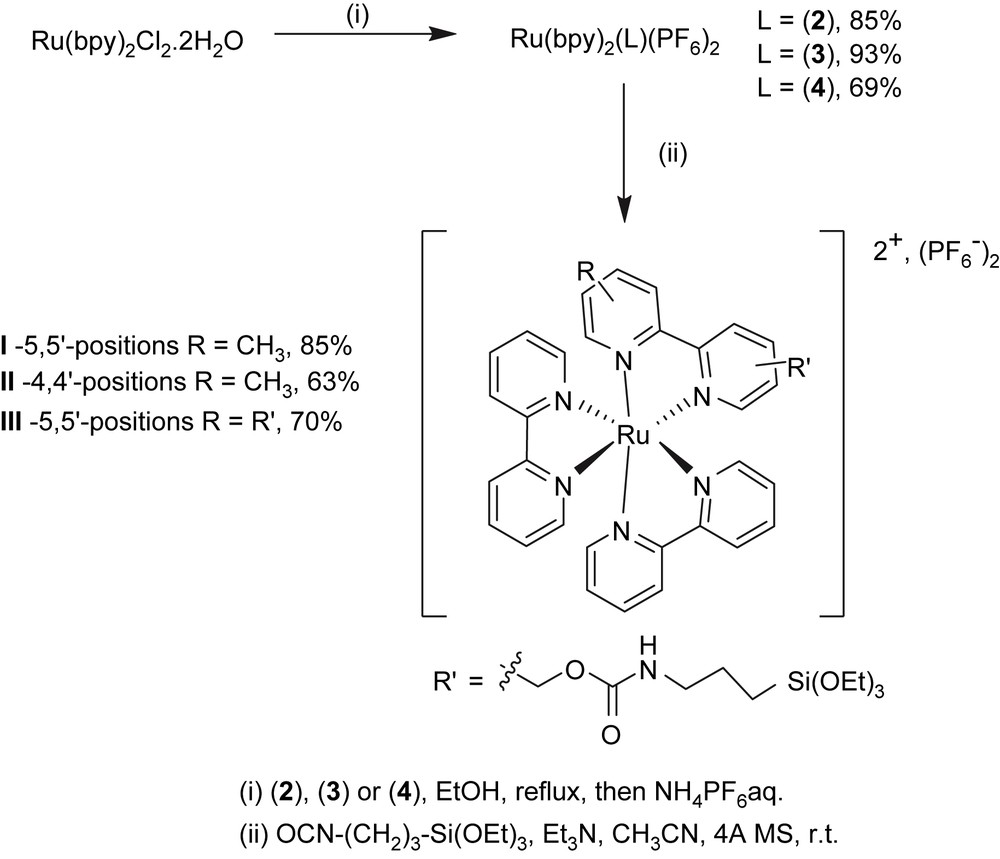
Oxidation of 4,4′-dimethyl-2,2′-bipyridine with 1.2 equiv of selenium dioxide [20,26–28] in refluxing 1,4-dioxane for 42 h led to 4-formyl-4′-methyl-2,2′-bipyridine which was directly reduced to its corresponding alcohol, 4-hydroxymethyl-4′-methyl-2,2′-bipyridine 3, using sodium borohydride in alkaline methanol. 5,5′-Bis(hydroxymethyl)-2,2′-bipyridine 4 was prepared by oxidation of the methyl groups of 5,5′-dimethyl-2,2′-bipyridine (CrO3/H2SO4) to carboxylic acid groups that were subsequently esterified (MeOH/H2SO4) and reduced with sodium borohydride in ethanol, following a protocol reported for the 4,4′ isomer [22] (Scheme 2). Complexes II and III were prepared similar to complex I, using ligands 3 and 4, respectively (Scheme 3).
3.2 Labeling of the Si–HPMC gels
Two batches of Si–HPMC aqueous solutions with different Si content (0.52 or 0.81 wt% Si) were mixed with various amounts of ruthenium complex (I, II or III) in basic conditions to form silanolate groups on both species. By increasing the pH close to neutral (pH = 7.4), condensation of the silanolate groups led to labeled reticulated Si–HPMC gels (Scheme 1). Those gels were dialyzed against water for removal of unbound label and the final ruthenium content within the gels was determined by UV–visible spectroscopy. Transferring the resulting gels from dialysis tubes to UV–visible cuvettes was difficult due to their high viscosity. The UV–visible measurements (453 nm) were thus performed on the dialysis washing solutions to quantify the amount of unbound label released, and the final ruthenium contents of the gels were calculated by difference from the initial label loading. The results showed that, whatever the nature of the label (I, II or III), the final ruthenium contents increased linearly with the initial label loading (Figs. 1 and 2), and the labeling yield with respect to the label was found to be much higher when the bisfunctionalized complex III (ca. 50%) was used, as compared to its monofunctionalized analogues I and II (ca. 20–30%). For the two latter ones, the incorporation of the ruthenium complex was slightly better when the alkoxysilane group was on the 5-position (versus 4-position) of the functionalized bipyridine. Surprisingly, the Si content of the Si–HPMC gels over the studied range (0.52 and 0.81 wt% Si) had no significant influence on the labeling yield whatever the nature of the label (see Table S-I, Supporting information).
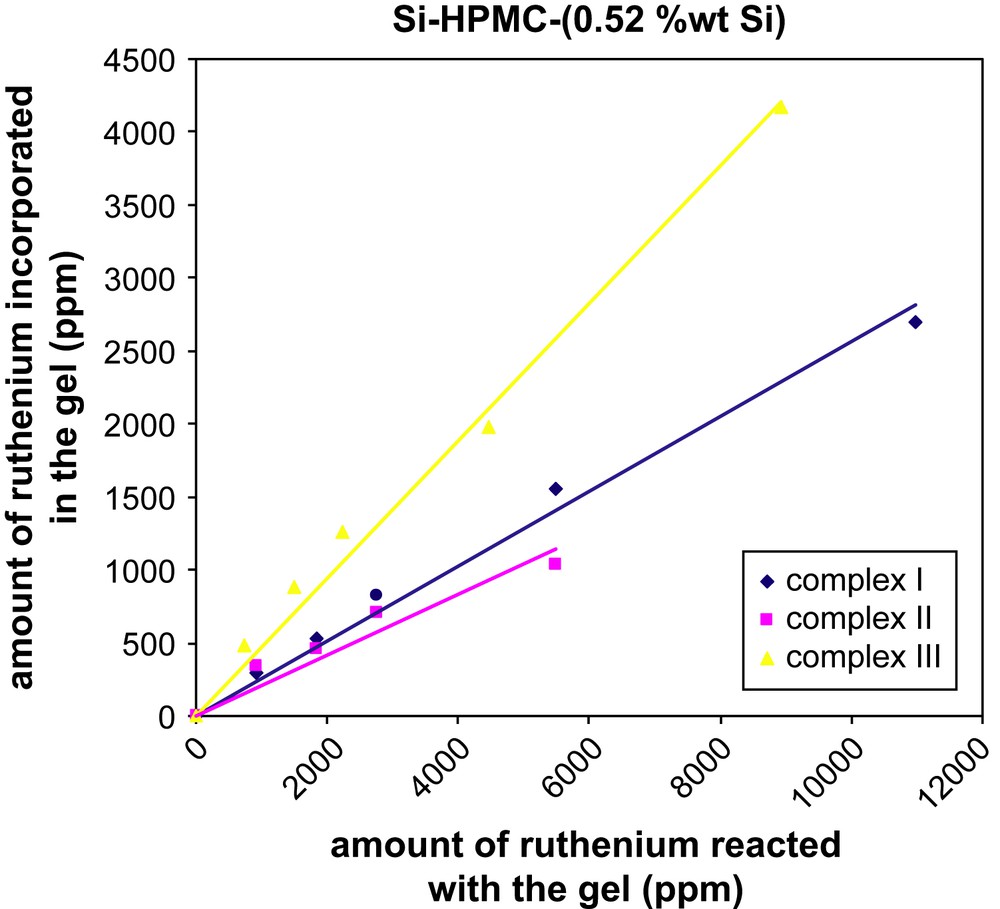
Amount of ruthenium incorporated in the reticulated Si–HPMC (0.52 wt% Si) hydrogel as a function of the initial ruthenium loading.
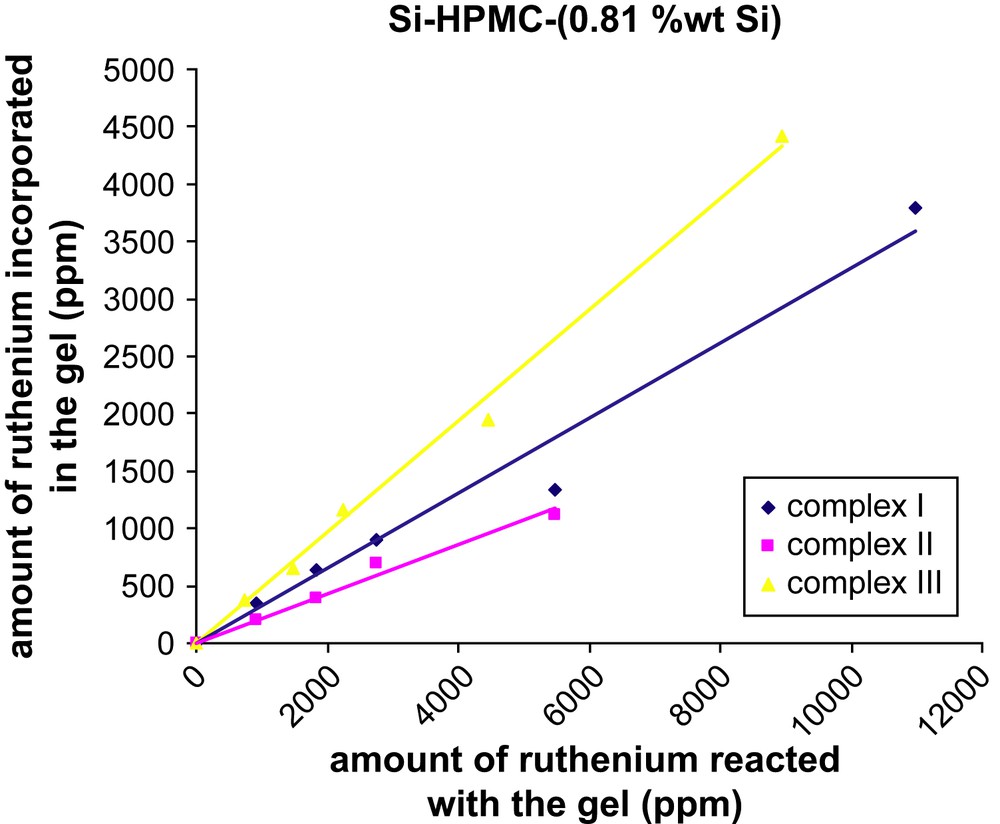
Amount of ruthenium incorporated in the reticulated Si–HPMC (0.81 wt% Si) hydrogel as a function of the initial ruthenium loading.
To examine whether the ruthenium probe present in the labeled reticulated gels after purification was covalently linked to the cellulose polymer: (i) the labeling protocol was carried out using Ru(bpy)3Cl2, for which no covalent attachment to the polymer was possible. Purification of the resulting gels by dialysis led to a colorless material and the entire amount of the initially loaded complex was found in the dialysis washing solutions; (ii) the same result was obtained when complex I or III were reacted with the non-silylated version of the polymer (HPMC E4M®). The latter experiments clearly suggested that no trapping of the ruthenium probe occurred within the gel, whether it is reticulated or not (see Table S-I, Supporting information).
3.3 Rheological properties
The rheological properties of the unlabeled and labeled Si–HPMC (0.52 wt% Si) aqueous solutions (3% w/w) were compared by recording typical parameters including the limiting Newtonian viscosity (η0), the storage and loss moduli (G′ and G″, respectively) and the gelation time (Table 1). The pristine Si–HPMC material showed a higher limiting viscosity (3 Pa·s) compared to samples labeled with ruthenium complexes I or III, however, with no influence of the nature of the label for a given concentration. The limiting viscosity was thus found to decrease with increasing the concentration of ruthenium (η0 ≈ 2.4 for 0.1% of I or III added; η0 ≈ 1.8 for 0.2% of I or III added). On the other hand, the gelation time of the various labeled Si–HPMC samples was found to be higher than in the case of unlabeled Si–HPMC and to increase with the number of alcoxysilane groups present on the ruthenium complex (III > I). In both cases, variation of the amount of label added to the hydrogel did not influence the gelation time. Finally, the storage modulus after 3 weeks of cross-linking was found to be similar for the unlabeled and labeled Si–HPMC samples, with a ∼0.1 wt% ruthenium complex loading, while significantly higher values were recorded by raising the loading to ∼0.2%. Accordingly, the experimental data suggest that the rheological properties of the hydrogel are not significantly affected, when low amounts (∼0.1%) of ruthenium label are used.
Newtonian viscosity (η0), gelation time, storage (G′) and loss (G″) moduli for different labeled Si–HPMC (0.52 wt% Si) samples
| Sample | Amount of complex (mg) | η0 (Pa s) | Gelation time (s) | Storage modulusa G′ (Pa) | Loss modulusa G″ (Pa) |
| Si–HPMC | 0 | 3.00 ± 0.04 | 830.00 ± 118.11 | 465.27 ± 54.70 | 20.26 ± 2.49 |
| Si–HMPC + I | 7.5 | 2.37 ± 0.02 | 1114.00 ± 176.31 | 358.70 ± 2.81 | 14.78 ± 0.09 |
| Si–HMPC + I | 15 | 1.77 ± 0.01 | 1101.00 ± 157.50 | 714.27 ± 23.08 | 41.85 ± 5.55 |
| Si–HMPC + III | 7.5 | 2.47 ± 0.02 | 2271.67 ± 324.97 | 453.50 ± 18.52 | 18.79 ± 0.59 |
| Si–HMPC + III | 15 | 1.75 ± 0.02 | 2639.67 ± 383.68 | 600.17 ± 6.68 | 24.71 ± 0.43 |
a Storage and loss moduli were obtained after 3 weeks of cross-linking.
4 Conclusion
In conclusion, we have demonstrated that ruthenium tris-bipyridine complexes functionalized by triethoxysilane groups allowed labeling a self-setting cellulose scaffold, used as a component of an injectable bone substitute. The labeling yield was found to be higher when two Si(OR)3 groups are present on the ruthenium complex backbone, and for low label loadings (less than ∼0.1 wt%), no significant modification of the rheological properties of the gel was observed. Implantations of labeled hydrogels in bone defects will be carried out in the next future, using animal models, in order to assess the biocompatibility and bioactivity of the materials and to investigate the in situ evolution of the gel by X-ray microfluorescence measurements on thin layers of bone explants.
Appendix Supporting information
Table S-I, describing the labeling yield of Si–HPMC (0.52 or 0.81 wt% Si) with ruthenium complexes I, II or III as a function of the initial label loading.