

1 Introduction
The notable similarity of a simple organometallic, (μ-pdt)[Fe(CO)3]2 (pdt = μ-S(CH2)3S), with the 2Fe subsite of the 6Fe6S hydrogen-producing or H-cluster of [FeFe]hydrogenase, [FeFe]H2ase, has led to an explosion of research aimed towards modifications which would better match the structure and ultimately the function of the enzyme's active site (eas). Critical to this goal are intrinsic properties that relate to the flexibility features of the (μ-pdt)[Fe(CO)3]2 molecule [2]. As shown in Fig. 1a, there are two possibilities for intramolecular site-exchange processes: (1) chair/boat interconversions in the iron–dithiacyclohexane rings; and (2) apical/basal CO site exchange resulting from rotation of the Fe(CO)3 units in the square pyramidal S2Fe(CO)3. For the latter process, computational studies have suggested the transition state shown in Fig. 1b, a structure which bears obvious similarity to the active site of the enzyme [3]. The structure of the eas was established by protein crystallography on the as-isolated paramagnetic redox level, presumed to contain FeIIFeI, as well as the reduced state, presumably FeIFeI [4–6]. It is expected that the unusual square pyramid/inverted square pyramid structure is fixed in the metalloprotein by second coordination sphere interactions. As electron/proton processes relating to the mechanistic cycle that defines enzyme activity (H2 ⇌ 2H+ + 2e−) require an open site for both H2 binding to oxidized FeII and for proton oxidation at reduced Fe0, we have hypothesized that the stabilization of such a rotated or square pyramid/inverted square pyramid is key to the design of a synthetic electrocatalyst that operates at mild potentials [1]. In fact two groups have now been successful in the replication of the rotated structure in a mixed valent FeIFeII model complex [7,8]. However without the supramolecular control of the protein matrix which exists in the eas, the rotation is not maintained in the small organometallic models which, upon reduction, relax to the optimal FeIFeI level unrotated geometry. Hence the challenge of isolating a rotated reduced diiron complex remains.

Depiction of (a) the fluxional processes of (μ-pdt)[Fe(CO)2L]2 (L = CO or phosphine substituents, with the HOMO of the ground state structure of the all-CO complex below); (b) optimized structure of the transition state for rotation or intramolecular COap/COba site exchange with HOMO of the all-CO complex below [3].
In a computational (DFT) analysis of this problem, it was concluded that substitution of a CO by a better electron-donor ligand should stabilize the rotated transition state of the FeIFeI complex as increased electron density at FeB would enhance the bridging CO character of the rotated structure [3]. While such a result was indicated for the (μ-pdt)[Fe(CO)3][Fe(CO)2IMes] complex, i.e., solution studies found the barrier to intramolecular CO site exchange in the [Fe(CO)3] unit to be lowered, a stable rotated FeIFeI form in the solid state was not realized [9]. A more recent DFT study addressed the effect of substituents on the bridgehead carbon of the S-to-S linker [1]. This study concluded that “the combination of a sterically demanding S-to-S linker and asymmetric substitution of the CO ligands is predicted to be a particularly effective synthetic target” [1]. To this end we have prepared a series of (μ-S(CH2CR2CH2)S)[Fe(CO)3]2 complexes and certain derivatives in which a CO ligand is replaced by an N-heterocyclic carbene ligand or PPh3. We report that in no case is the rotated form of the FeIFeI complexes observed as the ground state structure. Nevertheless, analysis of variable temperature NMR spectra delineates the effect of linker and substituents on the intramolecular site-exchange processes, finding further corroboration of the computational results. This work is preliminary to a study of oxidized disubstituted analogues, {(μ-S(CH2CR2CH2)S)[Fe(CO)2PMe3][Fe(CO)2NHC]}+, i.e., mixed valent FeIFeII species which do show evidence of stable rotated structures [7].
2 Experimental section
2.1 Methods and materials
Unless specifically stated, all syntheses and manipulations were performed using standard Schlenk-line and syringe/rubber septa techniques under N2 or in an Ar atmosphere glovebox. Solvents were purified according to published procedures, and freshly distilled under N2 prior to use or purified and degassed via a Bruker solvent system. The following materials were of reagent grade and used as received: Fe3(CO)12, IMes·HCl, PPh3, Me3NO (Aldrich Chemical Company), and deuterated solvents (Cambridge Isotope Laboratories). The dithiolate ligands depdt [10], dmpdt [10], bepdt [10], IMe [11], as well as the CDFCl2 used for low temperature NMR measurements [12] were synthesized according to the literature procedures. Anhydrous dimethylformamide (DMF) 99.9% was purchased from Acros Chemical Co.
Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, Georgia, United States. Infrared spectra were recorded on a Matteson Galaxy Series 6021 FTIR spectrometer or a Bruker Tensor 27 spectrometer in CaF2 solution cells of 0.1 mm path length. 1H and 13C NMR spectra were recorded on an Inova 500 MHz superconducting NMR instrument operating at 500.6, and 125.9 MHz, respectively.
2.2 Variable temperature NMR measurements
All samples used for variable temperature NMR measurements were isotopically enriched using a 275-W GE ultraviolet Sunlamp for the photolysis of the complexes in a solution of hexanes under a 13CO atmosphere. The solutions of hexanes were passed through a plug of silica gel and the solvent was removed. The resulting solids were used without further purification. All variable temperature NMR spectra were recorded on a Unity+ 500-MHz superconducting NMR instrument operating at 125.9 MHz.
2.3 Electrochemical studies
Cyclic voltammograms were recorded on a BAS-100A electrochemical analyzer using a three-electrode cell: a glassy carbon disk (0.071 cm2), the working electrode; reference electrode, Ag/AgNO3; and a coiled platinum wire, the counter electrode. Solutions were deaerated by an Ar purge for 5–10 min and a blanket of Ar (or CO as noted) was maintained over the solution while performing the measurements. All experiments were performed in CH3CN solutions containing 0.1 M nBu4NPF6 at room temperature. Ferrocene, Fc, served as the internal reference and all potentials are reported versus Fc/Fc+ as a standard (E1/2 = 0.00 V vs Ag/AgNO3 in CH3CN). Comparisons to earlier work in CH3CN may be accomplished by adding 0.40 V to the potential presented herein. Glacial acetic acid was added in molar equivalent increments via microsyringe.
2.4 X-ray structure analysis
Low-temperature (110 K) X-ray diffraction data were collected on a BRUKER SMART 1000 CCD-based diffractometer (Mo Kα radiation, λ = 0.71073 Å) for the (μ-dmpdt)[Fe(CO)3]2, (μ-depdt)[Fe(CO)3]2, (μ-bepdt)[Fe(CO)3]2, (μ-depdt)[(Fe(CO)3)(Fe(CO)2IMes)], (μ-depdt)[(Fe(CO)3)(Fe(CO)2IMe)] complexes [13]. For the (μ-dmpdt)[(Fe(CO)3)(Fe(CO)2IMes)] and (μ-dmpdt)[(Fe(CO)3)(Fe(CO)2PPh3)] complexes, data were obtained on a Bruker-AXS APEXII three-circle X-ray Diffractometer (Mo Kα radiation, λ = 0.71073 Å), also operating at 110 K.
The structures were solved by direct methods. Hydrogen atoms were added at idealized positions and refined with fixed isotropic displacement parameters equal to 1.2 times the isotropic displacement parameters of the atoms to which they were attached. Anisotropic displacement parameters were determined for all non-hydrogen atoms. Programs used: for data collection and cell refinement, SHELXTL; absorption correction [14], SADABS; structure solution, SHELXS-97 (Sheldrick)1; structure refinement, SHELXL-97 (Sheldrick) [15]. Molecular graphics and preparation of material for publication used SHELXTL-PLUS, version 5.1 or the latter (Bruker). X-seed was employed for the final data presentation and structure plots [16].
2.5 Synthesis
2.5.1 Preparation of (μ-dmpdt)[Fe(CO)3]2 (1)
To a 50-mL Schlenk flask containing 2,2-dimethyl-1,3-propanedithiol (1.5 g, 0.011 mol), toluene (15 mL) was added. This light yellow solution was transferred via cannula to a 200 mL Schlenk flask containing Fe3(CO)12 (5.56 g, 0.011 mol) in 100 mL of toluene. The resultant blue-green solution was stirred at 60 °C and monitored by IR spectroscopy. After approximately 3 h the reaction was cooled to room temperature. At this point silica gel (2 g) was added to the flask and the solvent removed by rotary evaporation. The resulting solid containing 1 and silica gel was loaded onto a 4 cm diameter column packed with 20 cm of silica gel. The product, which appeared as a bright red band, was eluted with hexanes. Upon removal of solvent by rotary evaporation, pure microcrystalline 1 was obtained. Yield: 1.56 g (34.1%). Crystals of X-ray quality were obtained by placing concentrated solutions of 1 in hexanes in the freezer overnight. IR(hexanes) ν(CO), see table in Fig. 2; Elem. Anal. Calc'd (found) C11H10O6S2Fe2: C, 31.89 (31.93); H, 2.44 (2.44). 1H NMR (CDCl3): 0.99 (s, 6H), 2.08 (s, 4H) ppm. 13C NMR (CDCl3): 30.5, 31.8, 33.5, 207.9 ppm. Mp: 138–141 °C.
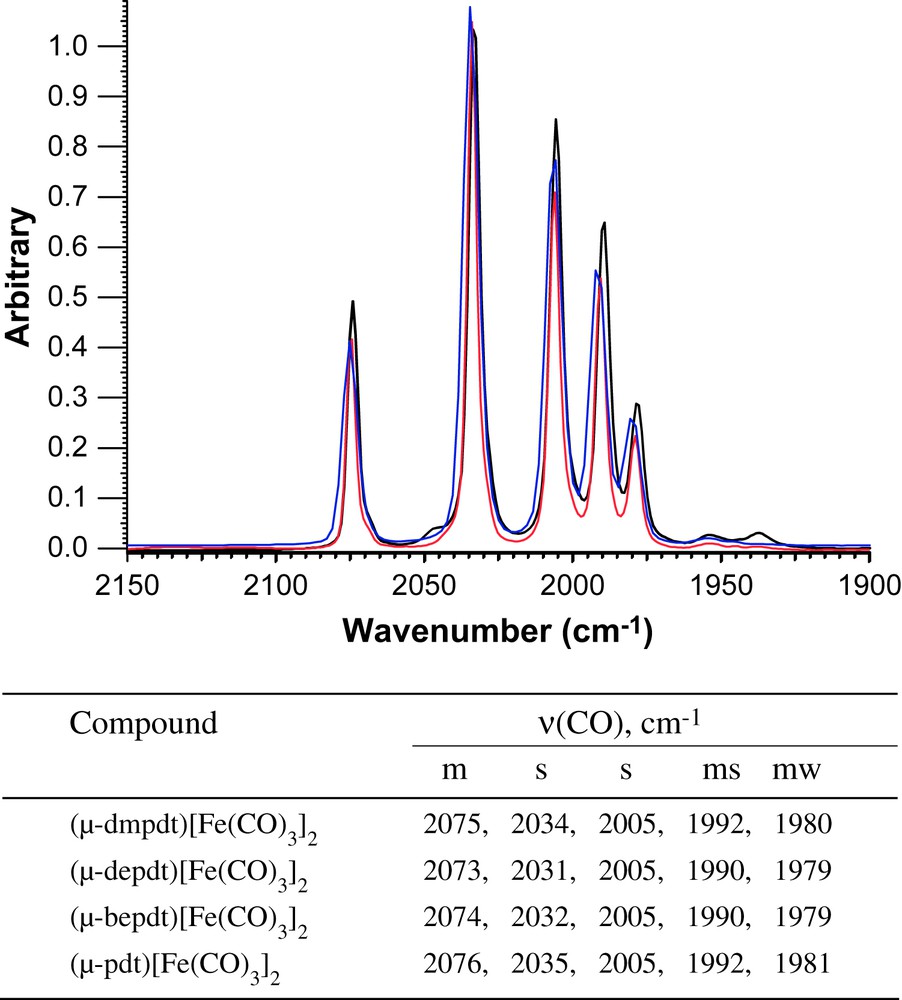
Overlay of ν(CO) IR spectra of (μ-Rdt)[Fe(CO)3]2 recorded in a solution of hexanes (blue: Rdt = dmpdt; red: Rdt = depdt; black, Rdt = bepdt) with a table listing the values and intensities for the bands. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
2.5.2 Preparation of (μ-depdt)[Fe(CO)3]2 (2)
Complex 2 was prepared in a manner analogous to 1 using 2,2-diethyl-1,3-propanedithiol (2.0 g, 0.012 mol) and Fe3(CO)12 (6.14 g, 0.012 mol). Yield: 1.18 g (21.9%). IR (hexanes) ν(CO), see table in Fig. 2; Elem. Anal. Calc'd (found) C13H14O6S2Fe2: C, 35.30 (35.39); H, 3.19 (3.16); 1H NMR (CDCl3): 0.74 (t, J = 7.1 Hz, 6H), 1.35 (q, J = 7.37 Hz, 4H), 2.08 (s, 4H) ppm. 13C NMR (CD2Cl2): 7.6, 29.4, 30.1, 38.5, 207.8 ppm. Mp: 123.8–125.5 °C.
2.5.3 Preparation of (μ-bepdt)[Fe(CO)3]2 (3)
Complex 3 was prepared in a manner analogous to 1 using 2-butyl-2-ethyl-1,3-propanedithiol (2.0 g, 0.010 mol) and Fe3(CO)12 (5.25 g, 0.010 mol). Yield: 0.95 g (19.4%). IR (hexanes) ν(CO), see table in Fig. 2; Elem. Anal. Calc'd (found) C15H18O6S2Fe2: C, 38.30 (38.20); H, 3.86 (3.84); 1H NMR (CDCl3): 0.73 (t, J = 6.9 Hz, 3H), 0.89 (t, J = 6.9 Hz, 3H), 1.04 (m, 2H), 1.27 (m, 4H), 1.36 (q, J = 7.39 Hz, 2H), 2.08 (s, 4H). 13C NMR (CDCl3): 7.6, 14.3, 23.2, 25.4, 29.8, 30.5, 37.5, 38.4, 207.8, 207.8 ppm. Mp: 80.4–82.8 °C.
2.5.4 Preparation of (μ-dmpdt)[(Fe(CO)3)(Fe(CO)2IMes)] (4)
To a 200-mL Schlenk flask containing 1 (0.500 g, 1.21 mmol), IMes·HCl (0.411 g, 1.21 mmol) and K+[OtBu]− (0.162 g, 1.45 mmol), 100 mL of dry THF was added. The reaction was stirred at 50 °C and monitored by infrared spectroscopy. After approximately 2.5 h the dark red/purple solution was cooled to room temperature. At this point silica gel (2 g) was added to the flask and the solvent was removed by rotary evaporation. The resulting solid was loaded onto a 4 cm diameter column packed with 20 cm of silica gel. Excess 1 was first eluted using hexanes, and the product, which appeared as a purple-red band, was then eluted with diethyl ether. The ether was removed by rotary evaporation, leaving a dark purple-red solid. Yield: 0.307 g (36.8%). Crystals of X-ray quality were obtained by slow evaporation of solvent from a concentrated solution of 4 in ether at room temperature. ν(CO) IR (THF): 2030 m, 1971 s, 1947 m sh, 1913 w cm−1; Elem. Anal. Calc'd (found) C31H34N2O5S2Fe2: C, 53.91 (54.20); H, 4.97 (4.98). 1H NMR (CDCl3): 0.77 (s, 3H), 1.58 (s, 3H), 2.18 (s, 14H), 2.35 (s, 8H), 6.94 (s, 2H), 6.99 (s, 4H) ppm. 13C NMR (CDCl3): 29.4, 31.2, 32.6, 34.0, 124.1, 129.1, 136.6, 137.9, 139.2, 187.2, 211.7, 215.4 ppm.
2.5.5 Preparation of (μ-depdt)[(Fe(CO)3)(Fe(CO)2IMes)] (5)
Complex 5 was prepared in a manner analogous to 4 using 2 (0.500 g, 1.13 mmol), IMes·HCl (0.385 g, 1.13 mmol) and K+[OtBu]− (0.152 g, 1.36 mmol). Yield: 0.358 g (44.1%). ν(CO) IR (THF): 2025 m, 1971 s, 1946 m sh, 1911 w cm−1; Elem. Anal. Calc'd (found) C33H38N2O5S2Fe2 + 1(C2H5)2O: C, 56.05 (55.99); H, 6.11 (5.65). 1H NMR (CDCl3): 0.57 (m, 6H), 1.09 (q, 2H), 1.25 (q, 2H), 2.18 (s, 14H), 2.35 (s, 8H), 6.94 (s, 2H), 7.0 (s, 4H) ppm. 13C NMR (CDCl3): 7.1, 7.3, 18.7, 21.2, 27.6, 31.2, 32.2, 37.7, 125.1, 129.0, 136.6, 137.8, 139.1, 188.2, 211.7, 215.4 ppm.
2.5.6 Preparation of (μ-depdt)[(Fe(CO)3)(Fe(CO)2IMe)] (6)
Complex 6 was prepared in a manner analogous to 4 using 2 (0.500 g, 1.13 mmol), IMe·HCl (0.149 g, 1.13 mmol) and K+[OtBu]− (0.152 g, 1.36 mmol). Yield: 0.086 g (14.9%). ν(CO) IR (THF): 2025 m, 1971 s, 1946 m sh, 1911 w cm−1; Elem. Anal. Calc'd (found) C17H22N2O5S2Fe2: C, 40.00 (40.46); H, 4.35 (4.45). 1H NMR (CDCl3): 0.42 (t, J = 7.0 Hz, 3H), 0.67 (t, J = 7.0 Hz, 3H), 1.15 (q, J = 6.9 Hz, 2H), 1.22 (q, J = 6.9 Hz, 2H), 1.58 (d, J = 12.5 Hz, 2H), 1.82 (d, J = 12.5 Hz, 2H), 4.03 (s, 6H), 7.0 (s, 2H) ppm. 13C NMR (CDCl3): 7.6, 7.9, 26.3, 30.4, 33.2, 37.8, 39.9, 123.7, 191.5, 211.2, 215.2 ppm.
2.5.7 Preparation of (μ-dmpdt)[(Fe(CO)3)(Fe(CO)2PPh3)] (7)
To a 50-mL round bottom flask containing trimethylamine-N-oxide (20 mg, 0.266 mmol) was added 20 mL of CH3CN. The resulting solution was transferred via cannula to a 100 mL Schlenk flask containing 1 (100 mg, 0.242 mmol) dissolved in 30 mL of CH3CN. After the reaction mixture had stirred for 30 min, a solution of PPh3 (70 mg, 0.266 mmol) in 10 mL of CH3CN was added and the reaction progress was monitored by IR spectroscopy. Upon completion, requiring ca. 30 min, solvent was removed under vacuum. The resulting solid was dissolved in a minimal amount of ether and loaded onto a 4 cm diameter column containing 20 cm of silica gel. The product, which appeared as a dark red band, was eluted with ether. Upon removal of solvent by rotary evaporation pure 7 was obtained. Yield: 24 mg (15.3%). ν(CO) IR (THF): 2045 ms, 1981 s, 1961 m sh, 1927 w cm−1; Elem. Anal. Calc'd (found) C28H25O5PS2Fe2: C, 51.86 (51.71); H, 3.89 (3.83). 1H NMR (CDCl3): 0.93 (s, 3H), 1.16 (d, J = 12.9 Hz, 2H), 1.26 (s, 3H), 1.80 (d, J = 13.4 Hz, 2H), 7.44 (m, 3H), 7.51 (m, 6H), 7.71 (m, 6H) ppm. 13C NMR (CDCl3): 28.7, 29.9, 31.3, 32.8, 128.7 (JC–P = 12.7 Hz), 128.7 (JC–P = 9.8 Hz), 132.3 (JC–P = 10.8 Hz), 133.7 (JC–P = 10.9 Hz), 209.6, 214.2 (JC–P = 12.8 Hz) ppm.
3 Results and discussion
3.1 Synthesis and characterization of all-carbonyl (μ-SCH2CR2CH2S)[Fe(CO)3]2 complexes
Simple addition of the dithiols listed in Scheme 1 to solutions of Fe3(CO)12 in toluene produced the series of thermally and air stable, red-orange crystalline products of the form (μ-S(CH2CRR′CH2)S)[Fe(CO)3]2 (R/R′ = Me/Me, Et/Et, Bu/Et) in isolated yields of 19–34%. Their infrared spectral signatures in the ν(CO) stretching region are nearly identical to each other and to the parent (μ-pdt)[Fe(CO)3]2 complex, Fig. 2, indicating that the addition of alkyl groups to the bridgehead position does not drastically change the thiolate donor ability.

As derived from X-ray diffraction studies, ball and stick drawings of the molecular structures of the three diiron hexacarbonyl derivatives and (μ-pdt)[Fe(CO)3]2 [17], are shown in Table 1 along with selected metric data. Consistent with the similar spectral properties of (μ-pdt)[Fe(CO)3]2 and the three analogues as found in solution, there are only minor solid state structural differences. The most significant deviation from the μ-pdt complex is evident in the torsion angle defined by the apical CO groups across the Fe–Fe bond vector. As shown in Fig. 3 the steric bulk of the bridgehead substituent induces a slight staggering of the apical CO unit, with the greatest distortion seen in the diethyl derivative. In all cases the basal COs eclipse each other similar to the μ-pdt complex. As seen in the μ-depdt derivative (2), positioning of the ethyl group over C1 is observed in the asymmetrically substituted bridgehead complex, (μ-bepdt)[Fe(CO)3]2, leaving the butyl group oriented away from the Fe2S2 core. In the extended packing diagram, a portion of which is shown in Fig. 4, this configuration allows the hydrophobic butyl groups of nearby complexes to be stacked, decreasing interaction of the butyl group with the nearby CO and thus the apical C–Fe–Fe–C torsion angle relative to the depdt derivative. For all of the sterically bulky complexes the substituents at the bridgehead carbon of the S-to-S linker do not cause major distortions in the Fe(CO)3 units, nor are the parameters which define the Fe2S2C3 cyclohexane-like ring different.
Comparison of structures of (μ-pdt)[Fe(CO)3]2 [17] with (μ-dmpdt)-, (μ-depdt)-, (μ-bepdt)[Fe(CO)3]2
| Name | Structure | Fe–Fe, Å | Cl–Fe1–Fe2–C6, ° | Flap angle, °a | , °b |
| (μ-pdt)[Fe(CO)3]2 | 2.510(1) | 0.0 | 137.1 | 107.6/107.6 | |
| (μ-dmpdt)[Fe(CO)3]2 | 2.494(6) | 5.7 | 134.8 | 118.0/105.7 | |
| (μ-depdt)[Fe(CO)3]2 | 2.501(4) | 15.8 | 136.7 | 115.6/105.2 | |
| (μ-bepdt)[Fe(CO)3]2 | 2.500(1) | 6.2 | 137.7 | 116.2/105.1 |
a Refers to angle between plane made from bridgehead carbons–elbow carbons and the plane made from the elbow carbons–sulfurs.
b Refers to angle between apical carbonyl and the adjacent Fe–S–S plane. Bridgehead side (left side as shown)/nonbridgehead side (right side as shown).
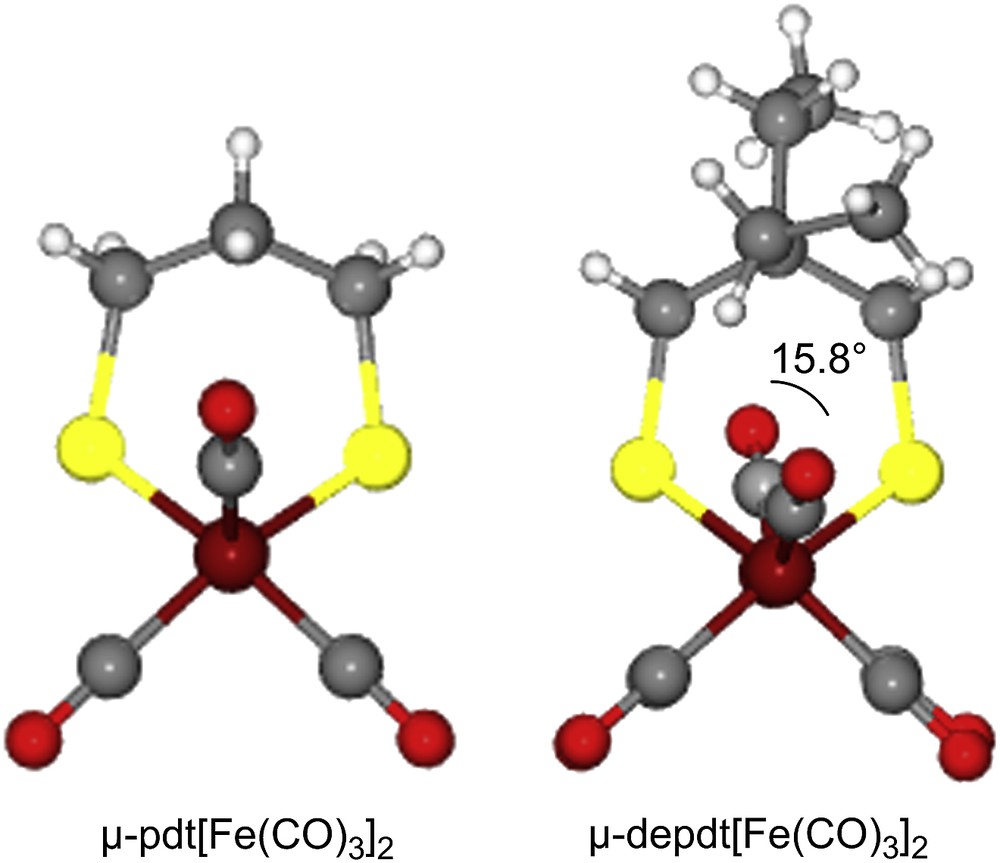
Ball and stick displays of the μ-pdt and μ-depdt derivatives showing the eclipse of (C1O1) and (C6O6) in (μ-pdt)[Fe(CO)3]2 [17] and the slight stagger of the (C1O1) and (C6O6) in the (μ-depdt)[Fe(CO)3]2 complex. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
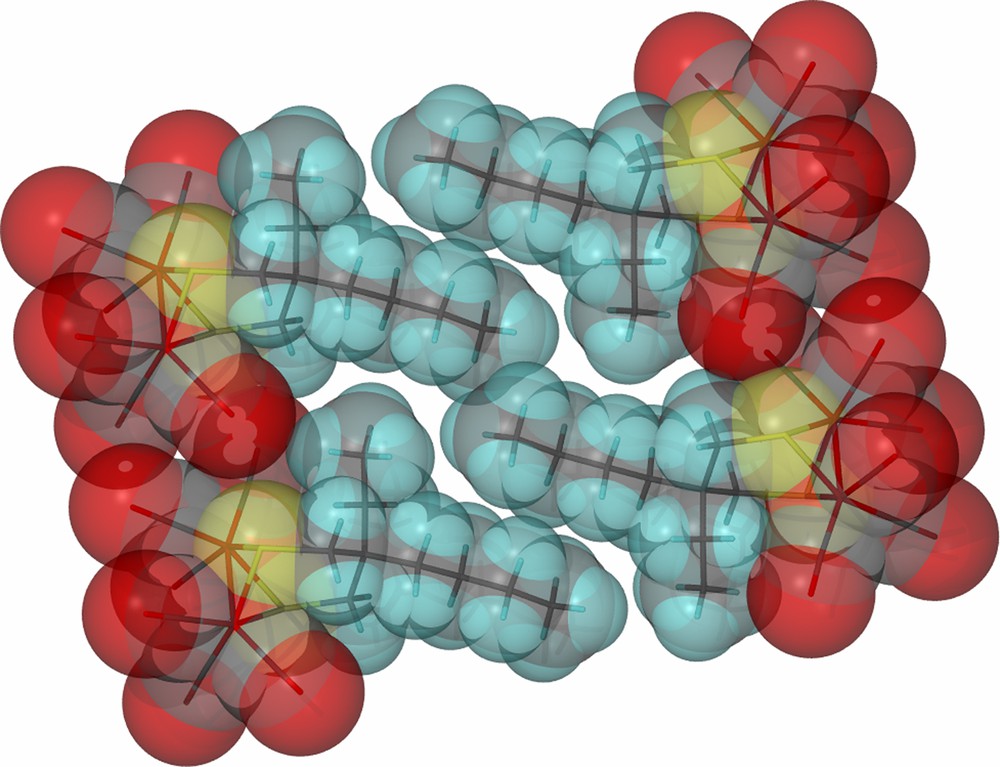
Space-filling, close-packing diagram of (μ-bepdt)[Fe(CO)3]2 showing the orientation of the hydrophobic butyl groups on the propane dithiolate linker. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2 Synthesis and characterization of monosubstituted (μ-SCH2CR2CH2S)[Fe(CO)3][Fe(CO)2L] complexes
The products of direct CO/L exchange, or oxidatively induced ligand exchange are displayed in Scheme 2, in the form of line-drawing structures, along with the conditions required for the syntheses. The particular orientations of the FeS2C3 bridgehead carbon and its substituents, as well as the isomeric form of the Fe(CO)2L unit are represented as found in the solid state molecular structures, described below. Upon substitution for L, two distinct changes can be seen in the NMR spectra. In the 1H spectra the resonance corresponding to the protons on the carbon α to the thiolates split into two doublets. This is consistent with the loss of the plane of symmetry perpendicular to the FeFe bond. Additionally, the 13C NMR spectra now show two separate signals of different intensities for the CO ligands indicating that substitution on one of the Fe units has occurred. Infrared spectra show a ν(CO) shift, ca. 30–40 cm−1, of the highest energy band to lower wave numbers supporting that CO has been replaced by a better donor. Vibrational spectroscopy results are also consistent with the poorer electron-donating ability of PPh3 relative to the imidazole NHC ligands. For example, Fig. 5 is an overlay of the ν(CO) IR spectra of (μ-dmpdt)[Fe(CO)3][Fe(CO)2PPh3] and (μ-dmpdt)[Fe(CO)3][Fe(CO)2IMes]. The IR bands for the former range from 12 to 23 cm−1 higher than those of the latter.
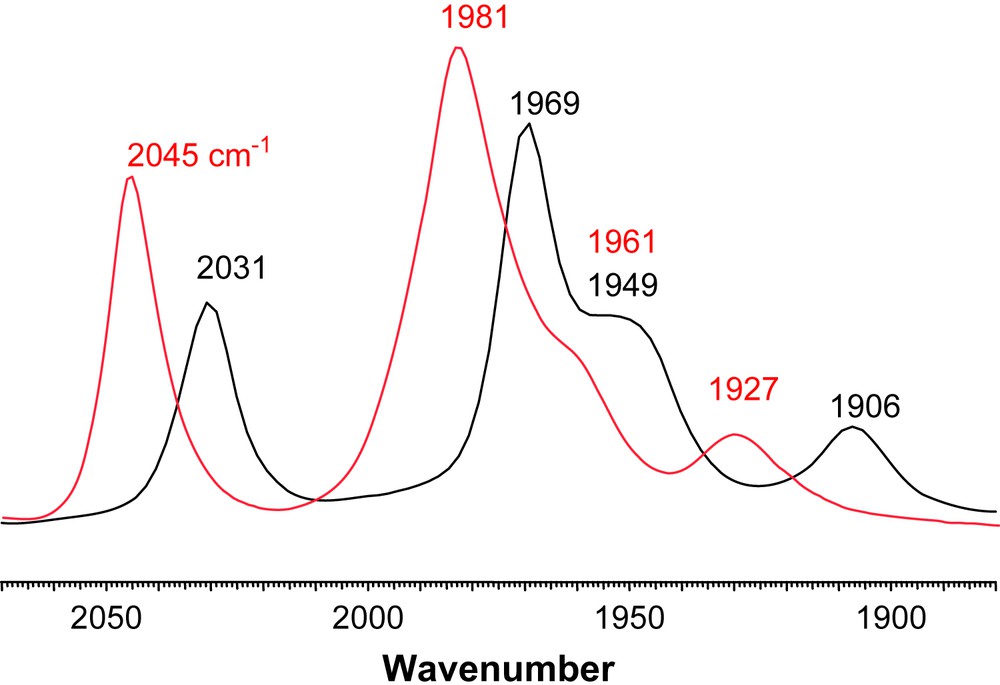
Overlay of ν(CO) IR spectra of (μ-dt)[Fe(CO)3][Fe(CO)2L] in CH3CN solution (black: L = IMes; red: L = PPh3) showing the weaker donating ability of PPh3 versus IMes. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
Chemdraw figures based on the X-ray diffraction studies of the monosubstituted diiron complexes are given in Table 2 along with selected metric data. While the overall structural features are the same as in the all-CO diiron complexes, comparison of the (μ-pdt)[Fe(CO)3][Fe(CO)2IMes] structure to those with steric bulk in the S-to-S linker finds differences in certain parameters. The IMes ligand in the μ-dmpdt and μ-depdt derivatives occupies the basal rather than apical position as was found for the μ-pdt complex [9]. Nevertheless, the bridgehead C of the FeS2C3 ring of both substituted derivatives is oriented towards the Fe(CO)2IMes unit as it is in the μ-pdt complex. This orientation of the bridgehead is also seen for (μ-depdt)[Fe(CO)3][Fe(CO)2IMe]. In contrast, the substituted bridgehead C is oriented away from the Fe(CO)2PPh3 and towards the Fe(CO)3 unit in (μ-dmpdt)[Fe(CO)3][Fe(CO)2PPh3]. This results in a different conformation from (μ-pdt)[Fe(CO)3][Fe(CO)2PPh3] where the bridgehead is oriented towards the Fe(CO)2PPh3 unit [18]. However, in both the IMe and PPh3 complexes the ligand assumes the apical position consistent with their respective pdt analogues [18,19]. These results show the importance of steric bulk in the ligands as well as in the bridgehead carbon of the S-to-S linker.
Structural comparison of complexes 4–7 with (μ-pdt) analogues
| Name | Structuresa | Fe–Fe, Å | Lap–Fe1–Fe2–Lap, ° | Flap angle, ° | Lap–FeSS, ° |
| (μ-pdt)[Fe(CO)3][Fe(CO)2IMes] [9] | 2.525 | 4.26 | 124.1 | 117.24/109.27 | |
| (μ-pdt)[Fe(CO)3][Fe(CO)2PPh3] [18] | 2.5247(6) | 9.31 | 133.8 | 116.67/106.10 | |
| (μ-pdt)[Fe(CO)3][Fe(CO)2IMe] [19] | 2.5333(7) | 5.29 | 126.9 | 108.24/106.77 | |
| (μ-dmpdt)[Fe(CO)3][Fe(CO)2IMes] | R = Me 2.572(4) | 40.7 | 136.9 | 116.45/101.83 | |
| (μ-depdt)[Fe(CO)3][Fe(CO)2IMes] | R = Et 2.552(2) | 27.8 | 136.1 | 117.37/102.76 | |
| (μ-depdt)[Fe(CO)3][Fe(CO)2IMe] | 2.512(1) | 18.3 | 133.6 | 117.30/105.94 | |
| (μ-dmpdt)[Fe(CO)3][Fe(CO)2PPh3] | 2.498(1) | 9.76 | 138.7 | 117.18/113.42 |
a Configurations drawn as in the solid-state X-ray structure.
The torsion angle, Cap–Fe–Fe–Lap, between the Fe(CO)3 and Fe(CO)2Lap units on the complexes with added steric bulk largely remains unchanged from that of the respective μ-pdt derivative with the exception of (μ-dmpdt)[Fe(CO)3][Fe(CO)2IMes] which is twisted by >36° relative to (μ-pdt)[Fe(CO)3][Fe(CO)2IMes]. The structure of (μ-dmpdt)[Fe(CO)3][Fe(CO)2IMes], Fig. 6, readily shows displacement of the apical CO while the ligands in the basal planes of each iron center remain eclipsed. This degree of distortion is however not seen for the μ-depdt derivative; the reason for this is presumed to be that the –CH3 unit of the ethyl groups in (μ-depdt)[Fe(CO)3][Fe(CO)2IMes] inhibits the approach of the mesityl group of the IMes ligand towards the apical CO, thus decreasing the amount of steric influence from the aryl methyl groups. In the μ-dmpdt derivative both the methyl groups on the S-to-S linker as well as on the mesityl groups assist in the deflection of the CO group.
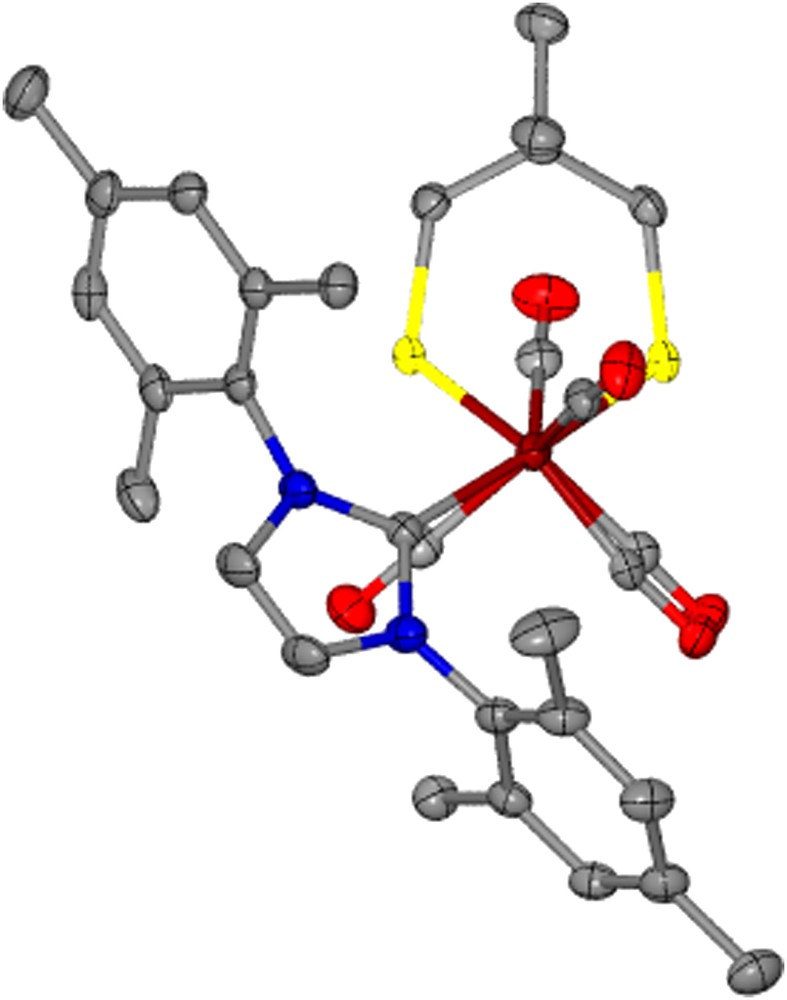
Thermal ellipsoid plot (50% probability) of (μ-dmpdt)[Fe(CO)3][Fe(CO)2IMes] showing the staggering of the apical CO units.
Interestingly, the dimesitylimidazolium substituted diiron complexes induce a lengthening of the Fe–Fe bond distance by ca. 0.05–0.07 Å relative to their respective all-carbonyl parent compound and ca. 0.03–0.05 Å relative to (μ-pdt)[Fe(CO)3][Fe(CO)2IMes]. The Fe–Fe bond length of 2.572 Å for the μ-dmpdt IMes complex is one of the longest reported for any monosubstituted or all-carbonyl μ-dithiolate diiron complex in the Cambridge crystal database of >150 hits for (μ-SR)2 or (μ-SRS)[Fe(CO)3]2. This extended bond distance is consistent with greater partial occupancy of the LUMO which is anti-bonding with respect to the Fe–Fe bond [3].
3.3 Electrochemical studies of complexes 1–3, 4, 6, and 7
Cyclic voltammograms of complexes 1–3 (Fig. 7a and Table 3) in CH3CN solution display similar events to each other as well as the (μ-pdt)[Fe(CO)3]2 derivative, with an irreversible oxidative wave in the range of +0.73 to +0.82 V and two reduction events: a quasi-reversible reduction at ca. −1.6 V and an irreversible reduction at ca. −2.4 V (vs Fc). Based on previously reported (μ-pdt)[Fe(CO)3]2 derivatives, the quasi-reversible reduction event can be assigned to the FeIFeI → FeIFe0 reduction and the second irreversible reduction event is assumed to be FeIFe0 → Fe0Fe0 [20]. Such assignments represent a simplistic scheme for the electrochemical processes of these systems, and the actual behavior of these models has been reported to be more complex [21]. The potentials listed in Table 3 are referenced to Fc/Fc+ = 0.00 V. In an earlier study of the electrocatalytic H2 producing ability of the (μ-pdt)[Fe(CO)3]2 complex, NHE was used as a reference; direct comparison may be made by subtracting 0.40 V from the original values reported. The fact that the redox events remain relatively constant in this series of complexes supports the conclusion from infrared data that the addition of alkyl substituents at the bridgehead carbon has little influence on the S-donor ability of the bridging dithiolate and hence, the electron density about the iron centers. This is in contrast to (μ-SC6H4S)[Fe(CO)3]2 where an aryl group is built into the linker causing the FeIFeI/FeIFe0 reduction potential to shift by ca. 400 mV more positive [22].
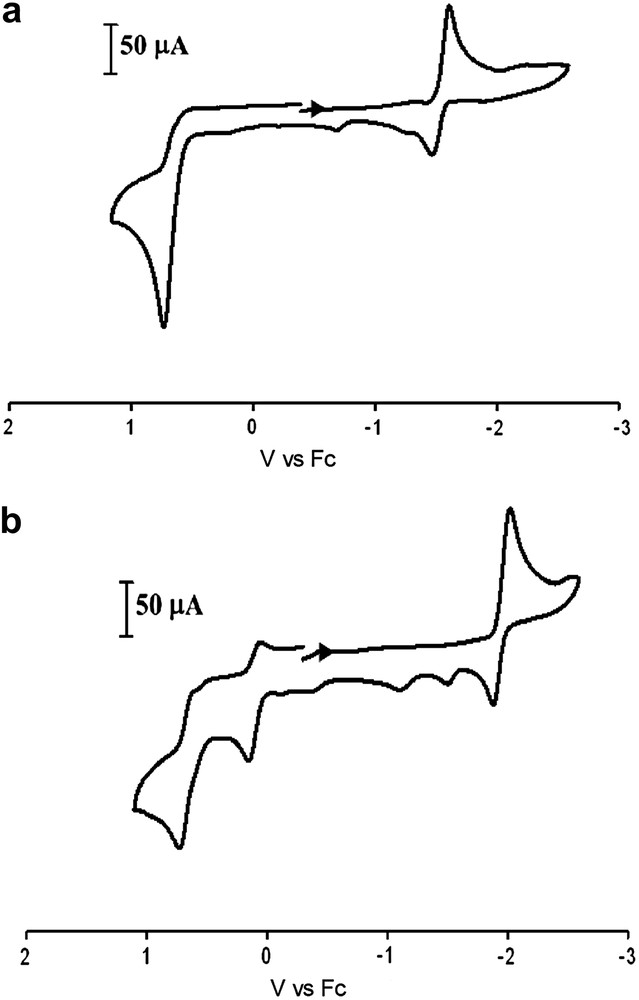
Cyclic voltammograms of (top) complex 1 and (bottom) 4 (2 mM) 0.1 M nBu4NBF4 in MeCN solution at a scan rate of 200 mV/s.
Electrochemical potentials (vs ferrocene) of complexes 1–4, 6, and 7
| Complex | Epa (V) | Epc (V) |
| (μ-dmpdt)[Fe(CO)3]2 (1)a | +0.73 | −1.61, −2.24 |
| (μ-depdt)[Fe(CO)3]2 (2)b | +0.82 | −1.67, −2.27 |
| (μ-bepdt)[Fe(CO)3]2 (3)b | +0.78 | −1.64, −2.27 |
| (μ-dmpdt)[Fe(CO)3][Fe(CO)2IMes]b (4) | +0.76, +0.05 | −2.01 |
| (μ-depdt)[Fe(CO)3][Fe(CO)2IMe]a (6) | +0.73, +0.16 | −2.02 |
| (μ-dmpdt)[Fe(CO)3][Fe(CO)2PPh3]b (7) | +0.69, +0.35 | −1.79, −2.29 |
| (μ-pdt)[Fe(CO)3]2 [17] | +0.74c | −1.74,c −2.35c |
| (μ-pdt)[Fe(CO)3][Fe(CO)2IMes] [9] | +0.72, +0.11c | −2.11c |
a Ar purged CH3CN solution (0.1 M nBu4NBF4).
b CO-saturated CH3CN solution (0.1 M nBu4NBF4). All experiments were recorded using a glassy carbon working electrode (A = 0.071 cm2) referenced to Cp2Fe/Cp2Fe+ as an internal standard and a Pt counter electrode at a scan rate of 200 mV/s.
c 0.40 V was subtracted from originally reported values for comparison.
Electrochemical data for the NHC-substituted complexes, 4 and 6, (Fig. 7b and Table 3) show ca. 300 mV cathodic shifts for the quasi-reversible FeIFeI/Fe0FeI reduction as compared to the all-CO complexes, consistent with the fact that CO has been exchanged for a more electron-donating ligand. As expected, due to the poorer electron-donating ability of PPh3 in complex 7, a shift of this magnitude is not seen (Fig. 8 and Table 3); however a second, irreversible reduction event at −2.29 V is present. The oxidation event presumed to be FeIFeI/FeIIFeI for complexes 4, 6 and 7 has become more accessible by ca. 600 mV for the NHC derivatives and ca. 300 mV for complex 7 as compared to the all-CO complexes. In addition to an anodic shift, this first oxidation wave has become partially reversible in the two NHC-substituted complexes but remains irreversible for the PPh3 complex. More interestingly, all of the monosubstituted complexes have gained a second irreversible oxidation event between 690 and 760 mV tentatively assigned to FeIIFeI/FeIIFeII. For all of the monosubstituted complexes with added steric bulk, the events observed are similar to those of their respective μ-pdt analogues [9,18,19].
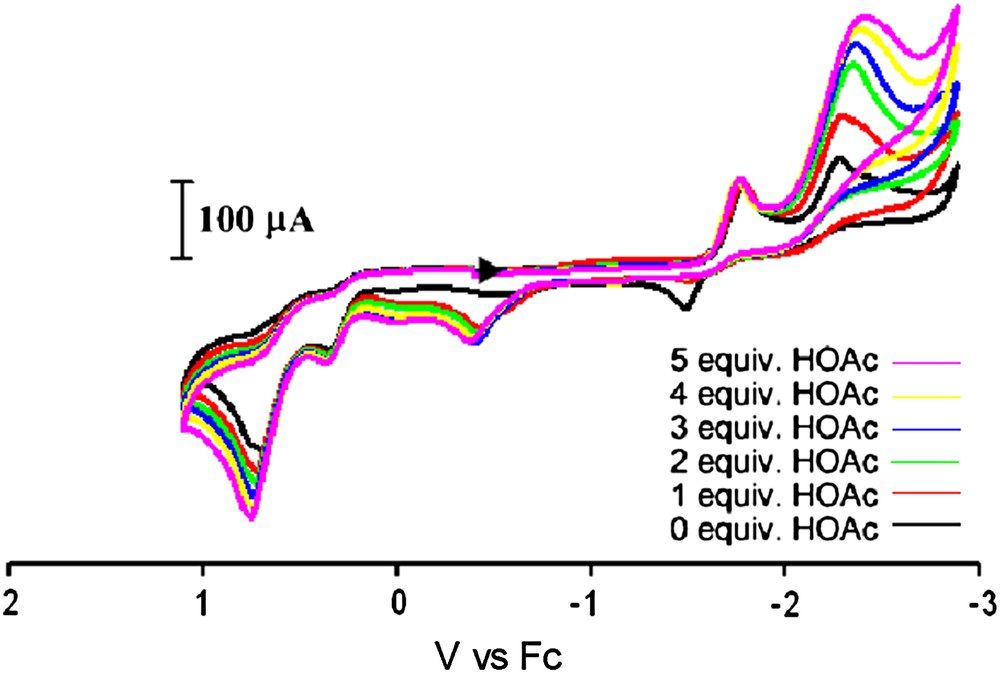
Cyclic voltammograms under CO of complex 7 and with increments of HOAc in an MeCN solution (0.1 M nBu4NBF4) using a glassy carbon electrode at a scan rate of 200 mV/s. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The electrocatalytic proton reduction activity of complex 7 was also tested. With added increments of HOAc, an increase in the peak current for the reduction wave at −2.29 V is observed while the peak current remains constant for the reduction at −1.79 V. This suggests that the Fe0Fe0 species (assuming this redox level for the more negative reduction event) is active towards electrocatalytic H2 production, whereas the Fe0FeI species is not. This behavior is more similar to that of the all-CO complexes than to the monosubstituted IMes complex reported in an earlier study [9]. This is most likely due to the fact that the weak donor ability of triphenylphosphine renders iron insufficiently electron rich to accommodate the oxidative addition of a proton.
3.4 Variable temperature NMR spectra of (μ-SCH2CR2CH2S)[Fe(CO)3][Fe(CO)2L] (L = CO or IMes) complexes
At 0 °C both the μ-dmpdt and μ-depdt derivatives show a single resonance in the CO region at δ = 208.3 and 207.8 ppm, respectively. These signals respond to temperature changes in a completely reversible way. As shown in Fig. 9, upon cooling (μ-depdt)[Fe(CO)3]2 to −110 °C the CO resonance splits into two singlets of differing intensities at δ = 208.2 and 206.1 with the intensity of the former roughly twice that of the latter. The δ = 208.2 ppm resonance is assigned to the basal COs and the δ = 206.1 ppm resonance to the apical COs in the regime where CO site exchange has ceased but the boat/chair interconversion of the FeS2C3 ring is still occurring. The spectra for the (μ-dmpdt)[Fe(CO)3]2 complex show a nearly identical trend.
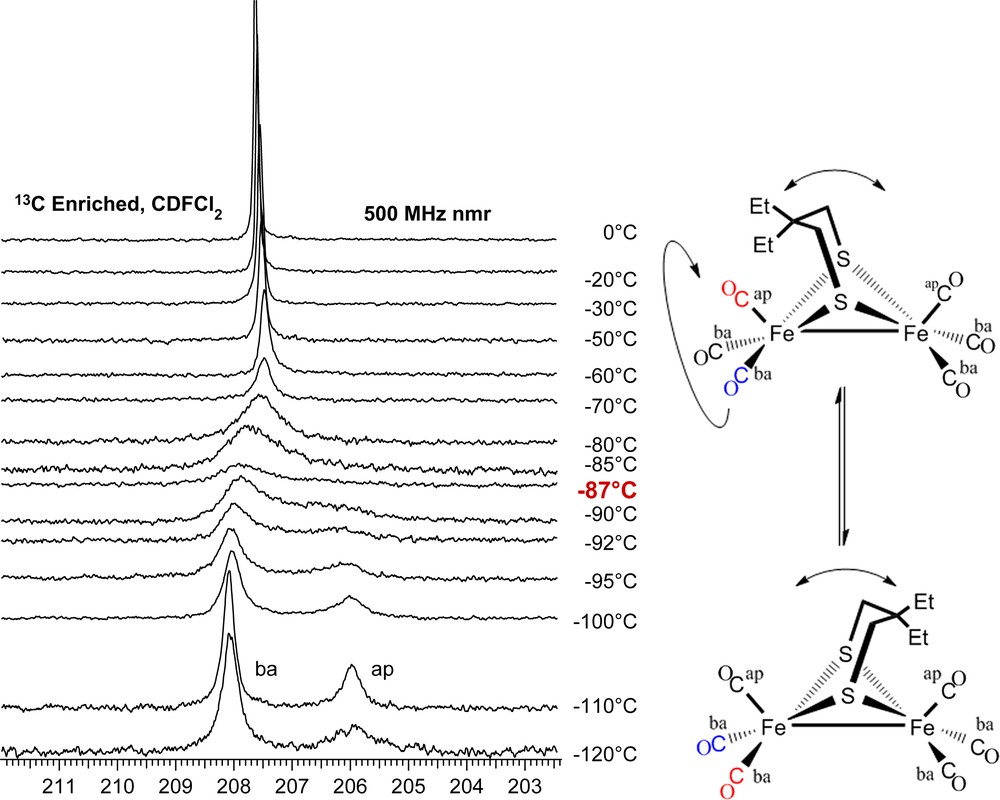
13C VT NMR spectra of (μ-depdt)[Fe(CO)3]2 showing separate resonances for the apical CO and basal COs at low temperature. Coalescence temperature indicated in red. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
The energy barrier for CO site exchange can be estimated using the peak separation (Δv) and coalescence temperature (Tcoal) with the equations ΔG‡ = −(RT)ln[kth/kbTcoal] and kt = (2πΔv)/20.5 [23]. Using these formulae, ΔG‡ of CO site exchange on the μ-depdt derivative is ca. 35 kJ/mol. This value is lower than the μ-pdt and μ-edt derivatives which have ΔG‡ = 43.5 and 50.7 kJ/mol, respectively [2]. A reasonable conclusion is that the addition of steric bulk to the bridgehead carbons has decreased the energy barrier for CO site exchange.
In contrast to the all-CO complexes, at 0 °C the NHC-substituted complex (μ-depdt)[Fe(CO)3][Fe(CO)2IMes] shows two resonances at δ = 215.8 and 212.1, assigned to the COs on the [Fe(CO)2IMes] and the [Fe(CO)3] units, respectively. As shown in the 13C VT NMR spectra of (μ-depdt)[Fe(CO)3][Fe(CO)2IMes] (Fig. 10) resolution of the COs on the [Fe(CO)2IMes] unit is obtained on cooling to −40 °C. This indicates, as expected, a significantly higher energy barrier for site exchange on the bulky IMes substituted side as compared to the all-CO complexes. For the [Fe(CO)3] unit, three separate resonances at 214.7, 213.7, and 208.6 ppm appear upon cooling to −100 °C. Because of the greater range in chemical shift for the resonances at −100 °C a larger rate constant is required to obtain coalescence. Since the temperature range between coalescence and resolution is similar to that of the all-CO complex this likely indicates a lower energy barrier for site exchange on the IMes derivative. Using the difference between the midpoint of the two peaks assigned to the basal COs and the resonance assigned to the apical CO for Δv and a coalescence temperature of ∼−80 °C, a ΔG‡ = 28.4 kJ/mol is obtained. Due to line broadening at low temperatures a full line shape analysis was not possible for these complexes, meaning that the error associated with the calculated values could make the difference between the substituted and unsubstituted complexes insignificant. The lower energy barrier observed here is supported in part by the spectral observations as well as earlier DFT studies which explored the effect of ligand substitution on transition state energies [3]. Nevertheless, these values can only be used as a rough estimate of ΔG‡.
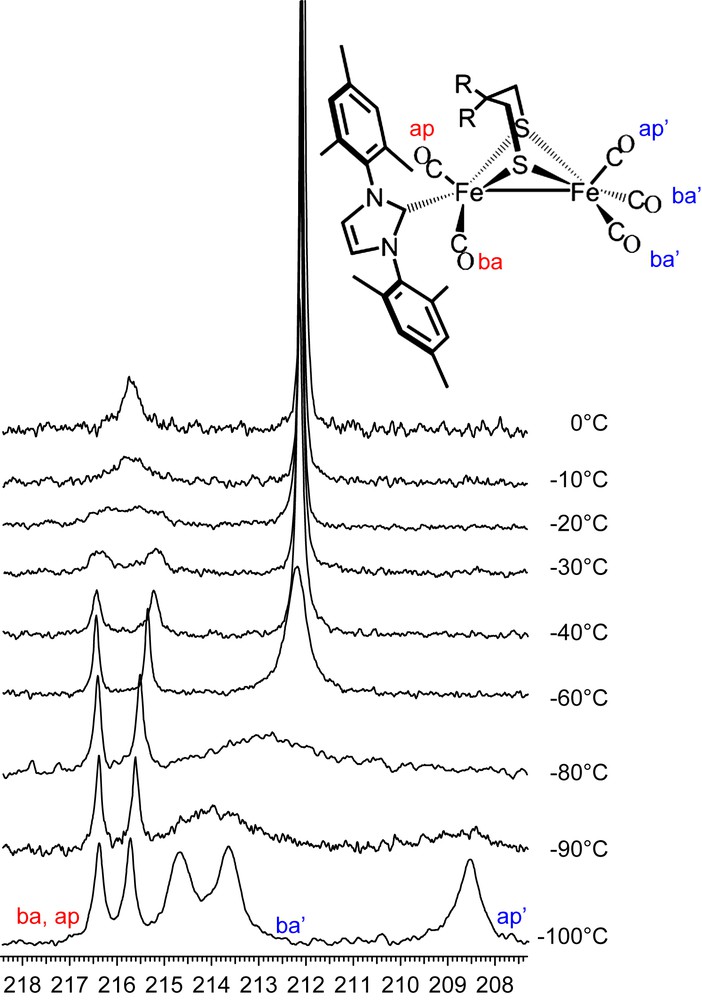
13C VT NMR spectra of (μ-depdt)[Fe(CO)3][Fe(CO)2IMes]. Bands for the IMes substituted side are labeled in red and those of the unsubstituted side are labeled in blue. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
4 Concluding comments
Overall, the addition of steric bulk to the μ-S-to-S linker of the all-CO complexes has not caused large changes in the electronic properties of the Fe(CO)3 units or significantly altered the solid state structure of these compounds relative to the μ-pdt analogue, and at no time was a rotated FeIFeI form observed. However, VT NMR studies of these complexes have shown that ΔG‡ for CO site exchange has been decreased by ca. 8 kJ/mol, indicating that the transition state, hypothesized to resemble the rotated form seen in the eas [4,5], has been stabilized as compared to the μ-pdt complex resulting in more rapid exchange of the CO groups. Interestingly the added alkyl groups on the S-to-S linker also increase the rate at which the FeS2C3 ring flip occurs. Because of the rapid boat/chair interconversion, the interaction of the alkyl groups with the COs may be lessened, however further exploration of the possibility of ring flip cessation would be required to determine the full extent to which this process affects CO site exchange.
Upon addition of the sterically bulky IMes ligand to the μ-dmpdt and μ-depdt complexes more substantial changes in the solid state structure are observed. The most striking difference is the preferential positioning of IMes at a basal site of the substituted S2Fe(CO)2NHC square pyramid. In addition to variation of the coordination site, the Fe–Fe bond distances of these complexes are lengthened by 0.05–0.07 Å giving values closer to that seen for the enzyme active site, typically 2.55–2.62 Å [4,5]. Also the Cap–Fe–Fe–Cap torsion angle has increased, with μ-dmpdt[Fe(CO)3][Fe(CO)2IMes] showing a distortion from eclipsed by ca. 41°. This degree of staggering is not seen for μ-dRpdt[Fe(CO)3][Fe(CO)2L], where R = Me or Et and L = PPh3 or IMe. The greater changes upon addition of the more sterically bulky ligands implies that the steric bulk of the ligand as well as the S-to-S linker are important properties in the design of electrocatalysts for H2 production/uptake based on [FeFe]H2ase.
5 Supplemental information
Data tables for X-ray structures of compounds 1–7 have been included. CIF files have been submitted to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK.
Acknowledgement
We acknowledge financial support from the National Science Foundation (CHE-0616695 to M.Y.D.) and the R.A. Welch Foundation (A-0924). We appreciate the X-ray crystallography facility at TAMU and the help of Dr. Joe Reibenspies. We would also like to thank Dr. Aurore Loudet and Dr. Sébastien Gauthier for the French translations of the abstract and title of this paper.
1 Analytical X-ray Inst. Inc., Madison, WI, U.S.A.