1 Introduction
An understanding of the factors controlling the flow of electrons and protons to and from substrates is essential for developing catalysts for multi-electron and multi-proton processes that are important in biological processes, fuel cells, and fuel production from non-fossil sources. For example, the reductions of CO2 to methanol and N2 to ammonia are both six-electron and six-proton processes. Water oxidation/O2 reduction involves a four-proton, four-electron conversion. The simplest multi-electron multi-proton process to understand is the two-electron, two-proton transfer involved in H2 production/oxidation. This reaction is of fundamental importance in developing a better understanding of the important features of multi-proton, multi-electron reactions.
1.1 Hydrogenase enzymes
Recent progress in the structural characterizations of several hydrogenase enzymes has led to greater efforts to understand the mechanisms of the reactions catalyzed at the active sites [1–8]. Structure 1 indicates some of the salient features of the Fe–Fe hydrogenase active site. Two iron atoms are coordinated by carbonyl and cyanide ligands and the two bridging sulfur ligands are connected through a three-atom chain. It has been suggested that the central atom in the dithiolate ligand is a nitrogen atom of a secondary amine [1]. This amine in the second coordination sphere is proposed to assist in the heterolytic cleavage of dihydrogen bound to the distal iron (Fed) and to relay the proton to a proton conduction channel. It is also thought that the active site of the enzyme lies at the intersection of this proton conduction pathway, a hydrogen channel, and an electron-transfer pathway.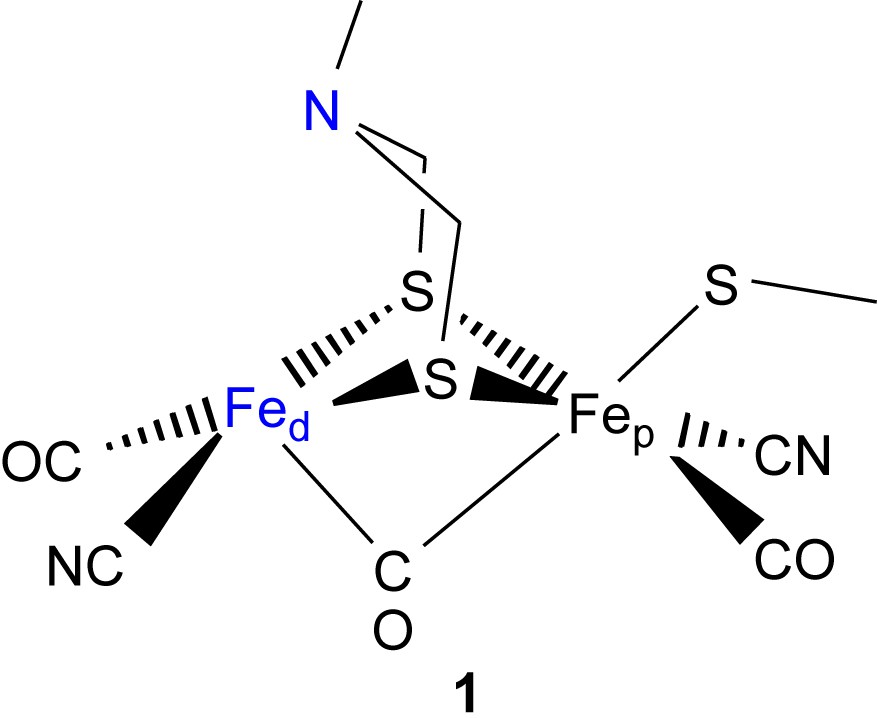
One possible catalytic mechanism for H2 oxidation is indicated in Scheme 1 [9,10]. In this scheme only the distal iron and the pendant amine of the bridging dithiolate ligand are shown. In the first step, H2 binds to iron to form a dihydrogen complex, and many examples of synthetic iron dihydrogen complexes are known. In step 2, the dihydrogen molecule is heterolytically cleaved to form an iron hydride and a protonated pendant amine [Fe(H)(NH)]2+. This cleavage reaction is followed by proton transfer from the pendant nitrogen atom to the proton conduction channel (step 3). Oxidation of the resulting [Fe(H)(N)]+ species to form an [Fe(H)(N)]2+ species in step 4 results in a large increase in the acidity of the hydride ligand. This makes the transfer of the hydride ligand (as a proton) from iron to the pendant nitrogen base favorable as shown in step 5. The resulting [Fe(NH)]2+ complex is deprotonated to form [Fe(N)]+ (step 6) which is oxidized back to the original [Fe(N)]2+ species in a second electron-transfer reaction (step 7). In the process of this cycle, H2 is oxidized, the two electrons are transferred to the electron transport channel, and two protons are transferred to the proton transport channel.
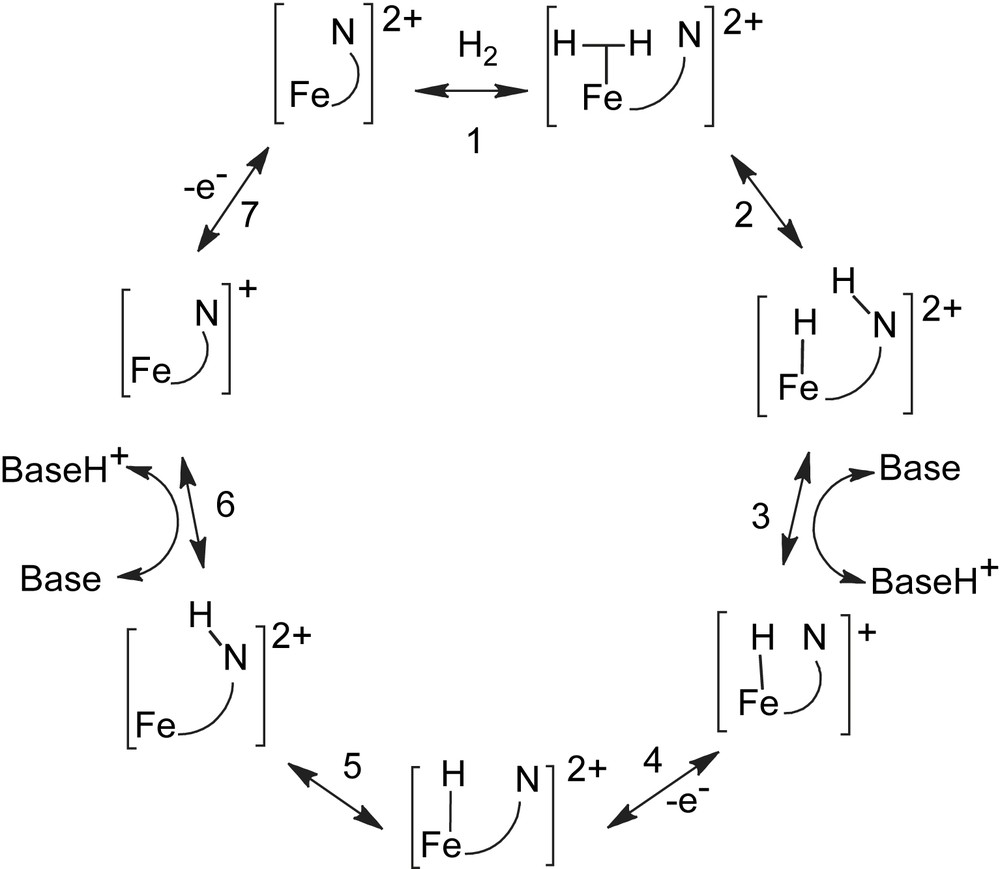
The pendant base plays a number of roles in this proposed catalytic cycle. It assists in the heterolytic cleavage of H2 (step 2), it serves to couple the proton-transfer and electron-transfer reactions (steps 4 and 5), and it is a relay for transferring two protons from the metal to the proton conduction channel (steps 3 and 6). Research carried out in our own [11–18] and other laboratories [19–27] has attempted to demonstrate that bases positioned in the second coordination sphere of synthetic complexes can play roles similar to those proposed in Scheme 1 for the Fe–Fe hydrogenase enzyme. This paper describes our work to date on simple mononuclear metal complexes of iron and nickel containing diphosphine ligands with pendant nitrogen bases. Our objective is to develop a better understanding of those factors that control the ability of bases in the outer coordination sphere to perform the functions required for efficient catalysis of dihydrogen oxidation and production and other multi-electron, multi-proton processes.
1.2 Synthetic systems: selection of ligands and metals to be studied
The presence of iron and nickel in the active sites of the different hydrogenase enzymes demonstrates that very active catalysts for hydrogen oxidation and production can be based on these relatively abundant and inexpensive metals [1–8]. There are a large number of dihydrogen and hydride complexes of iron that contain two diphosphine ligands [28–38] and work in our laboratories has shown that nickel diphosphine complexes can heterolytically cleave H2 in the presence of an appropriate base [34]. For these reasons, we were interested in studying the role that a pendant base incorporated into diphosphine ligands might play in the heterolytic cleavage of coordinated dihydrogen for this class of iron and nickel complexes.
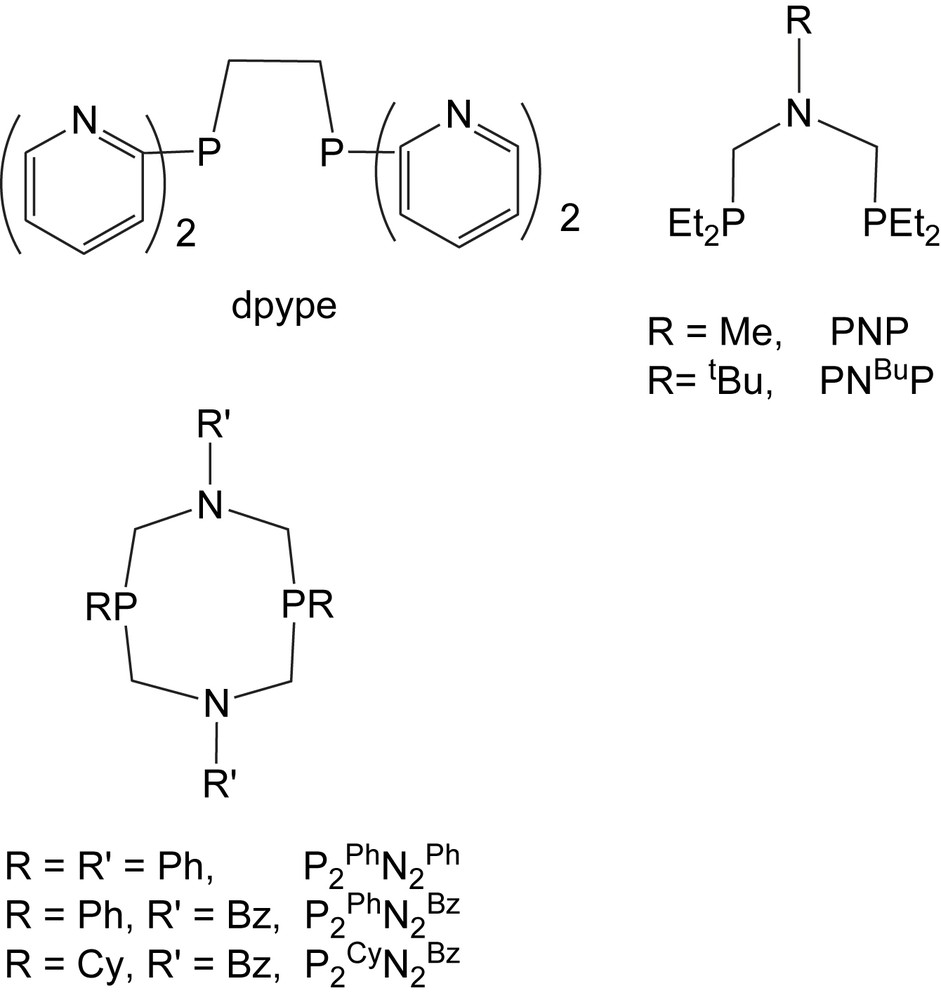
Chart 1 shows the diphosphine ligands that we have studied. These include ligands with pendant terminal pyridyl groups (dpype), acyclic diphosphine ligands with a tertiary amine incorporated into the ligand backbone (PNP and PNBuP), and cyclic diphosphine ligands with two pendant tertiary amines (PR2NR′2). Our initial studies of nickel with dpype indicated that although Ni(0) complexes with this ligand would bind via phosphorus as desired, Ni(II) preferred to bind to the harder nitrogen donors [11]. Incorporation of the pendant base into the backbone and the use of tertiary amines instead of pyridyl bases reduced the tendency for the nitrogen base to compete with phosphorus atoms for binding to the metal. It was found that both the acyclic PNP ligand and cyclic PR2NR′2 ligands bind to nickel as bidentate phosphine donors with non-coordinated nitrogen bases in the ligand backbones. For this reason, we have focused our studies on these two classes of ligand. The PNP ligands are more flexible than the cyclic diphosphines, but the latter allow for the positioning of the pendant base in close proximity to the metal, as will be discussed in much more detail below.
2 Studies of iron complexes
2.1 Syntheses
Syntheses of iron complexes containing two PNP ligands resulted in the formation of cis octahedral complexes of the formula cis-[Fe(PNP)2X2]n+ [17]. However, we were initially interested in obtaining trans complexes, because experimental and theoretical studies have shown that the acidity of the coordinated dihydrogen ligand in complexes of the type trans-[(H2)M(diphosphine)2(X)]n+ (where M = Fe, Ru, and Os) is strongly dependent on the nature of trans ligand X [39–41]. Strong π-acceptors, such as CO (n = 2), trans to the dihydrogen ligand result in very acidic complexes, whereas anionic π-donors (n = 1) result in weakly acidic dihydrogen complexes. These results suggested that it should be possible to tune the acidity of dihydrogen complexes containing pendant bases to favor either intramolecular heterolytic cleavage of hydrogen or the retention of the dihydrogen ligand.
An effective strategy to obtain the desired trans complexes was to use one PNP ligand and one diphosphine with a smaller chelate bite such as dmpm (where dmpm is bis(dimethylphosphino)methane) to prepare complexes such as trans-HFe(PNP)(dmpm)Cl. For example, exchange of the chloride ligand for acetonitrile followed by protonation with an appropriate acid leads to the formation of endo and exo isomers of trans-[HFe(PNHP)(dmpm)(CH3CN)]2+, 2 [17,18]. Similar methods were used to prepare trans-[HFe(PNHP)(dmpm)(CO)]2+, 3, and trans-[HFe(PNP)(dmpm)(H2)]+, 4. In 4, an anionic hydride is positioned trans to the dihydrogen ligand, and as a result the dihydrogen ligand is not sufficiently acidic for intramolecular heterolytic cleavage of H2 to occur. In contrast, complexes 2 and 3 have protonated pendant amines, while the terminal hydride ligands remain unprotonated. Clearly the trans ligand can be used to tune the acidity of the dihydrogen ligand to promote intramolecular heterolytic cleavage of H2.
2.2 Intramolecular NH/FeH exchange
Variable temperature 1H and 31P NMR studies of complexes 2 and 3 demonstrated the existence of both endo and exo isomers [18]. Spin transfer experiments were performed on complex 2 in which the spin of the hydride ligand for each of the endo and exo isomers was selectively inverted and the transfer of this polarization to the NH proton of each isomer was measured. At −60 °C, an exchange rate of 7 s−1 was calculated for the exchange between the endo proton and the corresponding endo hydride. This corresponds to an exchange rate at 25 °C of approximately 2 × 104 s−1. No exchange was observed for the exo isomer. These results are consistent with an exchange process (Scheme 2) in which the PNP ligand adopts a boat conformation to bring the NH proton into close proximity to the hydride ligand. This is followed by proton transfer to form a dihydrogen complex, fast rotation of the dihydrogen ligand, and cleavage of the H–H bond to reform the endo complex in which the original hydride ligand and NH proton have been exchanged. The failure of the NH and hydride protons of the exo isomer to exchange is consistent with the NH proton being unable to approach the hydride ligand in an intramolecular process and much slower or no intermolecular NH/FeH exchange between complexes.
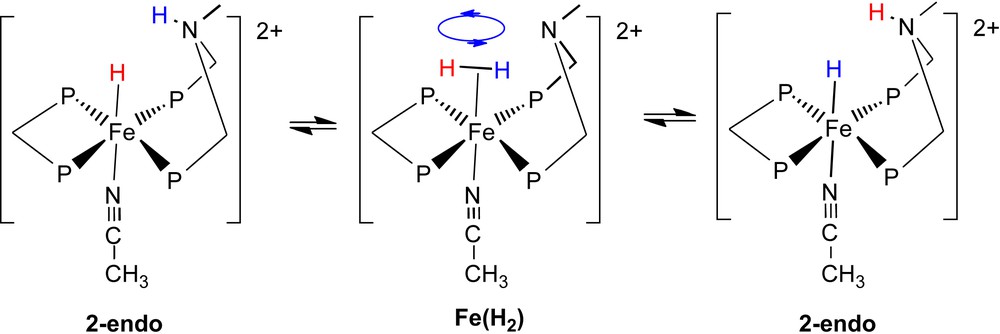
It was also observed that there was no intramolecular exchange of the hydride ligand and the protonated amine for the endo isomer of 3 in which the hydride is trans to CO. In this case, intramolecular transfer of the NH proton to the hydride ligand to form a dihydrogen complex is energetically unfavorable because of the high acidity of the resulting dihydrogen complex. This explanation is supported by the observation that protonation of 2 with strong acids such as triflic or tetrafluoroboric acid results in the formation of a dihydrogen complex. In contrast, 3 is not protonated under the same conditions to form a dihydrogen complex. This illustrates the importance of achieving a close matching of the acidities of the dihydrogen complex and the hydride ammonium complex, e.g., Fe(H2) and 2-endo in Scheme 2.
Although very rapid intramolecular NH/FeH exchange has been characterized for trans-[HFe(PNHP)(dmpm)(CH3CN)]2+, 2-endo [18], a similar rapid exchange was not observed for the complex with two PNHP+ ligands, cis-[HFe(PNHP)2(CH3CN)]3+, 5 [15]. The relatively slow rate of intramolecular N–H⋯H–Fe exchange for 5 relative to 2 results from unfavorable steric interactions in the cis derivative. In order for either of the cis or trans complexes to undergo the intramolecular exchange reaction, one PNP ligand must convert from the more stable chair form to the boat conformation. Molecular mechanics calculations have provided insight into this process for both the trans and cis derivatives. The relative energy barrier to form the boat conformer from the more stable chair conformer in a single PNP ligand is 26 kcal/mol for the cis complex, 5, versus only 9.5 kcal/mol for 2, the related trans derivative. In 2 there is little steric hindrance to this process. However, in 5 both molecular models and the calculations show that steric interactions between ethyl substituents on the cis PNP ligands make the interconversion between chair and boat conformers difficult. This result supports our experimental observations that rapid intramolecular exchange in octahedral complexes containing two cis PNP ligands does not occur and suggests that developing H2 oxidation catalysts based on such geometries with these ligands is not likely to be fruitful.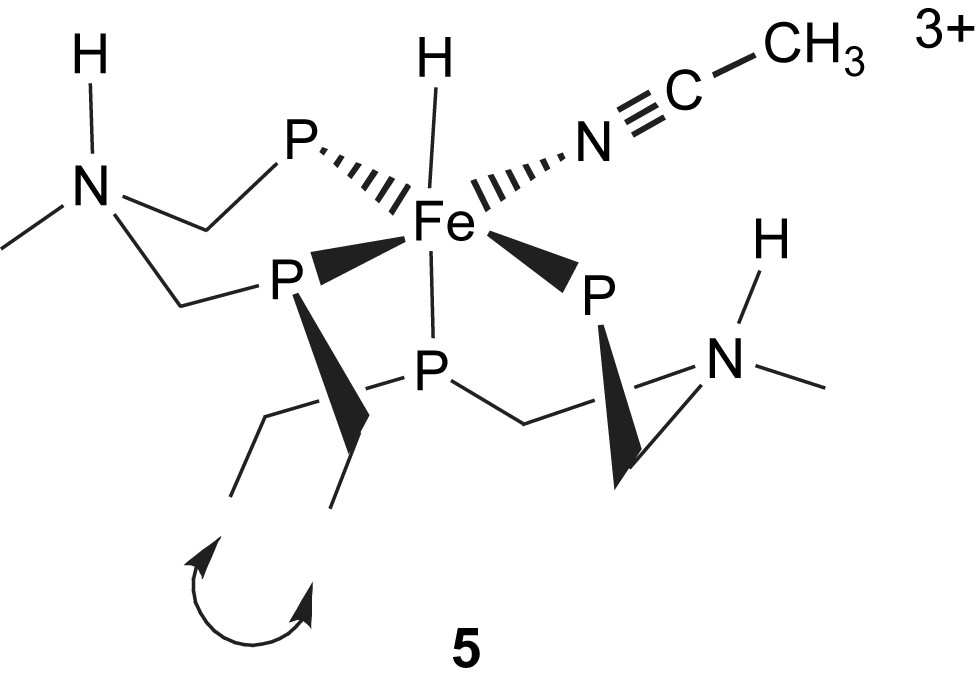
The iron hydride complex containing two cyclic ligands, trans-[HFe(CH3CN)(PPh2NPh2)2]+, 6, has a structure similar to that of 2, but in this case two of the chelate rings of the ligands are in boat conformations, placing two pendant amines at a distance of 2.6 Å from the hydride ligand [16]. Very facile Fe–H/N–H exchange was expected upon protonation of the complex. However, rapid exchange was not observed by NMR spectroscopy. In this system addition of acid promoted a rearrangement of chelate ring conformations to form product 7 shown in Eq. (1). In this doubly protonated product in which each proton interacts with two amine bases (confirmed by 15N labeling studies at −60 °C), the protons are positioned away from the hydride ligand in a manner very similar to the exo isomer of 2. Upon warming, coupling between the hydride ligand and phosphorus is lost, consistent with exchange rates of ≈10 s−1 at room temperature. Structure 7 helps to explain why much slower Fe–H and N–H exchange and dihydrogen elimination are observed for this system compared to 2. Although flexibility of the pendant base is a desirable feature for an effective proton relay, the stability of the product in Eq. (1) illustrates a potential limitation of this particular cyclic ligand design for catalyst development.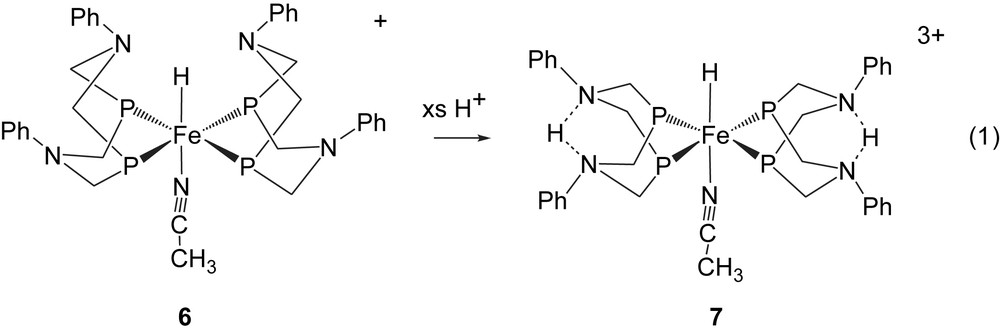
These studies on the intramolecular NH/FeH exchange clearly indicate that fast intramolecular heterolytic cleavage processes will require the careful matching of the pKa values of the metal dihydrogen complex and the protonated pendant base. In addition to these energy requirements arising from electronic effects, attention must be paid to the structures of the complexes so that the ammonium proton can approach the hydride ligand without encountering large energy barriers arising from unfavorable steric interactions.
2.3 Intermolecular exchange
Intermolecular exchange of protons in solution with metal complexes such as trans-[HFe(PNHP)(dmpm)(CH3CN)]2+, 2, can involve the hydride ligand, the protonated pendant base, or both [18]. Our studies of this system showed that intermolecular exchange of the NH protons of the pendant base with protons in solution led to the interconversion of the endo and exo isomers of 2 but did not involve exchange of the hydride ligand. This was established by low temperature NMR studies (−40 °C) of trans-[HFe(PNP)(dmpm)(CH3CN)]+, 8, in the presence of an anisidine/anisidinium mixture. Coupling of the hydride ligand to the four phosphorus nuclei was maintained while an average chemical shift was observed for the exchanging endo and exo isomers of 2. This result is consistent with the rate of intermolecular exchange between the protonated pendant base and the amine in solution exceeding the rate of intramolecular exchange of 2-endo, which, as described above, proceeds at a rate of approximately 2 × 104 s−1 at 25 °C. The rapid exchange of protons between protonated and unprotonated amines in solution is known to be fast for organic compounds and the experiments described above indicate that this is also true for these iron complexes with pendant amines.
Intermolecular exchange of protons in solution with the hydride ligand can also occur and a convenient method for probing this process is to study the incorporation of deuterium into the hydride position when the complex is exposed to D2O in solution. For example, treatment of trans-[HFe(depp)(dmpm)(CH3CN)]+, 9, (where depp is bis(diethylphosphino)propane) with D2O resulted in the conversion of Fe–H to Fe–D with a half life of 1.7 h. Under the same conditions, trans-[HFe(PNP)(dmpm)(CH3CN)]+, 8, undergoes complete exchange in less than 1 min, Scheme 3. These results clearly demonstrate that replacement of a methylene in the backbone of the depp ligand with NMe, i.e. the incorporation of a pendant base, results in a dramatic increase in the rate of intermolecular Fe–H/D2O exchange compared to the analogous complex without the pendant base.
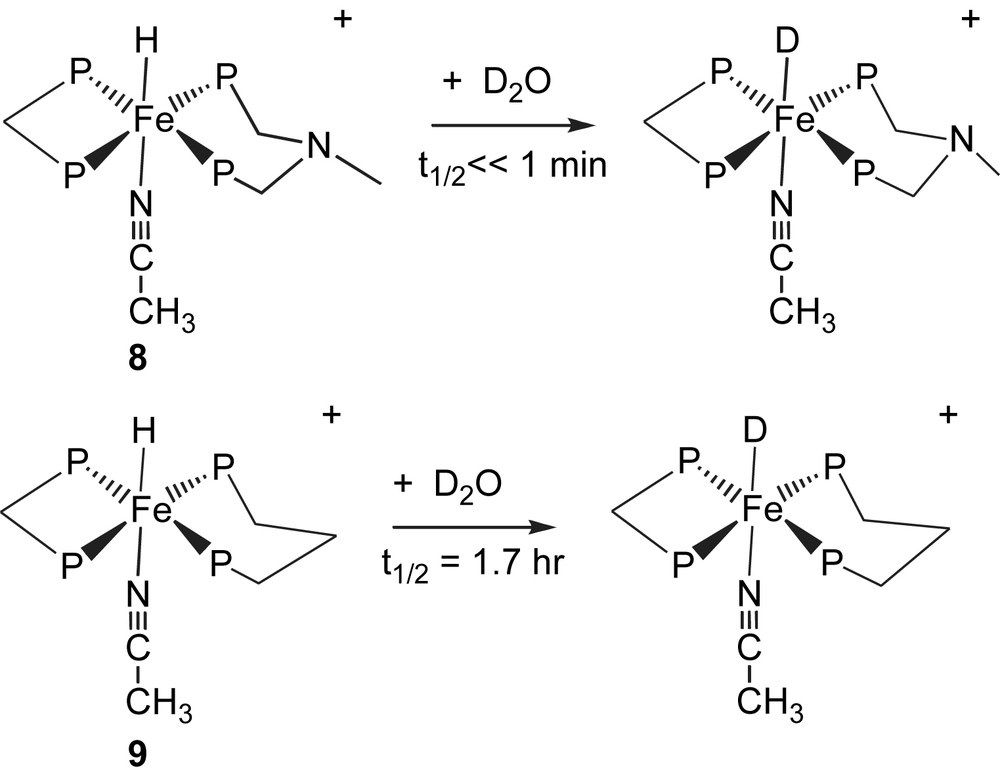
As with intramolecular NH/FeH exchange in 2, intermolecular D2O/FeH exchange is strongly affected by the electronic properties exerted by other ligands in the primary coordination sphere [18]. For example, while 8 undergoes complete exchange in less than 1 min, no exchange is observed for the analogous complex trans-[HFe(PNP)(dmpm)(CO)]+ after 24 h under the same conditions. For cis-[HFe(PNP)2(L)]+ complexes, the rate of intermolecular exchange in the presence of 125 equivalents of D2O is sensitive to the nature of L, with the rate of exchange decreasing as the electron donor ability of L decreases (t1/2 = 2 min for L = CH3CN, 30 min for L = P(OEt)3, and >24 h for L = CO) [15]. The complex cis-[HFe(PNP)2(CH3CN)]+ undergoes D2O/FeH exchange more slowly than the related complex trans-[HFe(PNP)(dmpm)(CH3CN)]+. This may be due to either electronic or steric effects and detailed mechanistic pathways for these D2O/FeH exchange processes are not known. However, it is clear that pendant bases can markedly accelerate deuterium incorporation into these complexes.
2.4 Proton coupled electron transfer
The results described above clearly indicate that a base in the second coordination sphere can play a significant role in intra- and intermolecular proton-transfer events. Another question that we wanted to explore was whether the pendant base could couple proton- and electron-transfer events as proposed for steps 4 and 5 of Scheme 1. To explore this possibility, we examined the electrochemical oxidation of trans-[HFe(PNP)(dmpm)(CH3CN)]+, 8, and trans-[HFe(depp)(dmpm)(CH3CN)]+, 9 [18]. The cyclic voltammograms of these two complexes are shown in Fig. 1. For 9 the Fe(II/III) couple is reversible over a broad range of scan rates (20–2000 mV/s) and a simple uncomplicated cyclic voltammogram is observed. In contrast, for 8 the Fe(II/III) couple is not reversible even at scan rates up to 75 V/s. This is due to a chemical reaction that rapidly follows electron transfer. The reaction occurs as a result of the pendant base and is thought to involve the rapid transfer of a proton from the acidic Fe(III) hydride to the amine in the second coordination sphere. The new oxidation wave that is observed in the CV of 8 near 0.0 V is assigned to the Fe(I/II) couple of the Fe(I) complex produced by this proton-transfer reaction. Scan rate dependent cyclic voltammetry measurements and chronoamperometric studies were used to determine a rate of 1.1 × 102 s−1 for this proton-transfer process. These results demonstrate that a pendant base can play an important role in coupling electron-transfer and proton-transfer events.
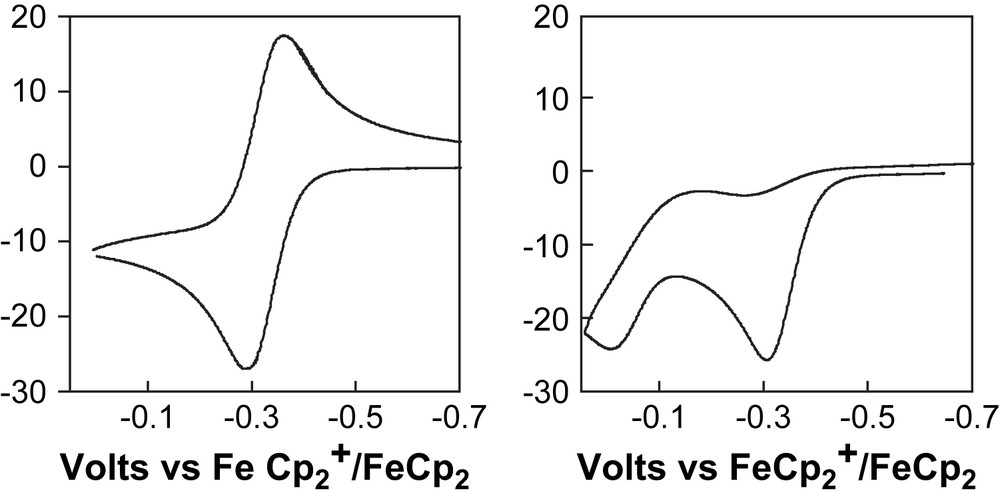
Cyclic voltammograms for [HFe(depp)(dmpm)(CH3CN)]+, 9, (left) and for [HFe(PNP)(dmpm)(CH3CN)]+, 8, (right) recorded in 0.3 M Bu4NBF4/acetonitrile at a glassy carbon electrode at a scan rate of 0.2 V/s.
2.5 Summary of results for iron complexes
The results for these iron systems confirm that pendant bases can play the roles shown in Scheme 1. The reversible intramolecular NH/FeH exchange observed for 2 involves the reversible heterolytic cleavage of H2 as shown in step 2 of Scheme 1. The ability to perform this key step depends on both an energy matching of the pKa values of the dihydrogen complex and the protonated pendant base and the ability of the diphosphine ligand to achieve the boat conformation necessary for a proton to transfer from the dihydrogen ligand to the base in the outer coordination sphere (Scheme 2). These bases also facilitate intermolecular exchange between the protons in solution, protons on the pendant base, and the hydride ligand. In this function the base in the second coordination sphere serves as a proton relay from the solution to the metal center without rearrangement or disruption of the primary coordination sphere. Finally, the pendant base provides the structural component needed for allowing the coupling of proton- and electron-transfer processes. The iron complexes studied here provided an ideal synthetic system for characterizing proton relay properties, but the rates of dihydrogen binding and release in these complexes appear to be too slow for the development of functional hydrogen oxidation/production electrocatalysts. Further work on the development of electrocatalytic systems was undertaken using nickel diphosphine complexes with pendant bases incorporated into the ligand chelate backbones.
3 Studies of nickel complexes
3.1 Complexes containing the PNP ligand
Nickel is the second of the two transition metals found in hydrogenase enzymes. Although there is debate about whether it is iron or nickel that interacts with H2 in NiFe hydrogenases, previous results in our laboratories clearly demonstrate that [Ni(diphosphine)2]2+ complexes are capable of heterolytically cleaving H2 in the presence of bases [34]. In addition, extensive thermodynamic studies have elucidated those factors controlling the ability of this class of compounds to act as hydride acceptors during the heterolytic cleavage of H2 [42–49]. As a result, we were interested in combining this thermodynamic knowledge with our understanding of how pendant bases facilitate many important steps in the catalytic oxidation and production of H2 to design nickel-based catalysts for this important reaction.
Our initial efforts to develop Ni-based electrocatalysts for hydrogen oxidation and production utilized the PNP ligand discussed above for iron complexes. In this context, we prepared [Ni(PNP)2]2+, 10, and [Ni(PNP)(dmpm)]2+, 11, for evaluation as catalysts [11].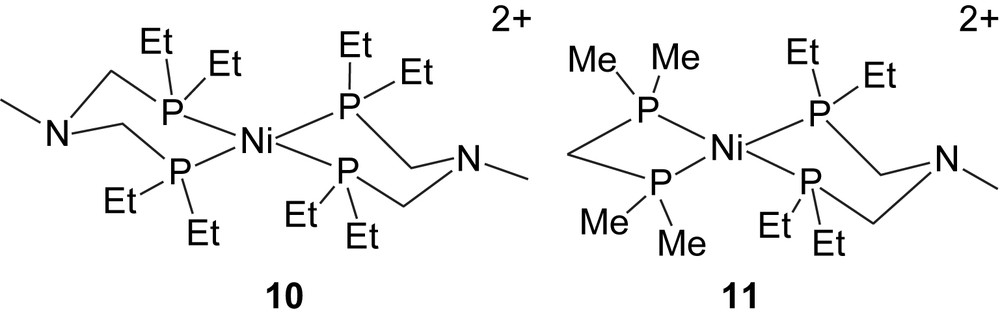
These complexes are analogous in many respects to the iron complexes described above. However, because of our knowledge of the factors controlling the hydride acceptor abilities of [Ni(diphosphine)2]2+ complexes, it was anticipated that [Ni(PNP)2]2+ (which has a large tetrahedral distortion from a square-planar geometry) would be able to heterolytically cleave H2 to form [HNi(PNP)(PNHP)]2+ in acetonitrile (reaction (2)). In contrast, [Ni(PNP)(dmpm)]2+ (which is almost perfectly planar) would be biased toward H2 production; that is, heterolytic cleavage of H2 to form [HNi(PNHP)(dmpm)]2+ should be unfavorable. These expectations were confirmed by thermodynamic studies which demonstrated the free energy associated with reaction (2) is −6 kcal/mol for [Ni(PNP)2]2+ and +6 kcal/mol for [Ni(PNP)(dmpm)]2+.
| [NiLL′]2+ + H2 → [HNi(LH)L′]2+ | (2) |
| ΔGo | ||
| 10 | L = L′ = PNP | −6.0 kcal/mol |
| 11 | L = PNP, L′ = dmpm | +6 kcal/mol (estimate) |
3.1.1 Intramolecular and intermolecular exchange processes
Variable temperature NMR experiments on [HNi(PNP)(PNHP)]2+ (the product of reaction (2)) demonstrated that intramolecular NH/NiH exchange is fast with a rate of approximately 104 s−1 at 25 °C [11]. NOESY NMR data showed that intermolecular exchange between D2O and the hydride ligand of [HNi(PNP)2]+ (>10 s−1) is also fast. In contrast, when D2O was added to [HNi(depp)2]+ under the same conditions, less than 10% deuterium incorporation was observed after 48 h. These results again demonstrate the importance of the pendant base in promoting heterolytic cleavage of H2 and in facilitating intermolecular exchange between the hydride ligand and protons in solution. That the presence of two PNP ligands in [HNi(PNP)(PNHP)]2+ does not retard intra- and intermolecular proton exchange as observed for cis-[HFe(PNHP)2(CH3CN)]3+ is attributed to decreased steric interactions in the five coordinate nickel species compared to the six-coordinate iron complex.
3.1.2 Proton coupled electron transfer and catalysis
To probe the role of the pendant base in coupling proton- and electron-transfer processes, the cyclic voltammograms of acetonitrile solutions of [HNi(depp)2]+ and [HNi(PNP)2]+ were compared in the presence of triethylamine as a proton acceptor [11]. For [HNi(depp)2]+, which contains no base in the second coordination sphere, an irreversible two-electron oxidation wave was observed at +0.1 V versus the ferrocenium/ferrocene couple. In contrast, this two-electron oxidation occurs at −0.6 V for [HNi(PNP)2]+. The large shift in potential (−0.7 V) for the oxidation wave of [HNi(PNP)2]+ indicates an extremely fast proton transfer from nickel to the amine of the PNP ligand upon oxidation of the metal from Ni(II)–H to Ni(III)–H.
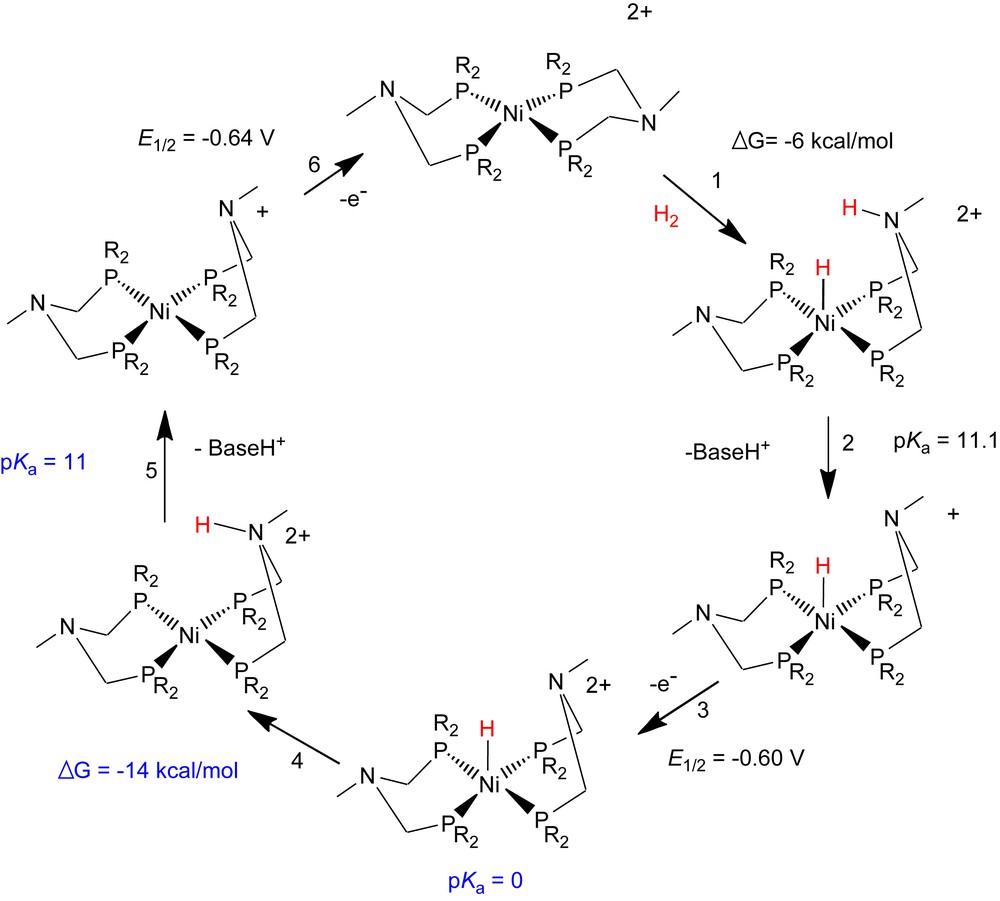
[Ni(PNP)2]2+ is an electrocatalyst for H2 oxidation, as expected for a complex that can heterolytically split H2, undergo intra- and intermolecular proton transfer, and coupled proton- and electron-transfer processes. Detailed spectroscopic and electrochemical studies of this system support the mechanism shown in Scheme 4, where the thermodynamic values shown in black represent measured values and those in blue (For interpretation of the references to color in text, the reader is referred to the web version of this article.) are estimated [11].1 As a result of the rapid proton transfer discussed in the preceding paragraph and shown in step 4 of Scheme 4, [Ni(PNP)2]2+ catalyzes the oxidation of H2 to protons at very low overpotentials (less than 100 mV). However, the overall catalytic rate for H2 oxidation is low (<0.5 turnovers/s) because the rate of H2 addition to [Ni(PNP)2]2+ to form [HNi(PNP)(PNHP)]2+ (step 1) is rate-determining. Since we have shown that the intramolecular exchange of the NH proton and the NiH ligand, is fast (≈104 s−1) for [HNi(PNP)(PNHP)]2+, the rate-determining reaction in this system must involve the initial coordination of H2 to [Ni(PNP)2]2+ to form either a dihydrogen or a dihydride complex, H2–[Ni(PNP)2]2+ (not shown in Scheme 4).
3.2 Studies of nickel complexes containing cyclic PR2NR′2 ligands
An examination of the structure of the active site of the Fe–Fe hydrogenase enzyme indicates that the six-membered ring formed by the distal Fe and the bridging dithiolate ligand adopts a boat conformation to minimize steric interactions with ligands of the second metal ion. This conformation positions the pendant base in close proximity to the distal Fe atom and may facilitate the binding of H2 to this complex in addition to promoting the proton-transfer functions described above. If this is true, it suggests that positioning pendant bases in close proximity to nickel in the synthetic complexes described in the preceding paragraphs might stabilize the formation of dihydrogen or dihydride complexes resulting from reaction with H2. This could lead to faster catalysts for H2 oxidation and production. Nickel complexes of the type [Ni(PR2NR′2)2]2+ were prepared to test this hypothesis [12]. The cyclic diphosphine ligands form two six-membered chelate rings upon coordination to nickel and steric factors favor the formation of at least one chelate in each ligand in the boat conformation. Structural studies have established that each of the [Ni(PR2NR′2)2]2+ complexes displays at least two non-bonding Ni⋯N distances of 3.2–3.4 Å.
The substituents on the cyclic PR2NR′2 ligands were chosen such that they would provide different thermodynamic driving forces for the addition of H2 to these complexes, in a manner similar to our efforts to control the thermodynamic driving forces for H2 addition to [Ni(PNP)2]2+ and [Ni(PNP)(dmpm)]2+. Recent thermodynamic studies in our laboratories have confirmed this to be the case as indicated by the free energies shown below in reaction (3) [14]. Because the addition of H2 to [Ni(PCy2NBz2)]2+, 14, is favorable, this reaction provided the opportunity to obtain detailed information about the nature of the complex formed upon H2 addition.
| [Ni(PR2NR′2)2]2+ + H2 → [Ni(PR2NR′2H)2]2+ | (3) |
| ΔGo | ||
| 12 | PR2NR′2 = PPh2NPh2 | 9 kcal/mol (estimate) |
| 13 | PR2NR′2 = PPh2NBz2 | +2.7 kcal/mol |
| 14 | PR2NR′2 = PCy2NBz2 | −3.1 kcal/mol |
3.2.1 Hydrogen activation
As indicated above, the heterolytic cleavage of H2 by [Ni(PNP)2]2+ formed a nickel hydride and a protonated amine in the ligand, step 1 of Scheme 4. In contrast, in the reaction of hydrogen with [Ni(PCy2NBz2)]2+, 14, the first complex observed at low temperature is the hydrogen oxidation product, 16A, in Scheme 5 [13]. The nature of the product has been established by extensive variable temperature 1H, 2D, 31P, and 15N NMR studies of isotopically labeled species. The product is a tetrahedral Ni(0) derivative in which one amine in each ligand has been protonated. The product suggests an alternate heterolytic cleavage mechanism in which two amines serve as proton acceptors while the metal ion serves as an acceptor for two electrons. Spectroscopic data support a structure in which the protonated chelate rings are in boat conformations with the protons endo with respect to nickel. At room temperature this complex is in equilibrium with two other isomers (structures 16B and 16C) with related tetrahedral structures. The average pKa value for the three isomers of 16 has been measured by NMR spectroscopy and a value of 13.4 in acetonitrile was determined. This pKa value is somewhat larger than that expected based solely on the electron donor properties of the amine substituents. Positioning the NH group in close proximity to Ni(0) (as in 16A) or to a second base (as in 16C) appears to increase the energy required to deprotonate 16A–16C by approximately 5 kcal/mol.
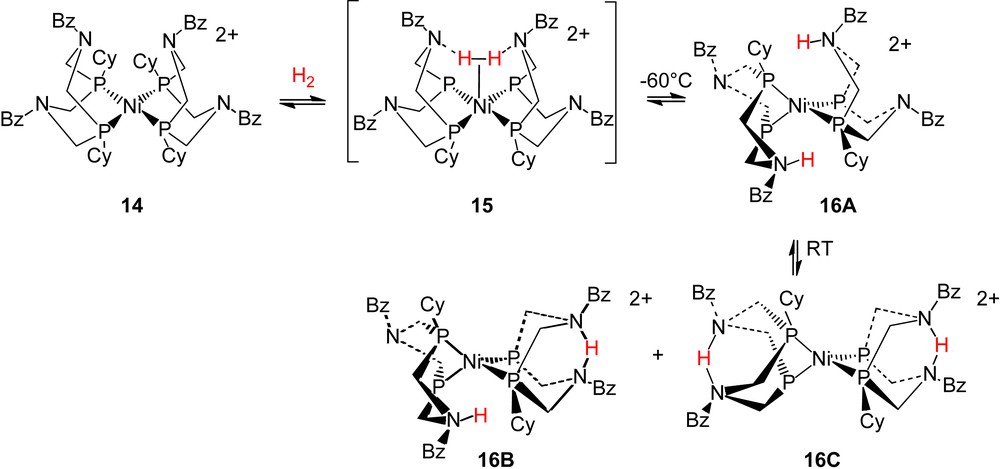
Our spectroscopic studies of the reaction of H2 with [Ni(PCy2NBz2)2]2+, 14, provided no direct evidence for a dihydrogen intermediate. However, DFT calculations indicate such an intermediate in which two nitrogen atoms interact with the dihydrogen ligand as shown in structure 15 [12]. The structure of the proposed intermediate is certainly consistent with the geometry of the first observed product (16A). The positioning of the two amines in close proximity to dihydrogen appears to stabilize the binding of this ligand to Ni(II). Stabilization of other ligands at nickel by weak interactions with two of the pendant bases in 14 has been suggested by structural studies described below.
3.2.2 Electrocatalytic oxidation of H2
Complex 14 is an electrocatalyst for the oxidation of H2 in acetonitrile solutions with a turnover frequency of 10 s−1 in the presence of excess triethylamine and 1 atm of H2. This rate is significantly higher than that observed for [Ni(PNP)2]2+ (<0.5 s−1) which has a larger driving force for H2 addition (6 kcal/mol). Thus kinetic studies of H2 oxidation, theoretical studies, and structural studies all support the view that positioning two pendant amines in close proximity to nickel is important for stabilizing dihydrogen intermediates and for high catalytic rates. The stabilization of dihydrogen binding is yet another role that pendant bases can play.
The combination of kinetic studies and the spectroscopic and theoretical studies described in the preceding paragraphs are the basis for the proposed mechanism for electrocatalytic H2 oxidation shown in Scheme 6. This scheme differs significantly from Scheme 4 with respect to the mechanism of H2 binding and cleavage (steps 1 and 2), as described above, and also in the mechanism of the metal hydride formation (step 3). Subsequent to H2 oxidation, the isomers 16A–16C undergo a reaction with the external base triethylamine in which one of the cyclic ligands is deprotonated. This results in increased electron density on Ni(0) and promotes proton transfer from the second protonated ligand to the metal ion, resulting in the formation of [HNi(PCy2NBz2)2]+ (step 3 of Scheme 6). This nickel(II) hydride complex has been characterized by 1H and 31P NMR spectroscopy. The rearrangement is reversible and addition of acid to the nickel hydride complex rapidly reforms the nickel(0) isomers of 16 [13,14]. This is an interesting and novel equilibrium in which the protonation/deprotonation of a base in the second coordination sphere results in a two-electron change in oxidation state at the metal center. Subsequent steps in the electrocatalytic cycle are similar to those suggested previously in Scheme 4 for [Ni(PNP)2]2+.
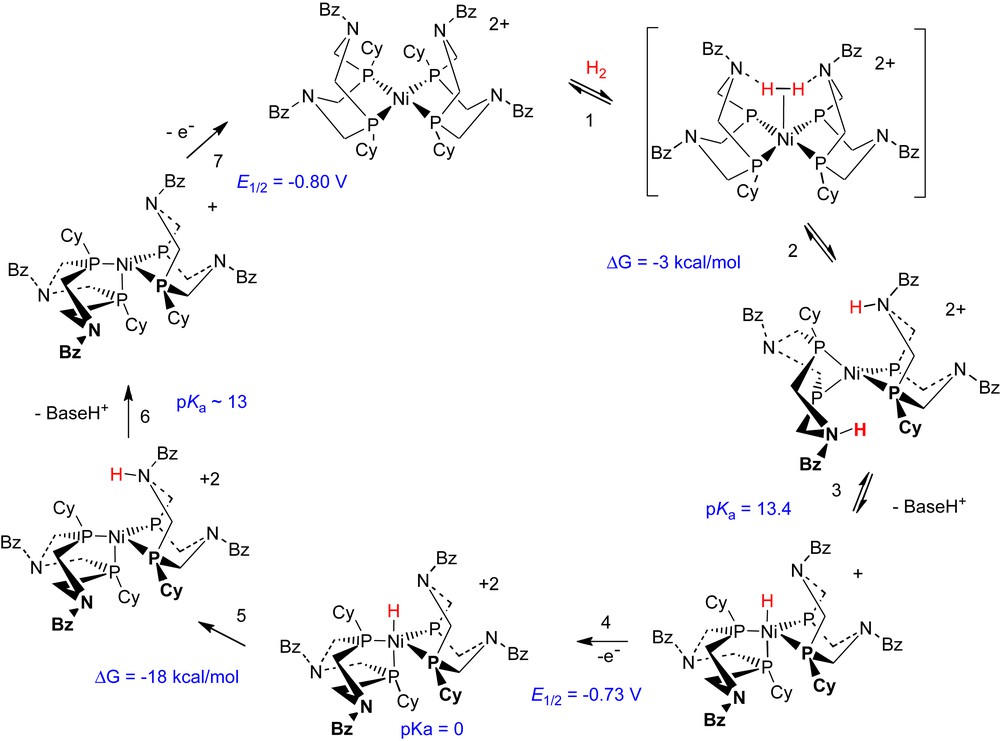
3.2.3 Studies of electrocatalytic hydrogen production
Addition of H2 to [Ni(PPh2NPh2)]2+, 12, and [Ni(PPh2NBz2)]2+, 13, is unfavorable as shown in reaction (3). As a result, these species are biased toward H2 production, and the electrocatalytic reduction of protons to H2 by these complexes has been studied. For 12 the catalytic rate shows a first order dependence on catalyst and a second order dependence on acid concentration at low acid concentrations and becomes independent of acid at high concentrations. In the high [acid] regime, a first order rate constant for H2 production of 350 s−1 at 22 °C has been observed with an overpotential of approximately 0.35 V [13]. This catalytic rate is similar to that reported for NiFe hydrogenase (700 s−1 at 30 °C) [50], although the overpotential is larger. In contrast when 13 is the electrocatalyst, the rate of hydrogen formation decreases by about two orders of magnitude, consistent with the smaller thermodynamic driving force associated with release of hydrogen from this complex. These results clearly demonstrate that electronic and steric properties of substituents on both phosphorus and nitrogen can be used to finely tune these catalysts. The balance of the hydride donor/acceptor properties of the complex and the proton donor/acceptor properties of the pendant base can be biased to favor either catalytic production or oxidation of H2.
3.2.4 Studies of potential CO inhibition
For practical catalysts for H2 oxidation in fuel cells, interactions with trace amounts of carbon monoxide can lead to rapid deactivation of the catalyst. The hydrogenase enzymes are also inhibited by CO. Carbon monoxide addition (1 atm) to a series of [Ni(diphosphine)2]2+ complexes at 25 °C has been surveyed by NMR spectroscopy. The complexes included diphosphine ligands with four, five and six-membered chelate rings, and electron-donating (Me, Et) and electron-withdrawing (Ph) groups. Complexes 12–14 with cyclic ligands and some derivatives with mixed ligand combinations were also included. Formation of a Ni(II) carbonyl complex was observed for only one of these complexes, the hydrogen oxidation catalyst [Ni(PCy2NBz2)]2+, 14. The CO adduct has been isolated and structurally characterized [51]. The structure of [Ni(CO)(PCy2NBz2)]2+, 17, is similar to that proposed above for the dihydrogen adduct. All four chelate rings of the cyclic ligands are arranged in boat conformations, bringing two of the pendant bases within 3.4 Å of the carbonyl carbon. The unique reactivity of this complex with carbon monoxide is attributed in part to a stabilizing ion–dipole interaction between the positively charged carbon and the pendant amines. The CO adduct provides an unusual example of how the second coordination sphere can exert a subtle influence on ligand coordination at a metal center even when no hydrogen-bonding interaction is involved. Thermodynamic studies of the reversible CO binding to [Ni(PCy2NBz2)]2+ compared to H2 binding to this complex indicate that H2 binding is favored by 1.8 kcal/mol. As a result, CO concentrations as high as 5% have no measurable inhibiting effect on the catalytic response of this nickel complex for hydrogen oxidation [51].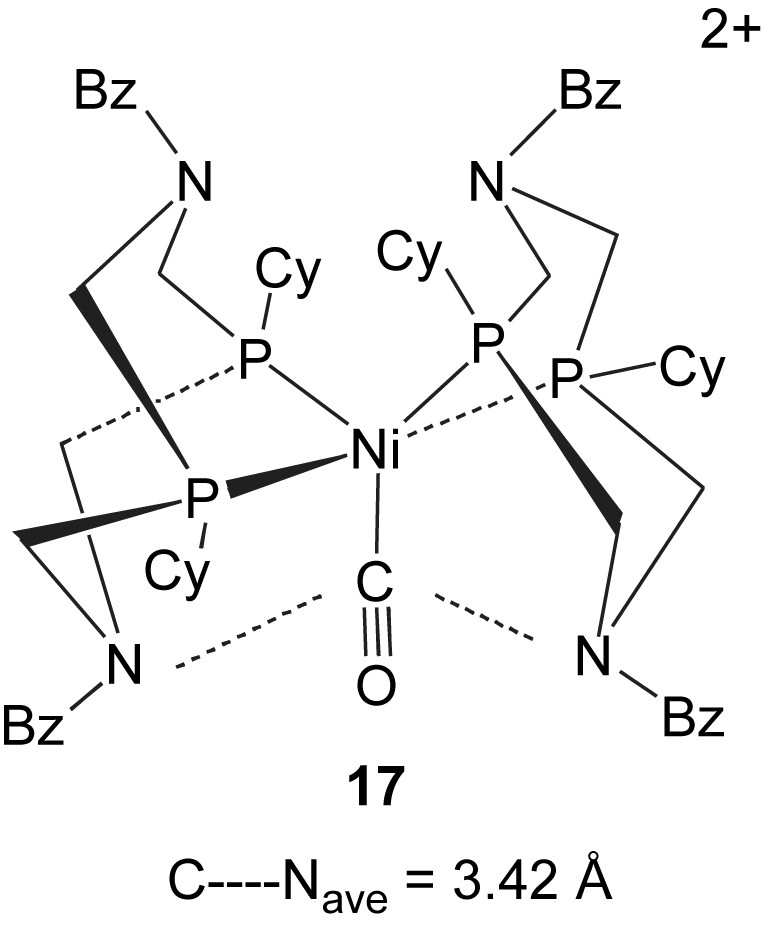
3.3 Summary of results for nickel systems
Studies of the [Ni(PNP)2]2+ and [Ni(PR2NR′2)]2+ systems indicate that the pendant bases can facilitate intermolecular exchange of protons between the protons in solution, protons on the pendant base, and the hydride ligand. In addition, the pendant base provides the necessary structural component for the coupling of proton- and electron-transfer processes. In these aspects, the nickel complexes are very similar to the iron derivatives. However, for the nickel systems containing cyclic diphosphine ligands with positioned pendant bases, relatively rapid electrocatalytic activity has been observed, and the catalysts can be tuned for either hydrogen oxidation or production by changing the substituents in the diphosphine ligands. Although dihydrogen complexes of nickel(II) are very rare, H2 binding appears to be stabilized for the nickel complexes containing two positioned pendant bases, and this is an important factor in achieving high catalytic rates for these derivatives.
Acknowledgments
We are grateful to the many students, postdoctoral coworkers, and colleagues who have participated in this project and whose names are listed in the referenced papers from our laboratories. This work was supported by Grant CHE-0240106 from the National Science Foundation. The support of the Office of Basic Energy Sciences of the Department of Energy, by the Chemical Sciences program is also acknowledged. The Pacific Northwest National Laboratory is operated by Battelle for the US Department of Energy.
1 The pKa values associated with steps 2 and 4 have been revised from our original publication [11]. The change in the pKa value of [HNi(PNHP)(PNP)]2+ (step 2) from 10.6 to 11.1 reflects an adjustment for a change in the literature pKa value of anisidinium which was used as a reference compound [14]. The pKa value of [HNi(PNP)2]2+ (step 4) has been revised downward from our original value of approximately 8 (based on a comparison to the changes of acidities of cobalt hydrides upon oxidation) to a value of approximately 0 (based on a more rigorous thermodynamic cycle). The conclusions are the same as those previously published, but step 4 has a significantly larger driving force in this revised scheme.