

{kind=link}
{kind=link}
{kind=link}
1 Introduction
Hydrogenases are enzymes that catalyze the reversible oxidation of molecular hydrogen (H2 ⇌ 2H+ + 2e−). Three main classes of hydrogenases have been identified according to the transition-metal components of the active site: [NiFe] hydrogenases [1] (including [NiFeSe] [2]), diiron [FeFe] [3] (previously called “Fe-only”), and monoiron or iron–sulfur–cluster free hydrogenases [4] (previously called “metal-free”). The [NiFe] hydrogenases are primarily utilized for hydrogen oxidation, whereas the [FeFe] hydrogenases are primarily utilized for proton reduction.
The [NiFe] hydrogenases obtained from Desulfovibrio gigas [5], Desulfovibrio fructosovorans [6], Desulfovibrio vulgaris (Miyazaki F) [7], and Desulfovibrio baculatus [2] have been investigated by a variety of experimental methods. The active sites of [NiFe] hydrogenases from these different sources are quite similar (Fig. 1). Three diatomic ligands, one CO and two CNs, are bound to the Fe atom, which is linked to the Ni by two cysteine bridges (Cys68 and Cys533 in D. gigas). The Ni is terminally bound by two cysteines (Cys65 and Cys530 in D. gigas), in which the sulfur atoms may or may not be protonated. A third bridging unit, X, between the Fe and the Ni is identified as either a hydroperoxide (–OOH) or a hydroxide (–OH) in the oxidized forms [5,8,9], and as a hydride or nothing in the reduced forms [2,7b].

Active site of the [NiFe] hydrogenases, where X can be –OH, –OOH, –H, or vacant. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Experimental studies have identified several forms of the [NiFe] hydrogenases: Ni–A, B, C, SI, SU, R, and L (SI and R in particular have been further subdivided). The catalytic behavior of [NiFe] hydrogenases can be explained in terms of interconversion between different forms and Scheme 1 shows how this might function. Ni–SI, Ni–C and Ni–R are the active forms and appear to participate in the cycle, while the role of Ni–L is less well understood. Ni–A, Ni–B and Ni–SU are inactive forms and Ni–A and Ni–SU (unready forms) require long incubation under H2 (or reductant) for activation, while Ni–B (the ready form) is rapidly converted to an active form by reductants. The Fe site is generally agreed to be low-spin (LS) Fe(II) as expected for its strong field CN− and CO ligands. However, the spin and oxidation state for the Ni site are still unclear for some of the proposed and observed forms. Previous experimental and theoretical studies suggest that the EPR-active species, Ni–A, Ni–B and Ni–C are low-spin (S = 1/2) Ni(III) forms [10] while the EPR-silent species Ni–SU, Ni–SI and Ni–R are Ni(II) but both low-spin (S = 0) [11] and high-spin (S = 1) [12] Ni have been proposed. Recent computational studies have suggested that the high-spin (triplet) Ni(II) is a better model for the observed geometry [13]. Ni–L has been proposed to be Ni(0) [14].
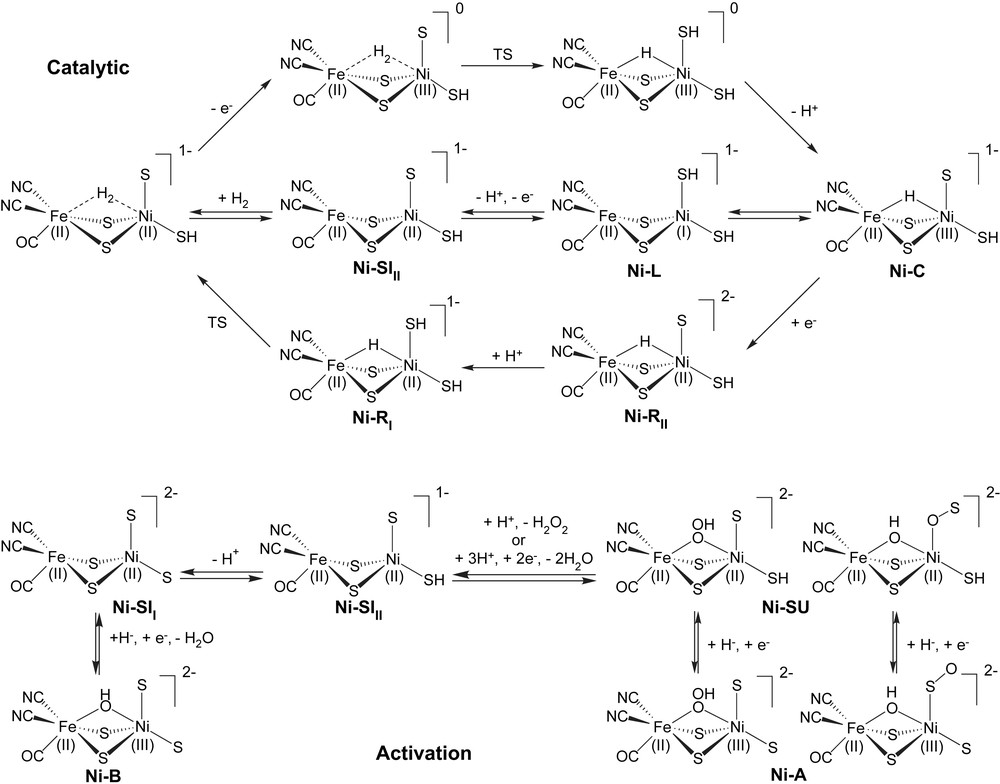
Proposed catalytic and activation cycles for [NiFe] hydrogenases. In the catalytic cycle, the upper half cycle has lower barriers for dihydrogen cleavage, while the lower half cycle has lower barriers for dihydrogen formation. The activation cycle shows alternative forms for both Ni–A and Ni–SU.
According to the crystal structure of D. gigas, the bridging cysteine residue (Cys533) is hydrogen bonded to a nearby histidine (His72) residue and the teminal cysteine residue (Cys530) forms a hydrogen bond to Glu18, which has been proposed as a proton transfer gate. The two CN− ligands coordinated to the Fe form hydrogen bonds with neighboring amino acid residues Ser486 and Arg463. Since the active site of hydrogenases is deeply buried inside the protein with four covalent bonds and four hydrogen bonds connecting it to the backbone, it is reasonable to suppose that the protein matrix will have a substantial influence on the active site. To investigate how the structural properties enforced upon the active site by the protein influence the activity, we have included various aspects of the protein environment in the computation models.
Even though extensive experimental and theoretical studies have been carried out for the investigation of the active site involved in the catalytic cycle of [NiFe] hydrogenases, there are still some disagreement and unsolved issues concerning the mechanism of the enzyme that will be addressed here. First, the spin state of the Ni is still controversial and the protein environment may play an important role. Second, the binding site of dihydrogen (H2) on the enzyme is still a mystery, does it bind to the Ni or the Fe atom, and is this binding influenced by the Ni spin state and surrounding protein. Last, the role of the neighboring protein environment on the catalytic cycle is not well documented. In the present study, hybrid DFT with the B3LYP functional is used to address these unsolved problems with five different models as shown in Scheme 2.
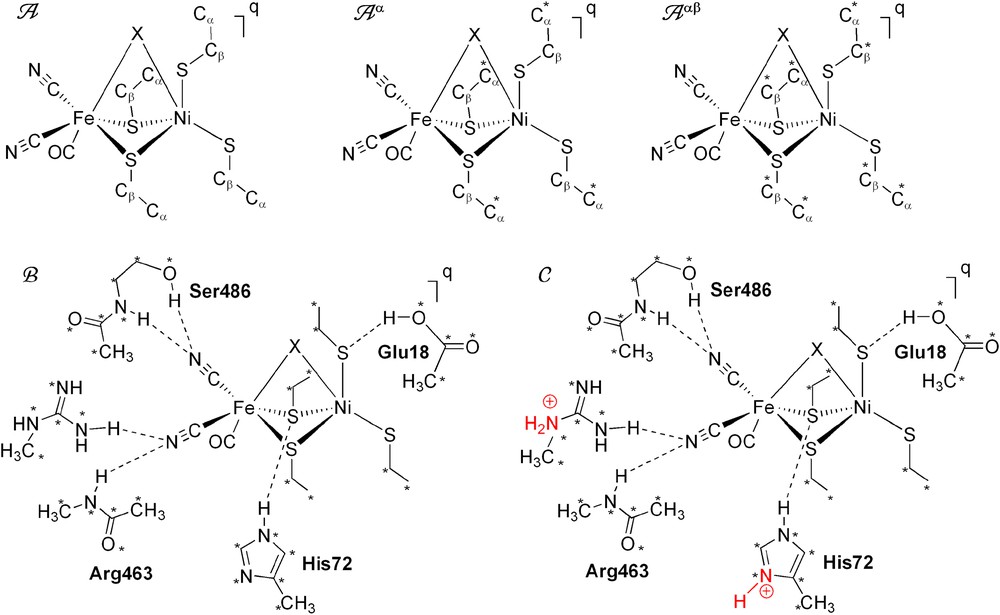
Schematic presentation of the five models used for the active site of [NiFe] hydrogenases. Positions fixed from the X-ray structure are marked with *. Residue labels correspond to D. gigas. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2 Computational details and model descriptions
All geometry optimizations and frequency determinations were carried out with the Gaussian 03 package of programs [15] at the B3LYP computational level [16]. Ni and Fe atoms were described by the Hay and Wadt basis set with effective core potentials (ECP) [17]; the 4p orbitals in the ECP basis set were replaced by optimized split valence functions from Couty and Hall [18]; these modified ECP basis sets of Fe and Ni were augmented by sets of f-polarization functions (exponent = 2.462 (Fe), 3.130 (Ni)) [19]. The LANL2DZdp [20] basis sets were employed for sulfur atoms. The 6-31G(d,p) basis sets [21] were applied for all carbon, nitrogen, oxygen and hydrogen atoms.
In the present study, the starting coordinates of the active site were taken from the X-ray structure of D. gigas [5b]. Five different models were used (Scheme 2). The simple models, A, Aα, and Aαβ, have the general form [(CO)(CN)2Fe(μ-SCβH2CαH3)2(μ-X)Ni(SCβH2CαH3)2]q, where X is the third bridging ligand when present, q is the charge of the model, and one or both of the termimal cysteine (modeled as SCH2CH3) entities may or may not be protonated. The charge q varies from neutral to −3 depending on the X ligand and the number of the protons on the terminal cysteines. The spin state is S = 0 for LS Ni(II), S = 1 for HS Ni(II), and S = 1/2 for LS Ni(III). To study the effects of backbone rigidity of the cysteine residues, modeled by ethylthiolate (–SCβH2CαH3), A was unconstrained, constraints were applied on only Cα atoms for model Aα, or on both Cα and Cβ atoms for model Aαβ. Note that the carbon atom to which the amino group, carboxyl group, and side chain (R-group) are attached is the alpha carbon (Cα).
We extend our simple model Aαβ by adding Glu18, His72, Ser486 and Arg463 functional groups as shown in model B (Scheme 2). In order to avoid artificial, large rearrangements, all heavy atoms (C, N and O) of each these amino acids were kept fixed at the corresponding position of the X-ray structure. Both α and β carbon atoms on the simplified ethylthiolate groups were also fixed at the X-ray positions. All other atoms in the complex were fully optimized. In model C, His72 and Arg463 were chosen to be protonated as shown in Scheme 2.
3 Results and discussion
3.1 Ni(II)–SI forms (Ni–SII and Ni–SIII)
The EPR-silent SI species has two forms, Ni–SII and Ni–SIII, which according to calculations differ by the protonation of a terminal cysteine and can be distinguished by their CO stretching frequencies: the CO stretching frequencies are 1914 cm−1 for the Ni–SII and 1934 cm−1 for the Ni–SIII in D. gigas [5,22]. The CO stretching frequencies from different enzymes are quite similar and this similarity of the CO frequencies provides further support for the structural similarity of the active site of the hydrogenases regardless of their source. Our former theoretical studies [13], in which the cysteine residues were modeled as –SCH3, show that the fully optimized geometries for low-spin (LS, singlet) and high-spin (HS, triplet) SI forms differ dramatically in the coordination sphere around Ni, which is a distorted square-planar structure for the LS and a distorted tetrahedral structure for the HS. The dihedral angle between the Ni–S2bridging plane and the Ni–S2terminal plane is the best way to quantify this difference. The geometry observed in the crystal structures of [NiFe] hydrogenases is a distorted tetrahedral Ni with dihedral angles of 73° in D. gigas [5], 72° in D. fructosovorans [6], and 68° in D. vulgaris [7]. Since our former calculations were carried out with the simplified model, which lacks a complete protein backbone, we would like to see whether or not the structural preference for the high-spin state would still exist when the rigidity of protein backbone and H-bonding of the active site with the protein environment are considered.
3.1.1 Models A, Aα, and Aαβ
Here, the models used for Ni–SII is [(CO)(CN)2Fe(μ-SCβH2CαH3)2Ni(SCβH2CαH3)2]2−, and for Ni–SIII is [(CO)(CN)2Fe(μ-SCβH2CαH3)2Ni(SHCβH2CαH3)(SCβH2CαH3)]1− although we cannot completely rule out a bridging H in the SI forms. Previous results and additional recent calculations show that this H must be lost before the incoming H2 binds. All three models, A (free optimization), Aα (Cα fixed), Aαβ (Cα and Cβ fixed), were calculated. Ni–SIII, which has one S protonated, was optimized corresponding to two locations on the two terminal S ligands. For better viewing, a schematic representation of Ni–SII and Ni–SIII forms (models A, Aα, and Aαβ) 1–12 is shown in Fig. 2 and their optimized geometries are presented in Fig. S1. Relative energies and important structural properties are listed in Table 1. With respect to the constraints themselves: fixing Cα costs on average ∼4 kcal/mol and fixing both Cα and Cβ costs on average ∼17 kcal/mol, energies that the entire protein could easily supply.
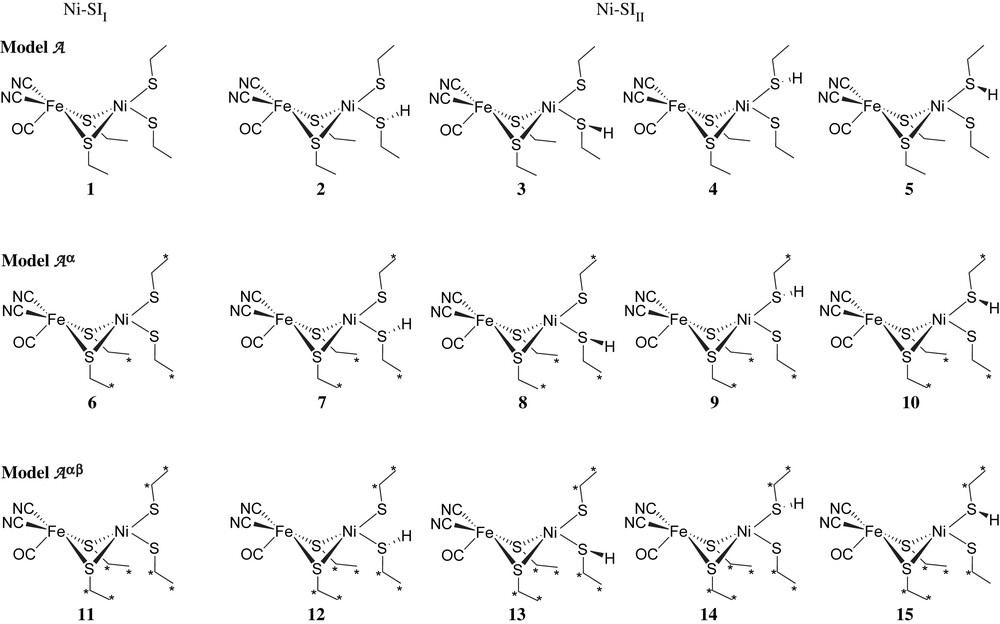
Schematic representation of Ni–SII and Ni–SIII forms (models A, Aα, and Aαβ). The hydrogens on ethyl groups are not shown for clarity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Relative energies and important structural properties for Ni–SII and Ni–SIII forms in their HS and LS states and LS Ni(III)–C form
| Model | ΔE (kcal/mol) | Fe⋯Ni (Å) | ∠(Ni–S2bridging) and (Ni–S2terminal) (°) | |
| Exp.a | 2.90 | 73 | ||
| Exp.b | 2.53 | 68 | ||
| Model A (full optimization) | ||||
| Ni–SII (1) | HS | 0.00 | 3.404 | 85.1 |
| LS | 8.94 | 3.323 | 19.9 | |
| Ni–SIII (2) | HS | 5.98 | 3.266 | 87.6 |
| LS | 11.69 | 3.166 | 9.6 | |
| Ni–SIII (3) | HS | 5.95 | 3.259 | 87.6 |
| LS | 12.25 | 3.192 | 6.9 | |
| Ni–SIII (4) | HS | 0.00 | 3.014 | 78.0 |
| LS | 8.33 | 2.719 | 34.4 | |
| Ni–SIII (5) | HS | 3.13 | 3.127 | 76.9 |
| LS | 9.33 | 2.892 | 19.3 | |
| Model Aα (partial optimization, fixing Cα atoms) | ||||
| Ni–SII (6) | HS | 0.00 | 3.433 | 76.8 |
| LS | 7.34 | 3.174 | 32.8 | |
| Ni–SIII (7) | HS | 3.21 | 3.229 | 79.1 |
| LS | 7.66 | 3.016 | 31.6 | |
| Ni–SIII (8) | HS | 2.86 | 3.270 | 74.6 |
| LS | 10.29 | 3.035 | 33.4 | |
| Ni–SIII (9) | HS | 0.00 | 3.157 | 75.8 |
| LS | 5.89 | 2.932 | 33.7 | |
| Ni–SIII (10) | HS | 0.00 | 3.101 | 72.0 |
| LS | 2.67 | 2.907 | 32.1 | |
| Model Aαβ (partial optimization, fixing Cα and Cβ atoms) | ||||
| Ni–SII (11) | HS | 0.00 | 3.258 | 65.8 |
| LS | 11.28 | 3.112 | 58.1 | |
| Ni–SIII (12) | HS | 0.00 | 3.203 | 67.3 |
| LS | 8.87 | 2.946 | 60.8 | |
| Ni–SIII (13) | HS | 1.52 | 3.241 | 69.3 |
| LS | 11.03 | 2.976 | 61.6 | |
| Ni–SIII (14) | HS | 2.99 | 3.073 | 89.6 |
| LS | 13.56 | 2.796 | 51.6 | |
| Ni–SIII (15) | HS | 5.17 | 2.829 | 56.7 |
| LS | 9.74 | 2.768 | 48.5 | |
| Ni(III)–C (20) | LS | 2.616 | 69.0 | |
| Model B | ||||
| Ni–SII (16) | HS | 0.00 | 3.053 | 62.5 |
| LS | 9.28 | 2.997 | 55.4 | |
| Ni–SIII (17) | HS | 0.00 | 2.960 | 65.5 |
| LS | 10.35 | 2.896 | 55.2 | |
| Ni(III)–C (21) | LS | 2.644 | 70.5 | |
| Model C | ||||
| Ni–SII (18) | HS | 0.00 | 2.996 | 63.4 |
| LS | 6.77 | 2.904 | 59.4 | |
| Ni–SIII (19) | HS | 0.00 | 2.956 | 64.6 |
| LS | 8.59 | 2.837 | 58.9 | |
| Ni(III)–C (22) | LS | 2.635 | 70.1 |
Consistent with previous theoretical studies [13], the fully optimized geometries (models A) for the SI forms show that the HS species have a distorted tetrahedral coordination around Ni, similar to that in the crystal structures, while the LS species have a nearly square-planar structure sphere. Also, the HS species are lower in energy than the LS species. However, this energetic preference for HS might be due to the known error for the B3LYP functional [23], which over-stabilizes HS states. Since there was not a third bridging ligand between the Fe and Ni atoms, the Ni–Fe distances were much longer than those observed for the crystal structures. The HS Ni–SIII (4) model has been excessively stabilized by a hydrogen bond between the hydrogen on one of the terminal cysteines and one of the CN groups, which also causes a decrease in the Ni–Fe distance.
When Cα atoms of the cysteine residue were fixed at their experimentally determined position (models Aα), the structures of both HS and LS species were closer to that of the crystal structures. For example, the twist angles (∠(Ni–S2bridging) and (Ni–S2terminal)) for HS states decreased and those for LS states increased. When constraints were applied to both Cα and Cβ atoms (models Aαβ), a situation that produces the strongest resemblance to the protein environment, both spin state species give better structural results. The twist angles of the HS states decrease and that of the LS states increase such that both are close to the crystal structure. For the most constrained model (Aαβ) the energetic differences between HS and LS were larger than those for full optimization (A) or for the partial optimization (Aα) and these differences exceed the known B3LYP functional error of <10 kcal/mol. Hence, due to both the energetic and structural preference, Ni–SI HS forms should be the dominant form in the enzyme. The most stable Ni–SIII (12), in which the S atom of Cys65 is protonated, is the model chosen for the complex models B and C. With respect to this choice, the other two low-lying isomers are 13 and 14; 13 has the same S protonated and would produce results similar to 12, while 14 has an S protonated that is a likely candidate for the base in the heterolytic cleavage reaction [13] so that the proton will migrate to form 12 or 13 first.
3.1.2 Complex models B and C
The optimized structures for the Ni–SII and Ni–SIII forms in both HS and LS using the larger models B and C are shown in Fig. 3. The overall structures are in good agreement with the crystal structure. The average calculated Ni–Fe distance is ∼3.0 Å for HS species and 2.9 Å for LS species (exp. 2.9 Å). The dihedral angles (∠(Ni–S2bridging) and (Ni–S2terminal)) are also close to the crystal structures for both spin states in both models. Although LS species are less stable than the HS species, the difference is close to the expected error for B3LYP. Most interestingly, as hydrogen bonding residues are added (model B) and as they are protonated to reduce the atomic charge of model B on the active site (model C), the structure of the HS and LS forms becomes more similar and the energy difference becomes smaller. Hence, it is reasonable to assume that both spin states could be involved in the [NiFe] enzyme.
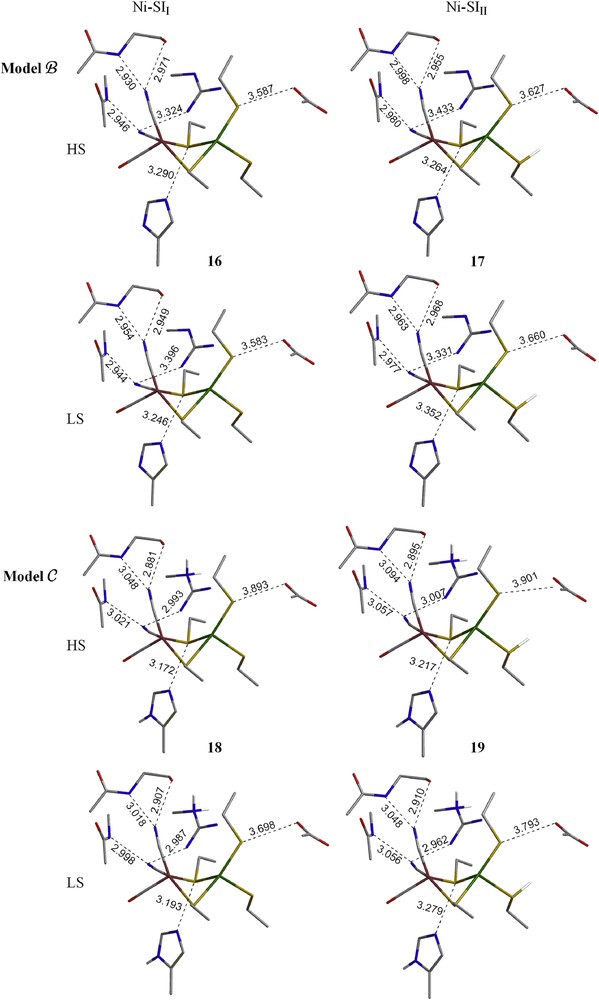
Optimized geometries of Ni–SII and Ni–SIII forms (models B and C) in their HS and LS states. The hydrogens on alkyl groups are not shown for clarity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2 Ni(III)–C form
EPR-active (S = 1/2) species Ni(III)–C, obtained by the reduction of Ni(II)–SI, is generally agreed to have a bridging hydride between the Fe and Ni atoms and protonation on one of the terminal cysteines. Hence, the formula of the model is [(CO)(CN)2Fe(II)(μ-H)(μ-SCβH2CαH3)2Ni(III)(S530CβH2CαH3)(S65HCβH2CαH3)]1−. Previous studies suggested that Ni–C forms from Ni–SI under H2 by H2 capture, e− loss and H+ transfer. Since all experimental and theoretical evidence shows that the Ni(III) species is LS rather than HS, we will only discuss LS (doublet) Ni(III) species in the current study. Geometries obtained from the partial optimization (fixing Cα and Cβ atoms) in model Aαβ and larger models B and C are shown in Fig. 4. The optimized structures have 6-coordinate octahedral geometry on the Fe center. The Ni center has a distorted square pyramidal geometry with the bridging S corresponding to Cys533 occupying the apical position. The bridging H is closer to the Ni atom (1.55–1.58 Å) than to the Fe atom (1.80–1.81 Å). The Fe⋯Ni distances (2.644 and 2.635 Å) are much shorter than that calculated for the Ni(II)-SI forms because of the bridge hydride and are similar to the experimental value of 2.53 Å from the reduced forms of [NiFe] hydrogenases [7b].
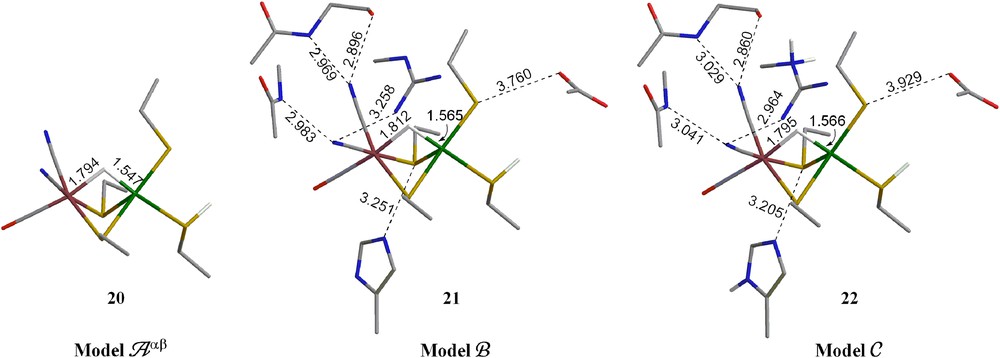
Optimized geometries of Ni–C form (models Aαβ, B and C) in its LS Ni(III) states. The hydrogens on alkyl groups are not shown for clarity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3 Dihydrogen (H2) complexes
Since the [NiFe] enzyme is usually used to oxidize H2, it would be reasonable for it to have an H2 binding site. Previous computational studies show that the position of H2 in the local minimum is on iron. Conversely, X-ray diffraction experiments suggest that an H2 gas channel connects the surface of the enzyme to the NiFe active site and the channel appears to be directed towards Ni [24,25], which was therefore suggested as the site for H2 binding and activation. Also, studies have shown that carbon monoxide (CO) acts as an inhibitor in the [NiFe] hydrogenases and the X-ray crystallography and Raman spectroscopy show that the added CO binds at the Ni, not at the Fe [7c]. The CO-inhibited hydrogenases cause inhibition by blocking the active site. With Fe only 2.6 Å away from Ni, it is difficult to rule out the participation of Fe. Furthermore, for studies of organometallic H2 complexes, the iron site would be the expected site for dihydrogen binding [26] due to an empty d-orbital of Fe. Therefore, H2 binding at the Fe atom cannot be neglected. The actual cleavage reaction could also be on one or both metals [13f]. Our previous work reported that at the B3LYP level for the H2 heterolytic cleavage, which involves both Fe and Ni atoms, barriers are 30.3 kcal/mol for LS Ni(II), 18.5 kcal/mol for HS Ni(II) and 8.6 kcal/mol for LS Ni(III). The barriers are much higher for the H2 cleavage on Ni alone (homolytic cleavage), 28.7 kcal/mol for HS Ni(II) and 29.7 kcal/mol for LS Ni(III). At the PBE level, the barriers for the homolytic cleavages (17.4 and 9.4 kcal/mol for HS Ni(II) and LS Ni(III), respectively) are close to those for the heterolytic barriers for B3LYP or even lower. In the current study, both Ni and Fe binding sites were therefore carefully investigated in these models.
Another question investigated was the oxidation state of the reactant for H2 activation. The choice of different oxidation states leads to different charge states of the [NiFe] core. The charge of the core is −2 and −1 for Ni(II)–SII and Ni(II)–SIII, respectively, and is neutral for Ni(III)–C in models A and B; and the charge of the core is 0 and +1 for Ni(II)–SII and Ni(II)–SIII, respectively, and is +2 for Ni(III)–C in model C. Earlier studies using smaller models suggested that the Fe(II)Ni(III) state was the active state [13,27], while a recent review using large models suggests that it is most likely the Fe(II)Ni(II) state that activates dihydrogen [28], which has also been suggested experimentally [1] and was for that reason chosen in some early theoretical studies of the hydrogenase mechanism [29]. Because of the apparent controversy, the model compounds starting from LS Ni(II)–H2, HS Ni(II)–H2 and Ni(III)–H2 precursors are all investigated in the current study Table 2.
Relative energies and important structural properties for the H2 complexes of Ni–SII, Ni–SIII and Ni(III)–C forms (“a” species refer to H2 or CO on Fe, while “b” species refer to H2 or CO on Ni)
| Model | ΔE (kcal/mol) | Fe⋯Ni (Å) | ∠(Ni–S2bridging) and (Ni–S2terminal) (°) | |
| Exp.a | 2.90 | 73 | ||
| Exp.b | 2.53 | 68 | ||
| Model A (full optimization) | ||||
| Ni–SII–H2 (23-a) | HS | 0.00 | 3.405 | 89.7 |
| LS | 9.57 | 3.476 | 25.3 | |
| Ni–SIII–H2 (28-a) | HS | 0.00 | 3.326 | 87.8 |
| LS | 7.19 | 3.338 | 13.0 | |
| Ni(III)–C–H2 (33-a) | LS | 3.246 | 67.3 | |
| Model Aα (partial optimization, fixing Cα atoms) | ||||
| Ni–SII–H2 (24-a) | HS | 0.00 | 3.465 | 78.9 |
| LS | 8.94 | 3.381 | 32.6 | |
| Ni–SIII–H2 (29-a) | HS | 0.00 | 3.164 | 77.0 |
| LS | 6.32 | 3.329 | 33.1 | |
| Ni(III)–C–H2 (34-a) | LS | 0.00 | 3.294 | 71.5 |
| Ni(III)–C–H2 (34-b) | LS | 4.86 | 2.523 | 65.0 |
| Model Aαβ (partial optimization, fixing Cα and Cβ atoms) | ||||
| Ni–SII–H2 (25-a) | HS | 0.00 | 3.329 | 65.9 |
| LS | 12.36 | 3.241 | 56.4 | |
| Ni–SIII–H2 (30-a) | HS | 0.00 | 3.202 | 67.3 |
| LS | 10.82 | 3.214 | 59.9 | |
| Ni–SIII–H2 (30-b) | LS | 10.22 | 2.760 | 66.5 |
| Ni(III)–C–H2 (35-a) | LS | 0.00 | 3.216 | 65.9 |
| Ni(III)–C–H2 (35-b) | LS | 4.99 | 2.604 | 67.9 |
| Model B | ||||
| Ni–SII–H2 (26-a) | HS | 0.00 | 3.309 | 60.7 |
| Ni–SII–H2 (26-b) | LS | 5.57 | 2.855 | 66.6 |
| Ni–SIII–H2 (31-a) | HS | 0.00 | 2.927 | 62.5 |
| Ni–SIII–H2 (31-a) | LS | 8.86 | 2.902 | 55.6 |
| Ni–SIII–H2 (31-b) | LS | 1.15 | 2.806 | 67.4 |
| Ni(III)–C–H2 (36-a) | LS | 0.00 | 2.898 | 62.4 |
| Ni(III)–C–H2 (36-b) | LS | 0.33 | 2.796 | 66.2 |
| Model C | ||||
| Ni–SII–H2 (27-a) | HS | 0.00 | 3.008 | 62.7 |
| Ni–SII–H2 (27-b) | LS | 4.83 | 2.864 | 68.0 |
| Ni–SIII–H2 (32-a) | HS | 0.00 | 2.907 | 63.3 |
| Ni–SIII–H2 (32-b) | LS | 1.82 | 2.770 | 65.0 |
| Ni(III)–C–H2 (37-a) | LS | 2.900 | 61.6 |
3.3.1 Simple models A, Aα and Aαβ
For the −2 charged Ni–SII form, as in the previous theoretical studies using smaller models [13], H2 prefers to bind at the Fe atom whether the backbones of the cysteine residues are free (A), semi-fixed (Aα) or totally fixed (Aαβ). No minimum with H2 binding on Ni could be located after several careful trials. H2 weakly binds on the Fe center (binding energies <3 kcal/mol), hence the complexes' geometries are similar to the Ni–SII species before H2 binding. HS states are still lower in energy than the LS states.
For the −1 charged Ni–SIII form, no H2 complexes, where H2 coordinates to Ni, were located as minimum energy structures for models A or Aα. However, when constraints were applied on both the Cα and Cβ atoms of the cysteine residues, in addition to two HS and LS H2 complexes binding to Fe, an LS H2 complex binding to Ni was located (30-b) and it was 0.6 kcal/mol lower in energy than the LS H2 complex binding to Fe.
For the neutral Ni(III)–C form, only a species where H2 coordinates to Fe located for the full optimization. While for the two partial optimizations (Aα and Aαβ), H2 was found to bind to either Fe or Ni. Here, binding to Fe is about 5 kcal/mol more favorable than binding to Ni (Table 2, Fig. 5).
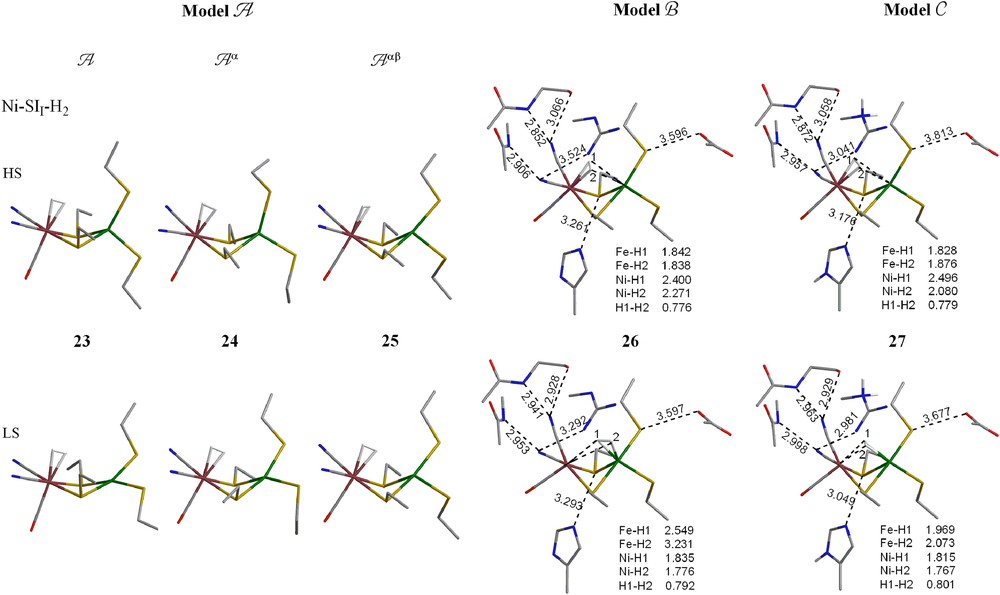

Optimized geometries of H2 complex of Ni–SII, Ni–SIII and Ni–C forms (models A–C). The hydrogens on alkyl groups are not shown for clarity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3.2 Complex models B and C
When the protein environment is added (models B and C), it appears that for both Ni–SI forms, H2 prefers to bind at Fe for HS species and to bind at Ni for LS species. In model B, there is an LS Ni–SIII H2 complex (31-a) where H2 coordinates to Fe, however it is 7.7 kcal/mol less stable than the one (31-b) where H2 coordinates to Ni. H2 may bind at either metal center for the Ni(III)–C form in model B (both H2 complexes are isoenergetic), while in model C, H2 only binds at Fe, not Ni.
Overall, it appears as if adding the protein environment brings the species with HS Ni(II) and H2 coordinated to Fe close in energy to species with LS Ni(II) and H2 coordinated to Ni. This result resembles what would be expected for the phenomenon known as two-state reactivity [30]. In this scenario, the resting form, before H2 enters the active site, is HS Ni(II). If H2 approaches the Ni, as suggested by the gas channel, and the Fe is not very accessible, then as the H2 enters the active site, the Ni changes from HS Ni(II) to LS Ni(II) in order to capture the H2.
3.4 Carbon monoxide (CO) complexes
As mentioned in the earlier text, studies have shown that carbon monoxide acts as an inhibitor in the [NiFe] hydrogenases [7c]. From X-ray studies the added CO appears to be coordinated to Ni and the Ni–CO unit shows a bent conformation with an angle of 158° and long Ni–C distance of about 1.77 Å. The coordination geometry of Fe or Ni–Fe distance did not show any distinct changes on interaction with the exogenous CO. The CO stretching frequencies are 2056 cm−1 for the exogenous CO and 1931 cm−1 for the intrinsic CO. The high frequency of exogenous CO and the small change in the intrinsic CO frequency suggest a weak interaction with the Ni atom, but sufficient to cause inhibition. Hence, understanding this CO-inhibited form is important and here we examine both Ni–SI and Ni(III)–C forms in five models as was done for the H2 complexes (Table 3, Fig. 6.).
Relative energies and important structural properties for the CO complexes of Ni–SII, Ni–SIII and Ni(III)–C forms
| Model | ΔE (kcal/mol) | Fe⋯Ni (Å) | (C–O)extri (Å) | ∠Ni–C–O (°) | ∠(Ni–S2bridging) and (Ni–S2terminal) (°) | |
| Exp.a | 2.62 | 1.14 | 158 | 71 | ||
| Model A (full optimization) | ||||||
| Ni–SII–CO (38-a) | HS | 0.00 | 3.522 | 1.149 | 177.0 | 86.9 |
| LS | 4.99 | 3.460 | 1.147 | 177.4 | 6.1 | |
| Ni–SII–CO (38-b) | HS | 24.25 | 3.462 | 1.143 | 171.5 | 45.0 |
| LS | 30.55 | 2.784 | 1.150 | 162.7 | 67.7 | |
| Ni–SIII–CO (43-a) | HS | 0.00 | 3.279 | 1.148 | 176.7 | 88.8 |
| LS | 6.59 | 3.318 | 1.146 | 177.6 | 13.0 | |
| Ni–SIII–CO (43-b) | HS | 30.40 | 3.287 | 1.137 | 173.0 | 81.1 |
| Ni(III)–C–CO (48-a) | LS | 0.00 | 3.331 | 1.136 | 177.9 | 84.6 |
| Ni(III)–C–CO (48-b) | LS | 15.89 | 2.599 | 1.138 | 177.7 | 80.6 |
| Model Aα (partial optimization, fixing Cα atoms) | ||||||
| Ni–SII–CO (39-a) | HS | 0.00 | 3.491 | 1.146 | 176.1 | 78.2 |
| LS | 13.43 | 3.483 | 1.147 | 176.3 | 36.2 | |
| Ni–SII–CO (39-b) | HS | 24.88 | 3.433 | 1.142 | 172.4 | 43.4 |
| LS | 30.66 | 2.777 | 1.150 | 163.6 | 70.3 | |
| Ni–SIII–CO (44-a) | HS | 0.00 | 2.952 | 1.146 | 169.7 | 73.0 |
| LS | 7.73 | 3.375 | 1.145 | 176.2 | 33.8 | |
| Ni–SIII–CO (44-b) | HS | 21.39 | 3.022 | 1.135 | 172.1 | 64.0 |
| Ni(III)–C–CO (49-a) | LS | 0.00 | 3.264 | 1.135 | 175.6 | 71.4 |
| Ni(III)–C–CO (49-b) | LS | 10.08 | 2.557 | 1.137 | 178.5 | 72.5 |
| Model Aαβ (partial optimization, fixing Cα and Cβ atoms) | ||||||
| Ni–SII–CO (40-a) | HS | 0.00 | 3.269 | 1.148 | 173.6 | 61.4 |
| LS | 10.53 | 3.281 | 1.147 | 174.3 | 52.6 | |
| Ni–SII–CO (40-b) | HS | 19.03 | 3.180 | 1.145 | 170.1 | 61.5 |
| LS | 18.53 | 2.729 | 1.151 | 156.7 | 67.4 | |
| Ni–SIII–CO (45-a) | HS | 0.00 | 2.895 | 1.147 | 169.4 | 63.4 |
| LS | 9.96 | 3.195 | 1.145 | 174.9 | 56.6 | |
| Ni–SIII–CO (45-b) | HS | 21.14 | 3.106 | 1.138 | 170.0 | 64.8 |
| LS | 14.35 | 2.778 | 1.142 | 177.2 | 70.1 | |
| Ni(III)–C–CO (50-a) | LS | 0.00 | 2.727 | 1.144 | 168.9 | 64.4 |
| Ni(III)–C–CO (50-b) | LS | 4.27 | 2.597 | 1.137 | 176.9 | 70.2 |
| Model B | ||||||
| Ni–SII–CO (41-a) | HS | 8.17 | 3.321 | 1.152 | 164.7 | 64.0 |
| LS | 19.39 | 3.324 | 1.152 | 164.8 | 58.5 | |
| Ni–SII–CO (41-b) | HS | 0.00 | 3.050 | 1.137 | 161.2 | 63.5 |
| LS | 4.59 | 2.801 | 1.144 | 155.4 | 68.0 | |
| Ni–SIII–CO (46-a) | HS | 8.71 | 3.109 | 1.149 | 164.3 | 65.8 |
| LS | 20.06 | 3.229 | 1.147 | 165.6 | 59.5 | |
| Ni–SIII–CO (46-b) | HS | 5.00 | 3.009 | 1.133 | 157.7 | 66.4 |
| LS | 0.00 | 2.790 | 1.137 | 168.0 | 69.9 | |
| Ni(III)–C–CO (51-a) | LS | 9.89 | 3.017 | 1.148 | 163.7 | 66.6 |
| Ni(III)–C–CO (51-b) | LS | 0.00 | 2.762 | 1.141 | 170.0 | 68.8 |
| Model C | ||||||
| Ni–SII–CO (42-a) | HS | 9.75 | 3.182 | 1.152 | 164.1 | 65.6 |
| LS | 4.62 | 3.670 | 1.149 | 166.8 | 85.1 | |
| Ni–SII–CO (42-b) | HS | 0.00 | 2.968 | 1.142 | 163.9 | 61.7 |
| LS | 2.07 | 2.751 | 1.151 | 155.7 | 66.6 | |
| Ni–SIII–CO (47-a) | HS | 2.11 | 3.072 | 1.147 | 164.7 | 84.4 |
| LS | 21.41 | 3.290 | 1.148 | 164.6 | 87.8 | |
| Ni–SIII–CO (47-b) | HS | 6.16 | 2.973 | 1.136 | 160.4 | 66.2 |
| LS | 0.00 | 2.747 | 1.141 | 170.9 | 67.3 | |
| Ni(III)–C–CO (52-a) | LS | 1.46 | 3.061 | 1.149 | 163.2 | 83.4 |
| Ni(III)–C–CO (52-b) | LS | 0.00 | 2.744 | 1.140 | 169.9 | 67.4 |
a Ref. [7c] (CO-inhibitor of [NiFe] hydrogenases).
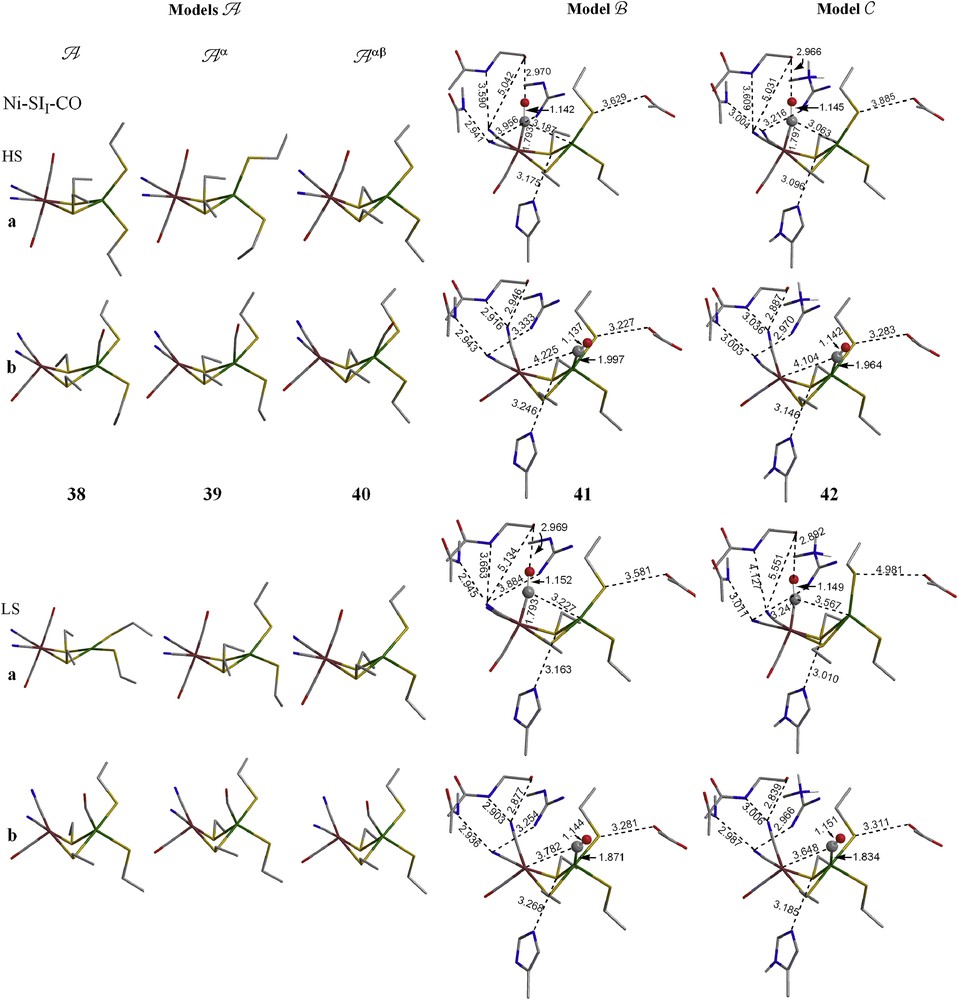
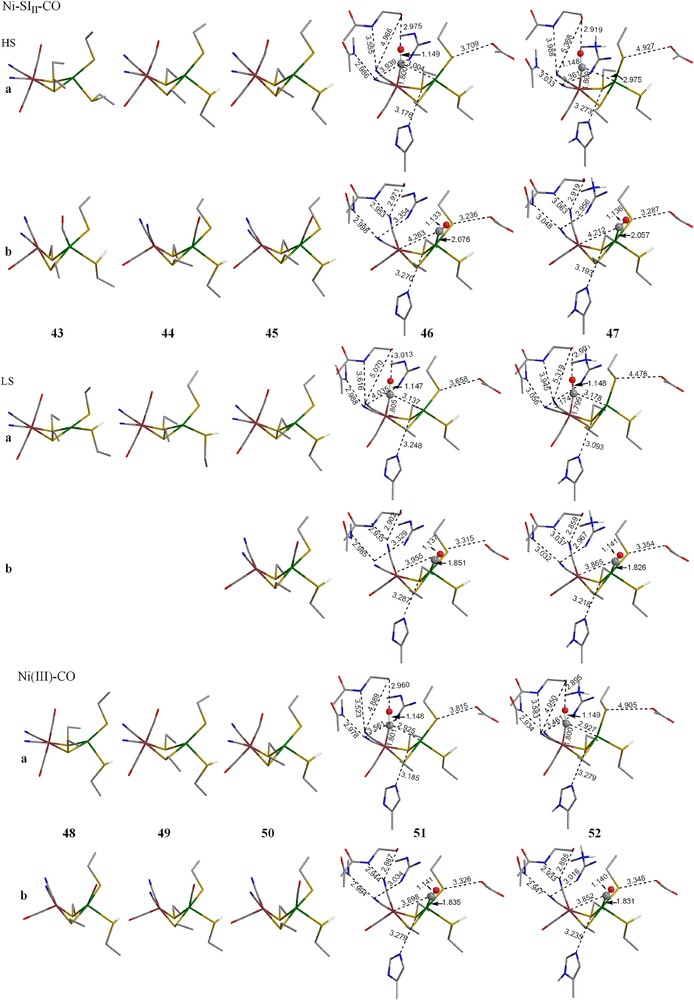
Optimized geometries of CO complex of Ni–SII, Ni–SIII and Ni–C forms (models A–C). The hydrogens on alkyl groups are not shown for clarity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4.1 Simple models A, Aα, and Aαβ
For the −2 charged Ni–SII form, CO may bind at either Fe or Ni atom and binding at the Fe atom is more favorable in all A models (A, Aα, and Aαβ), but this energetic preference decreases as more constraints were applied. Although the LS CO complexes with CO binding at Ni (LS 38-b, 39-b and 40-b) are generally the least stable species, their optimized geometries agree with the crystal structure best; the Fe⋯Ni, the ∠Ni–C−O and the ∠(Ni–S2bridging) and (Ni–S2terminal) are all close to the experimental values.
For the −1 charged Ni–SIII form, no LS CO complexes, where CO coordinates to Ni, were located as minimum energy structures for the A or Aα models. When constraints were applied on both the Cα and Cβ atoms of the cysteine residues (Aαβ), an LS CO complex (LS 45-b) binding to Ni was located and it is 6.8 kcal/mol lower in energy than the HS CO complex binding to Ni. Like the CO complexes of Ni–SII form, this LS 45-b is the better structural model.
For the neutral Ni(III)–C form, CO may bind at either Fe or Ni for all situations with energetic preference for binding at Fe. This energetic preference decreases as more constraints were applied on the molecules (15.9 kcal/mol for A, 10.1 kcal/mol for Aα, and 4.3 kcal/mol for Aαβ). Again, like the Ni–SI forms, the important structural properties (i.e. d(Fe⋯Ni) and ∠(Ni–S2bridging) and (Ni–S2terminal)) of the CO complexes that have CO binding at Ni are closer to the experimental values than those which have CO binding at Fe.
3.4.2 Complex models B and C
Although all the simpler A models, in which no protein environments are considered, are more stable for CO binding at Fe, when the protein environments are included, the preference changes to binding at Ni. Table 3 shows between 2 and 10 kcal/mol preference for Ni–CO for both models B and C. For the Ni–SI forms, the LS CO complexes with CO binding at Ni are the best models both structurally and energetically.
4 Conclusions
The hydrogenases are mechanistically complex and there are a number of important unanswered questions. We have addressed some of these in this study and also raised some new questions. For all of the species studied, adding the nearby protein residues to a fairly rigid active site framework stablizes the LS Ni(II) species. Although models for Ni–SI forms, with a vacant binding site, still prefer HS, addition of H2 or CO causes a switch to LS. Thus, access to this LS state and two-state reactivity may play a role in the mechanism, a situation that has not been considered before. Furthermore, the more complete protein models show that the energetic preference for the binding site for both H2 and CO changes from Fe to Ni. This change brings the computational results in closer accord with the experimental ones.
Acknowledgement
This work was supported by the National Science Foundation (CHE-0518074,0541587) and the Robert A. Welch Foundation (0648-A).