1 Introduction and history
About 20 years ago, two metabolites with antifungal activity, later named epothilones A and B (1a, 1b), were isolated at GBF (Gesellschaft für Biologisch-chemische Forschung at Braunschweig, Germany) from the soil bacterium Sorangium cellulosum, strain So ce90 (Fig. 1). Early application tests against plant pathogenic fungi failed due to phytotoxicity. The pharmaceutical industry, on the other hand, was not interested in a possible application as an anticancer agent, even though these compounds exhibited also significant toxicity in cell culture assays. Thus the compounds were abandoned by GBF in 1994 [1,2].

Most important epothilones from fermentation.
In 1993, MSD chemists set up a screening of natural products with taxane-like antitumor activity. They had one confirmed hit for an extract from an S. cellulosum strain coded SMP44. To their great surprise, the compounds responsible turned out to be 1a and 1b [3]. Compound 1b was about 10 times more active than 1a and even more active than taxol in the tubulin polymerisation assay; it replaced bound taxol from microtubules, and, most remarkably, its activity against cancer cells was hardly impaired by the resistance to taxol and other cytostatics. These exciting results immediately triggered a variety of activities in pharmaceutical companies and academia. The first total syntheses of 1a, 1b were published in 1996/1997 by the groups of Danishefsky, Nicolaou and Schinzer (see Section 5). Meanwhile GBF again improved the production strain, optimized nutrients and fermentation process, and streamlined the extraction process and large-scale purification by chromatography. From side fractions epothilones C and D (2a, 2b) were isolated as major biosynthesis by-products along with 36 minor epothilones, for instance epothilones E and F (1c, 1d) [4].
2 Mode of action
The observation that epothilones can displace tubulin-bound taxol [3] indicated a common pharmacophore. After the structure of the taxol–tubulin complex had been published, attempts were made to model epothilone within the taxol binding site. Thus, NMR experiments were performed with a soluble form of the epothilone A–tubulin complex [5]. The structure of epothilone A, bound to α,β-tubulin in zinc-stabilized sheets, was determined by a combination of electron crystallography at 2.89 Å resolution and nuclear magnetic resonance-based conformational analysis [6]. The complex explains both the broad-based epothilone structure–activity relationship and the known mutational resistance profile. Comparison with taxol shows that the longstanding expectation of a common pharmacophore is not met, because each ligand exploits the tubulin-binding pocket in a unique and independent manner.
However, no consistent pharmacophoric model could be derived from these controversial results so far.
3 Physical and chemical properties
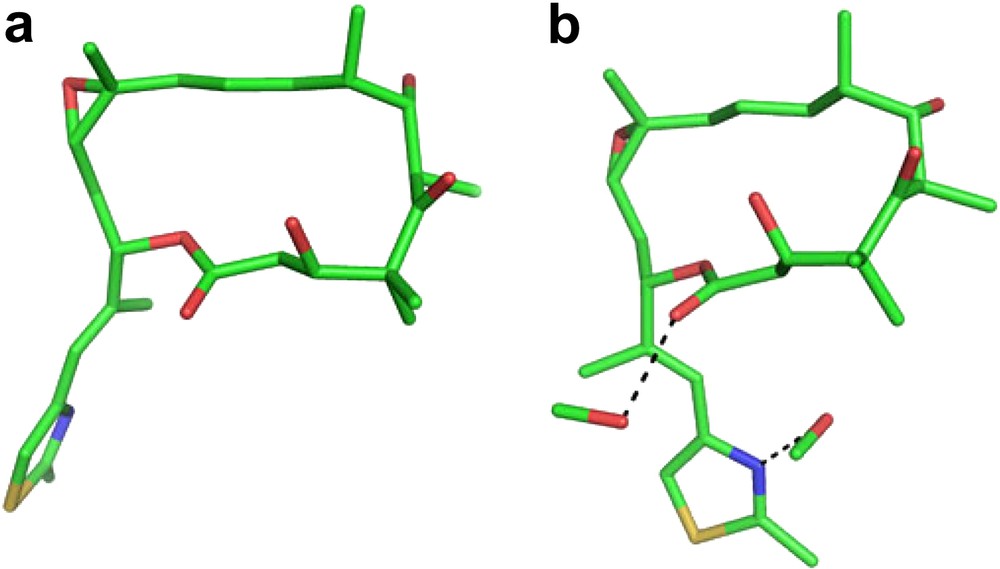
Compounds 1a, 1b and 2b have been crystallized. Epothilone A (1) is well soluble in polar organic solvents like methanol, ethyl acetate, acetone, diethyl ether and DMSO, whereas epothilone B is distinctly less soluble. Both are sparingly soluble in benzene, toluene and petroleum ether. A variety of melting points have been reported ranging from 76 °C to 128 °C depending on crystal form and nature of solvate. The X-ray crystal structures of 1b from dichloromethane/petroleum ether and methanol/water show only small conformational changes in the orientation of the side chain, whereas the macrocyclic ring is virtually superimposable (Fig. 2) [1,7].1
The preferred solution conformation of 1b, derived from NMR studies in DMSO, was found to be very similar to that in Fig. 2a [1]. In a detailed conformational analysis, Taylor and Zajicek [8] detected a second minor conformer in which the 3-OH is rotated from a pseudo-axial to a pseudo-equatorial position.
Chemically, lactone and epoxide are the most reactive groups in 1a and 1b [9]. As expected, the lactone group is rapidly hydrolyzed in aqueous medium above pH 11; the epoxide is hydrolyzed or rearranged below pH 3 [10]. At neutral pH, pig liver esterase rapidly cleaves the lactone [9,11]. Otherwise, epothilones are perfectly stable under normal conditions.
4 Biosynthesis
It was originally speculated that the epothilones are assembled jointly by polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS) multienzymes (Fig. 3). In such multimodular megasynthetases [12–14] the activated monomeric building blocks are selected as CoA esters in PKS by acyltransferase (AT) domains.
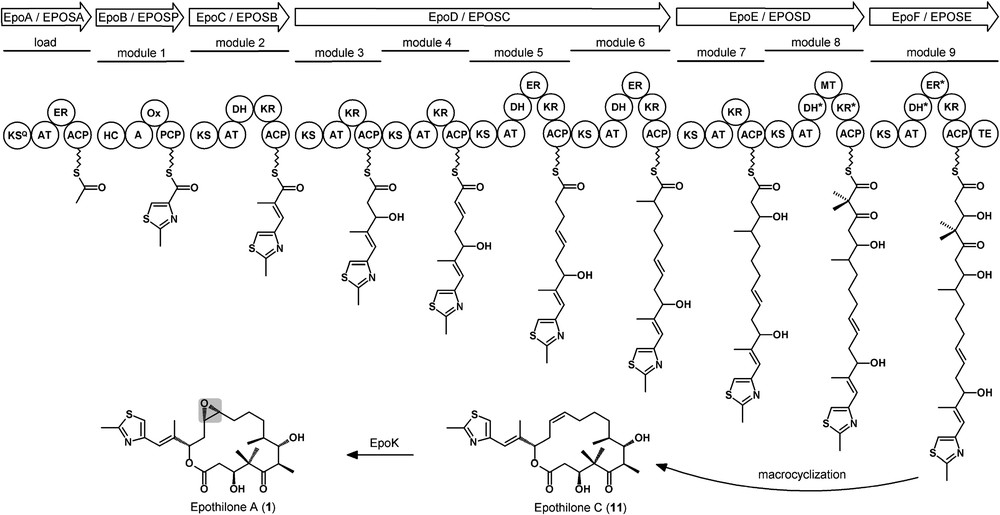
Epothilone biosynthetic assembly line exemplified for the formation of epothilone A.
The activated acids are then covalently tethered to carrier proteins (acyl carrier proteins – ACPs – in PKS and peptidyl carrier proteins – PCPs – in NRPS) and then condensed with each other via the action of ketosynthase (KS) domains in a Claisen type reaction (PKS) or by condensation (C) domains in NRPS forming peptide bonds. Additional domains are ketoreductase (KR) domains, dehydratase (DH) domains, enoylreductase (ER) domains, and O-methyltransferase (O-MT) domains in PKS. In NRPS, heterocyclization (HC) domains forming thiazoline and oxazoline rings from cysteine and serine, respectively, can be employed. Additional oxidation (Ox) domains generate the thiazole and oxazole structures. Additionally, N-methyltransferase (N-MT) domains or epimerization (E) domains forming d-amino acids from the natural l-forms are found frequently. Typically, the fully extended intermediate is released from the final carrier protein by the action of a thioesterase domain forming free acids, lactones, or lactams. In fact, feeding studies [15] in the natural host S. cellulosum So ce90 revealed that the carbon atoms in the epothilone backbone are derived from acetate (from malonyl-CoA; mCoA), propionate (from methylmalonyl-CoA; mmCoA), the methyl group of S-adenosyl-methionine (SAM), and cysteine (which also introduces the sulfur and nitrogen atoms). Compounds 1a and 1b are formed from the alternative incorporation of mCoA or mmCoA at position C11–C12. The epoxide is incorporated from molecular oxygen by a P450 type enzyme, EpoK, which was expressed in recombinant form. Its crystal structure has been solved [16,17].
It is noteworthy in this connection that Khosla et al. have synthesized epothilones using fermentation methods by cloning and heterologous expression of the epothilone gene cluster in Myxococcus xanthus [18].
5 Total synthesis
The total synthesis of epothilones A–F is a worthwhile objective as their potential for broad refunctionalization and structural modification is rather limited. Moreover, the epothilones are structurally much less complex than, for instance, the taxanes. Altogether, 16 complete and formal approaches have been reported for 1a, 2a and 20 for 1b, 2b (for reviews, see Refs. [19,22]). In the following section, an arbitrarily chosen selection will be presented. It has to be emphasized that many of these approaches use the same disconnection strategies and key fragments, so that there is a limited number of truly independent syntheses. Typically, most sequences intercept one of the key intermediates (vide infra) from the Nicolaou, Danishefsky or Schinzer syntheses.
From these pioneering investigations, a standard retrosynthesis has emerged over the years (Scheme 1) [20–24]. The last step in the synthesis is the more or less stereocontrolled epoxidation of the C12,13-double bond. As the penultimate step, the macrolactonization of seco-acid I is performed in 10−3 M to 10−4 M solution via the Yamaguchi protocol [25]. Seco-acid I stems from a stereocontrolled aldol addition of the C21–C7 aldehyde II and the (Z)-enolate of ketone III [26], the asymmetric induction of which crucially hinges on the presence and the configuration of the stereocenter at C3 and the functionalization of the C1–C3-region in III. Aldehyde II has been prepared along a variety of routes which characteristically differ in the introduction of the C12,13-double bond. A second, less general access to 2a–d is via RCM of di-olefin ester IV which is obtained from alcohol V and acid VI. For the preparation of VI, an aldol reaction between III and aldehyde VII is normally used, similar to the one between III and II.
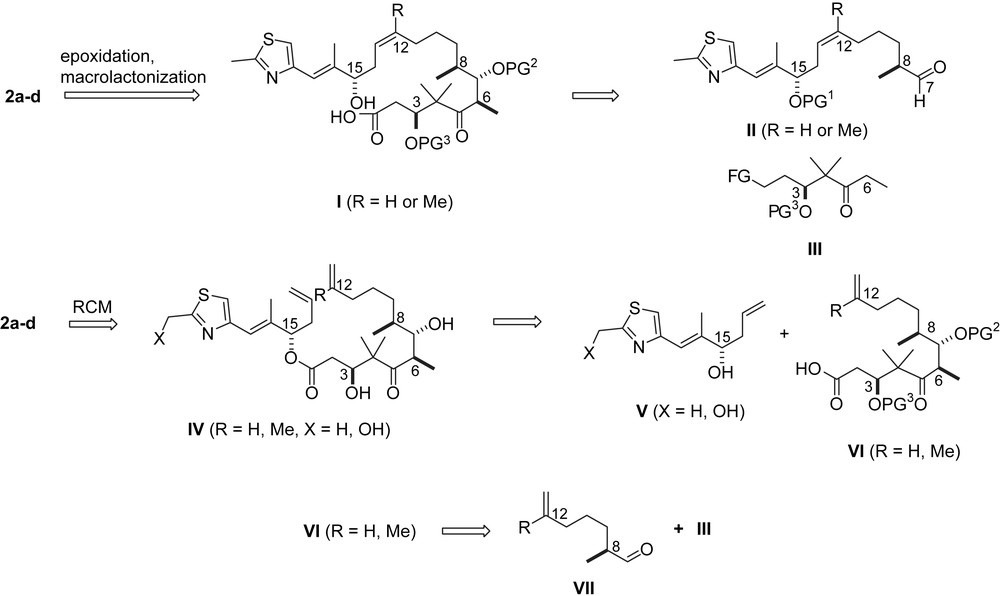
Retrosynthetic considerations.
The first three syntheses (Danishefsky, Nicolaou, and Schinzer), though initially all aiming for 1a, 2a, were flexible enough to target also 1b, 2b without major alterations.
5.1 Danishefsky syntheses (Schemes 2–11)
The Danishefsky group first [27–29] has pursued three different ring closure strategies (Scheme 2). First, they started with the preparation of the C21–C12 segments 9 and 10 (Scheme 3) both easily available from thiazolyl ester 3.
Overview of Danishefsky's syntheses.
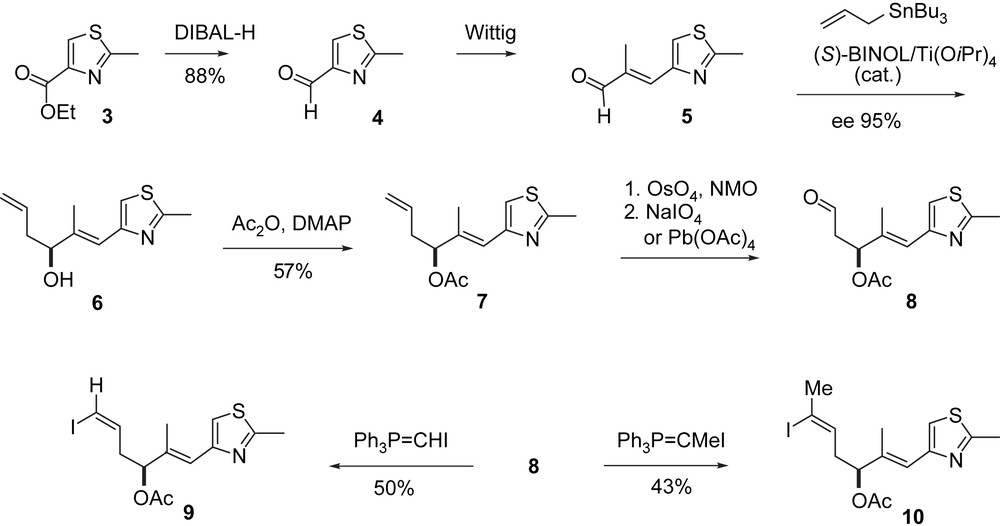
Danishefsky's synthesis of epothilone building blocks.
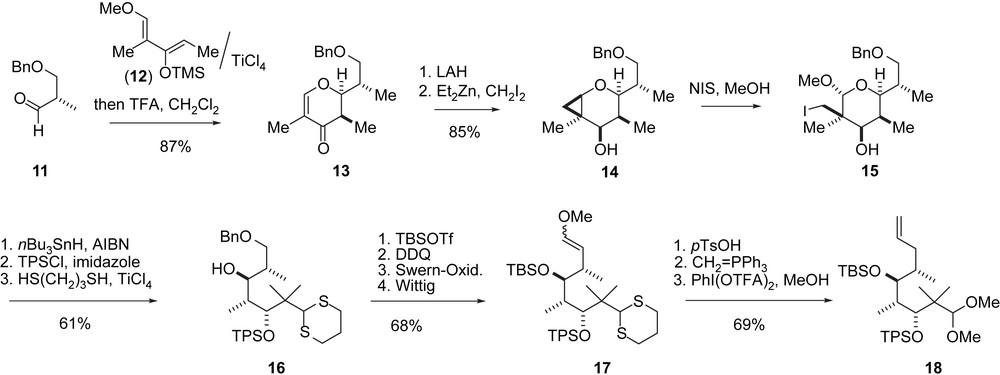
Danishefsky's macroaldolization synthesis of epothilones A and C, part I.
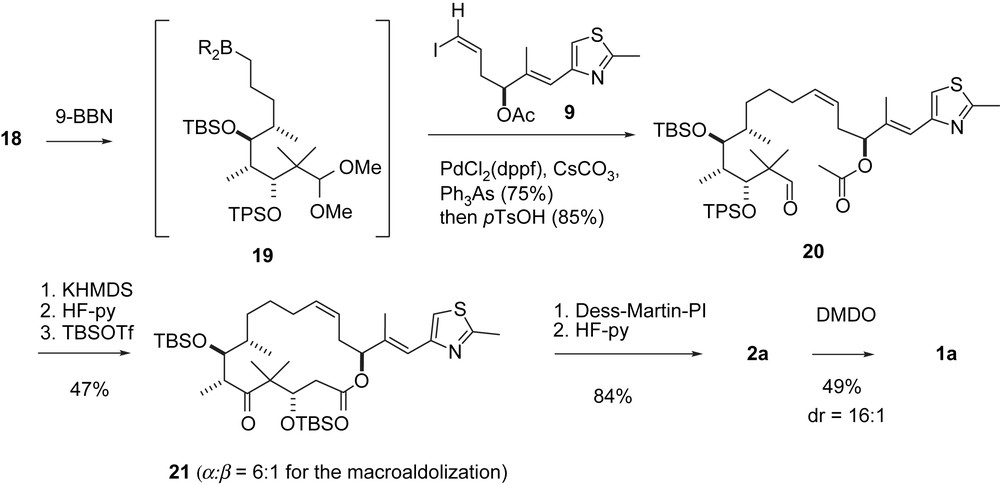
Danishefsky's macroaldolization of epothilones A and C, part II.

Danishefsky's macrolactonization synthesis of epothilones A and C.
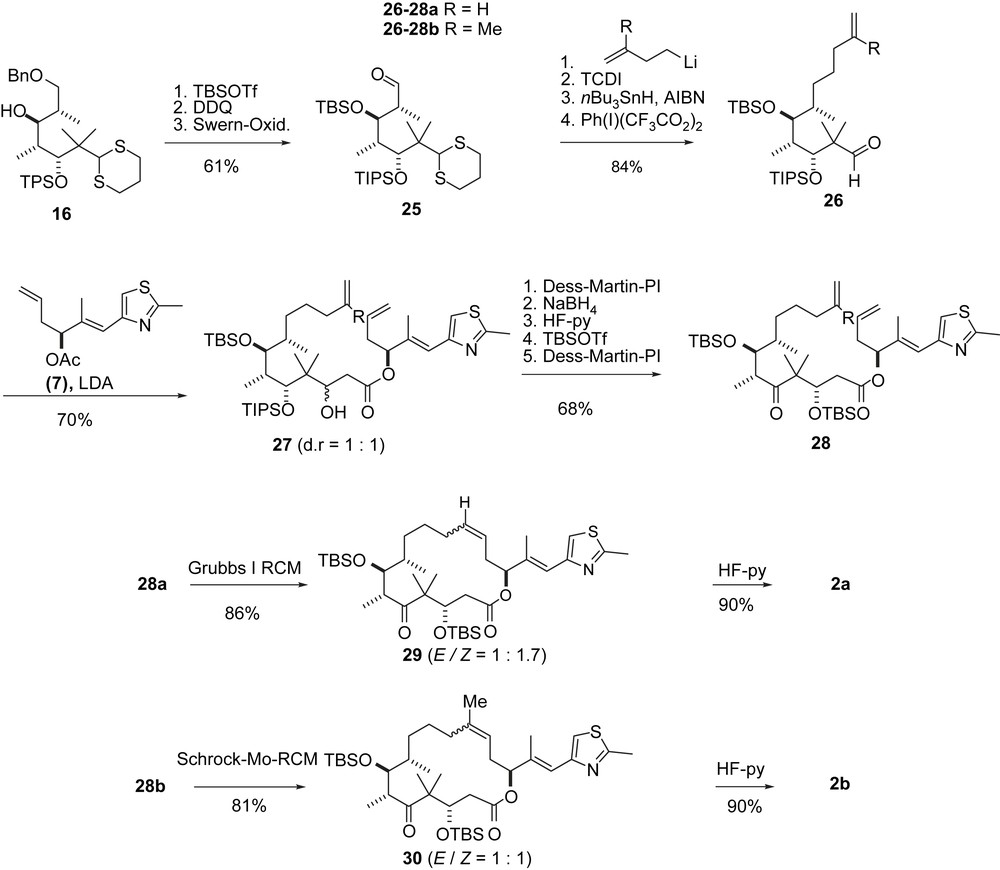
Danishefsky's first RCM synthesis of epothilones A–D.
Danishefsky's B-alkyl-Suzuki approach to epothilone B, part I.
Danishefsky's B-alkyl-Suzuki approach to epothilone B, part II.
Danishefsky's second RCM synthesis of epothilone D.
Danishefsky's third RCM synthesis of epothilone D.
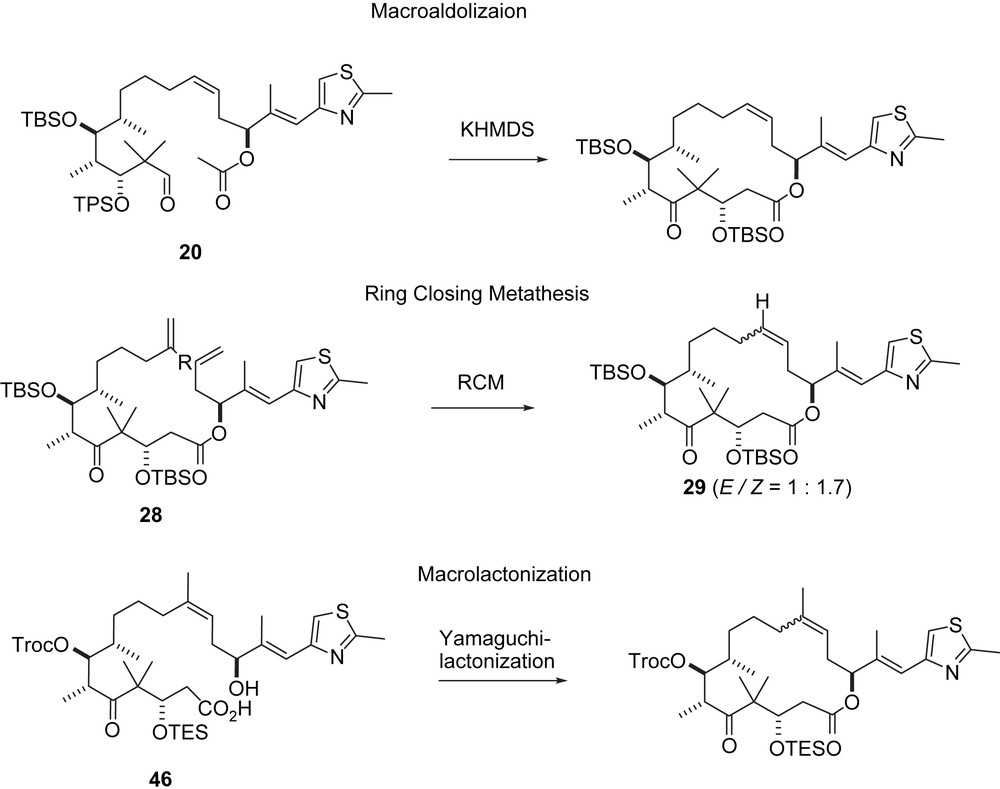
The C3–C11-segment 18 was obtained along a route (Scheme 4) that differs from the general strategy described in Scheme 1. A hetero-Diels–Alder reaction between chiral aldehyde 11 and Danishefsky diene 12 furnished dihydropyranone 13 with high Felkin–Anh selectivity. Cyclopropanation to 14 and ring opening to iodide 15 gave the acyclic C3–C9-fragment 16 after deiodination. Elaboration into the C3–C11-fragment 18 and B-alkyl-Suzuki coupling with vinyl iodide 9 completed the epothilone A/C seco-intermediate 20 (Scheme 5). Macroaldolization gave 21 with a 6:1 diastereomeric ratio at C3. Oxidation at C3 and desilylation led to 2a. The regio- and stereoselective epoxidation of the C12,13-double bond to 1a was achieved with DMDO [30].
A second approach to 2a [31] used macrolactonization as the ring closing step (Scheme 6). Hence, acetal 18 was converted into the aldehyde, which was subjected to an aldol addition with t-Bu-acetate to give, after deprotection, compound 22 as a separable 2:1 diastereomeric mixture at C3. After oxidation and silylation 23 was obtained which was hydroborated and subjected to a Suzuki coupling with 9 to furnish seco-acid 24. Yamaguchi lactonization and desilylation gave 2a. Although this second route is four steps longer, the overall yield is about the same (23% vs 25%).
Ring closing metathesis (RCM) was tested as a third mode of macrocyclization [32], this time for both 2a and 2b (Scheme 7). In fact, the epothilones were the first relatively complex substrates for an RCM cyclization. In the event, intermediate 16 was converted into C9-aldehyde 25, which after chain elongation gave olefinic aldehydes 26a, 26b. Aldol addition with 7 furnished 27a, 27b as 1:1 diastereomeric mixtures at C3. The configuration at C3 was rectified to (3S) via an oxidation–reduction sequence after which some further modifications led to the diastereomerically pure ketones 28a, 28b. Epothilone A precursor 28a was cyclized with Grubbs' first generation RCM catalyst to 29 as a 1:1.7-E/Z-mixture which was separated and desilylated to give 2a. In contrast, the RCM of epothilone B intermediate 28b had to be performed with Schrock's catalyst to furnish 30 as a 1:1-E/Z-mixture. Separation and desilylation gave 2b.
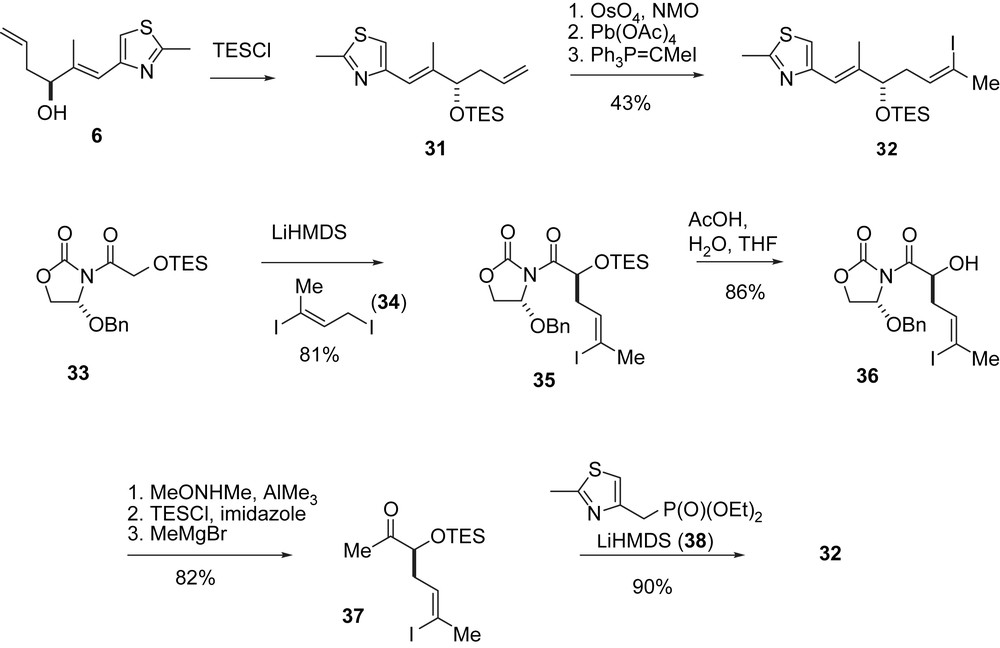
In an updated approach, OTES-protected vinyl iodide 32 was prepared from 31 (Scheme 8) [33] or, better, via Evans' allylation of 33 with di-iodide 34.
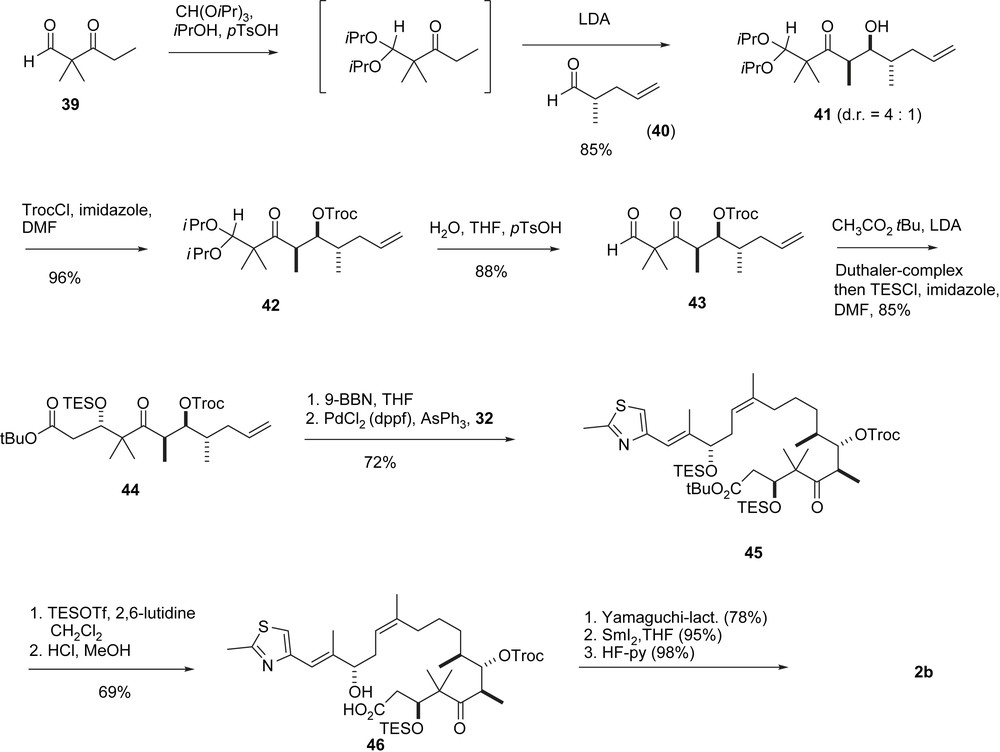
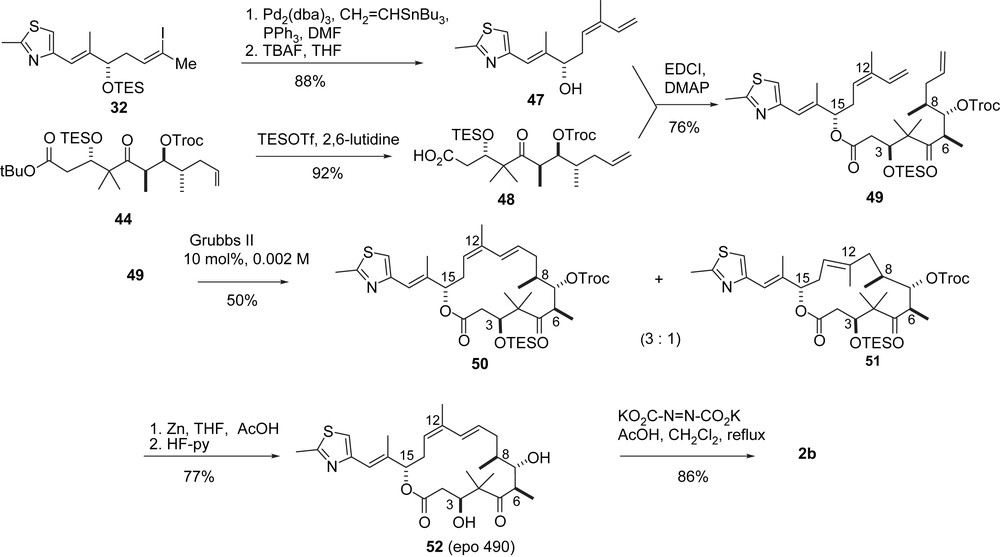
For the B-alkyl-Suzuki coupling olefinic ester 44 was prepared via an aldol addition between aldehyde 40 and the isopropyl acetal of ketone 39, which gave 41 with moderate diastereoselectivity (Scheme 9). Isomer 41 was separated, converted into aldehyde 43, which was subjected to a Duthaler aldol addition to give 44 with high stereocontrol. Suzuki coupling of 44 with vinyl iodide 32 furnished seco-intermediate 45. Deprotection gave the seco-acid 46 which was macrolactonized and deprotected to 2b.
A variation of the original RCM approach (Scheme 10) [33] was initiated by preparing the terminal olefins 47 and 48 from 32 and 44, respectively, and connecting them via esterification to the seco-intermediate 49 which on RCM with Grubbs' second generation catalyst gave a 3:1 mixture of the desired macrocycle 50 and the ring-contracted derivative 51. After separation, 50 was deprotected to give epothilone 490 (52) as a highly active non-natural epothilone derivative. Selective hydrogenation of the C10,11-double bond with diimide led to 2b.
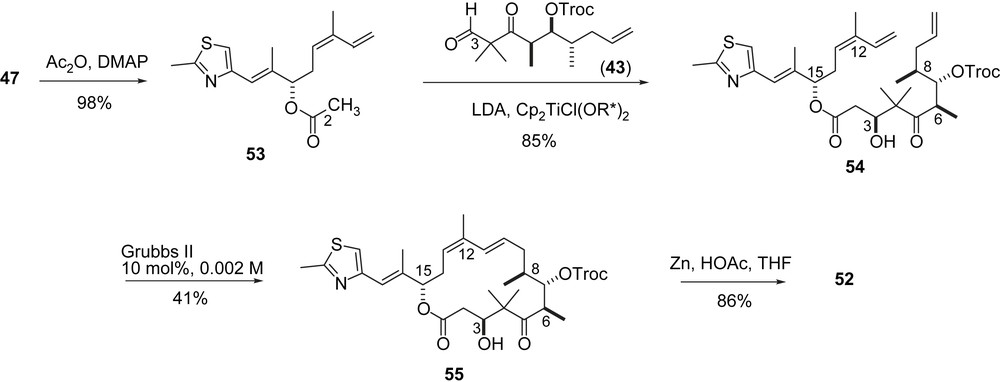
In a second approach to 52 (Scheme 11) the Duthaler aldol addition of acetate 53 to aldehyde 43 was used. Compound 54 was obtained stereoselectively and macrocyclized by RCM to furnish 55. In this case, no ring contraction was observed. After removal of the Troc-group 52 was obtained.
5.2 Nicolaou syntheses (Schemes 12–20)
The Nicolaou group was second to complete total syntheses of both 1a and 1b. Their strategy (cf. Scheme 12) was focused on an aldol addition between a C6 ketone enolate (as in 57, 59) and a C7 aldehyde 63. The C12,13-double bond was formed via metathesis in the A series [34] and via Wittig olefination in the B series [35].
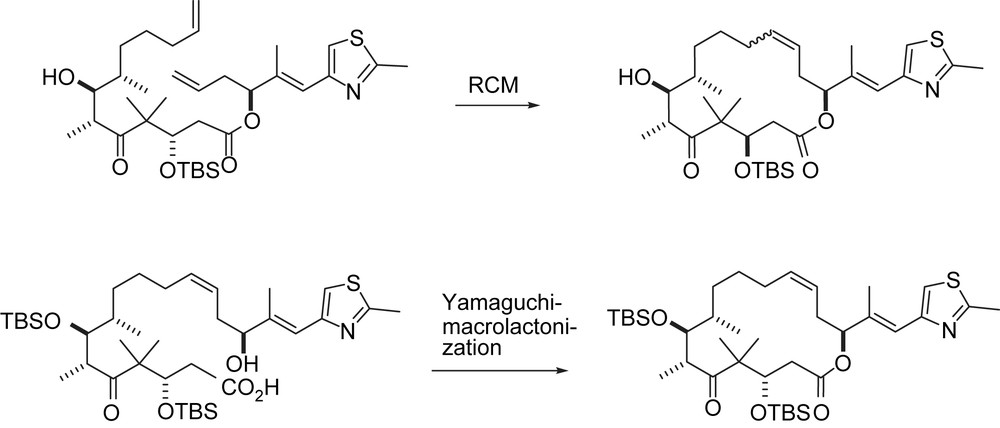
Overview of Nicolaou's syntheses.

Nicolaou's synthesis of epothilone building blocks.
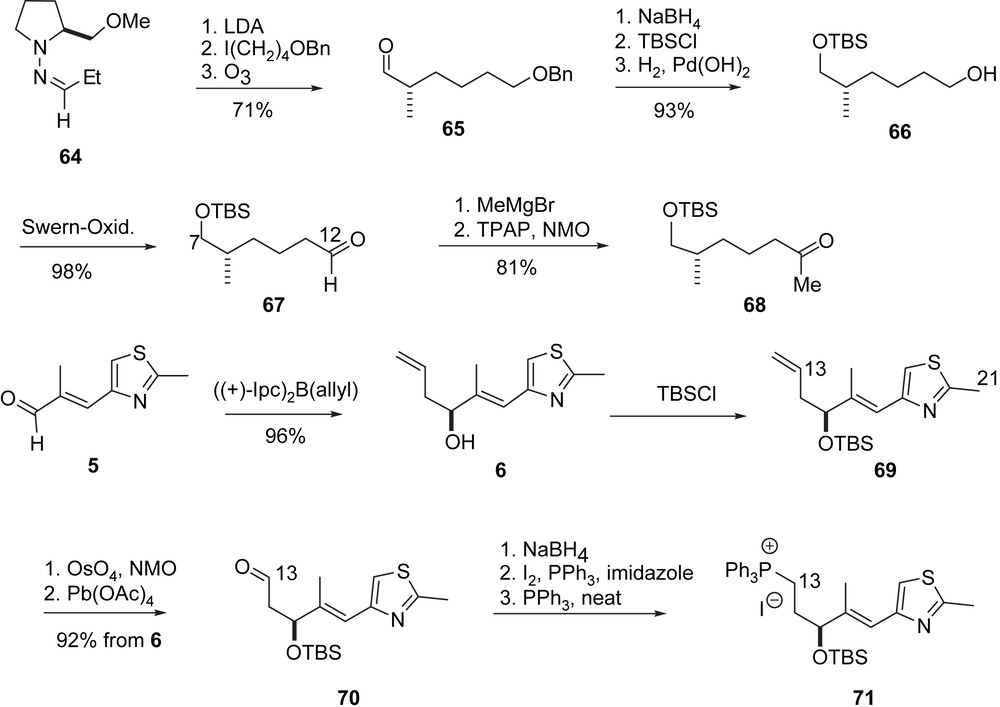
Nicolaou's synthesis of epothilone fragment 71.

Nicolaou's RCM synthesis of epothilones A and C.
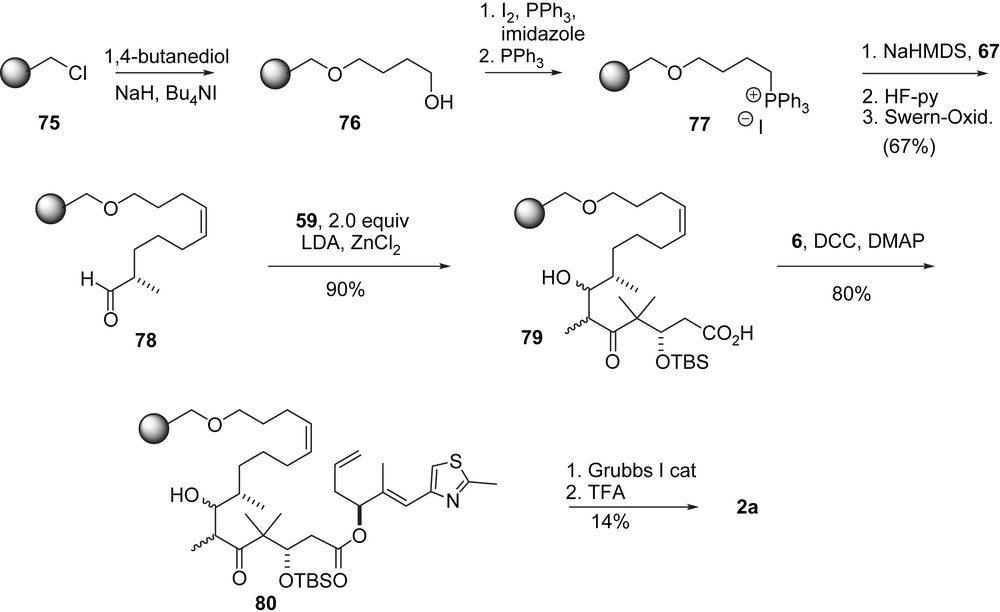
Nicolaou's solid phase synthesis of epothilone C.
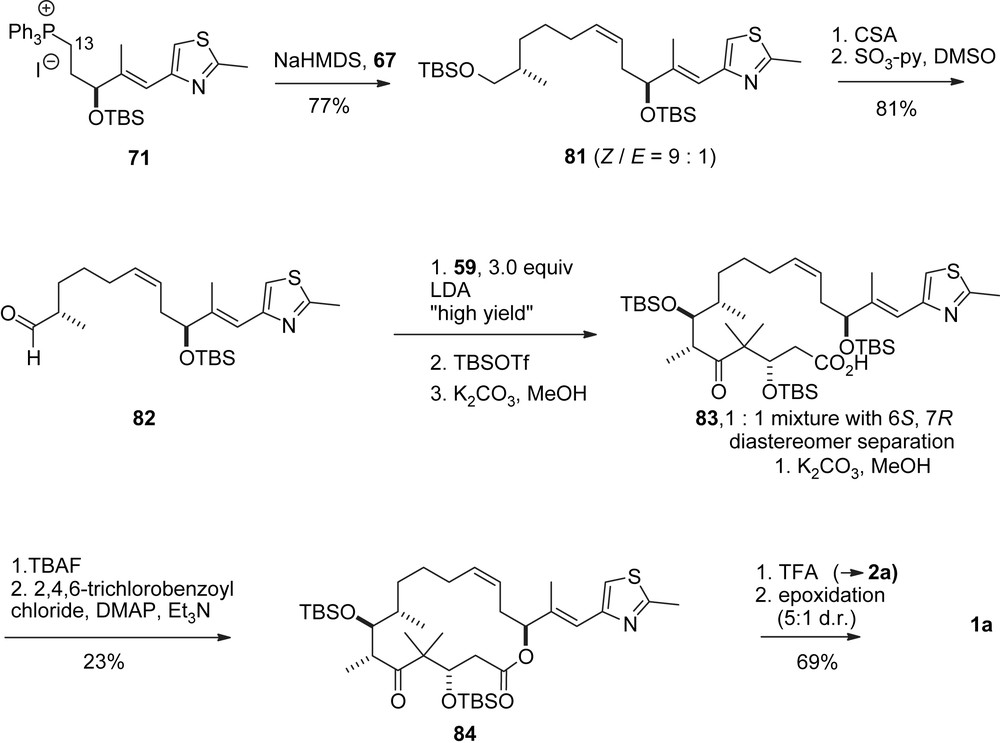
Nicolaou's macrolactonization approach to epothilones A and C.
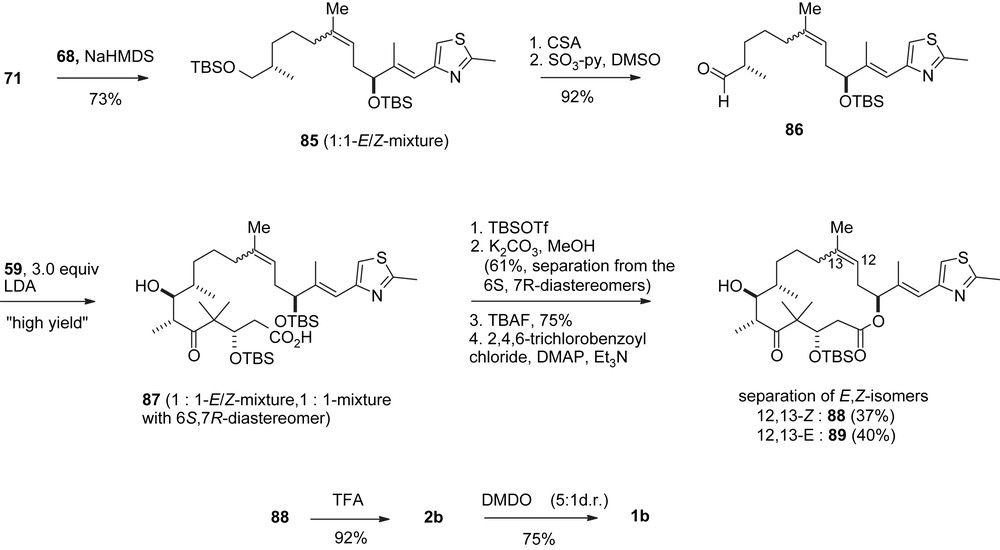
Nicolaou's first synthesis of epothilones B and D.
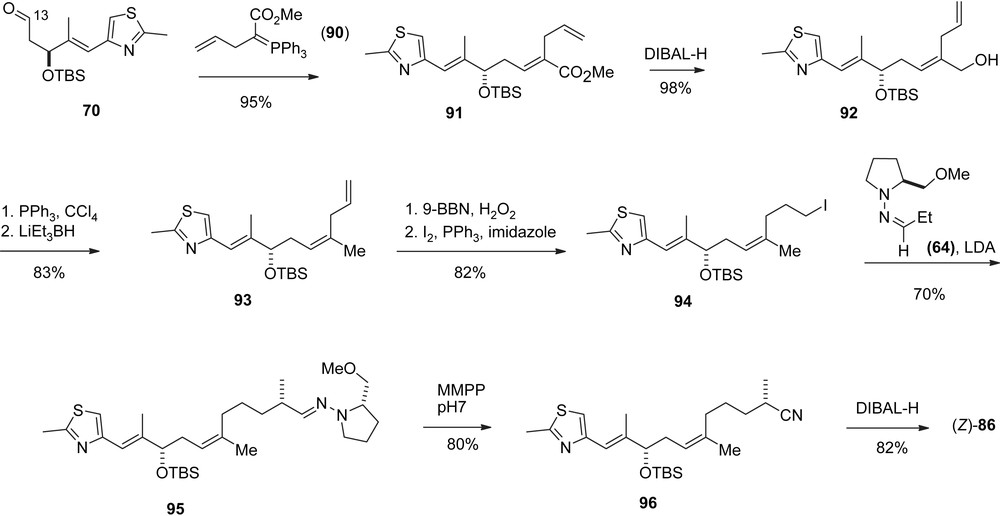
Nicolaou's synthesis of key intermediate (Z)-86.
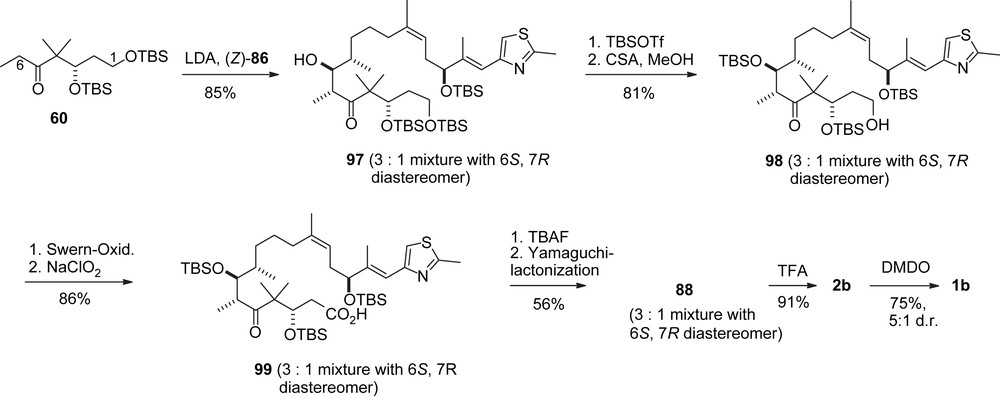
Nicolaou's second synthesis of epothilones B and D.
The preparation of the building blocks was different from the Danishefsky approach. Thus (Scheme 13), the C1–C6 fragment 59 was prepared via a regio- and enantiocontrolled Brown allylation of aldehyde 39 to form 57 after silylation. Ozonolysis led to 58 which was either oxidized to carboxylic acid 59 or reduced to diol 60. The C7–C12-aldehyde 63 was obtained via alcohol 62 by an alkylation with Oppolzer's sultam 61.
An alternative route to the C7–C12-fragment (Scheme 14) started with an Enders alkylation of 64 to form 65 enantioselectively, which was converted into aldehyde 67 (epothilone A series) and ketone 68 (epothilone B series), respectively. The C13–C21-phosphonium iodide 71 was prepared via a Brown allylation of aldehyde 5 to 6, from which 71 was obtained in five steps.
Nicolaou's first route to 1a [34] started with a non-stereoselective aldol type addition of the dianion of 59 to aldehyde 63 (Scheme 13). RCM of olefin 73a with Grubbs' first generation catalyst furnished an E/Z-mixture of the macrolides 74 which was separated and desilylated to give 2a. Epoxidation with DMDO as before led to 1a.
The RCM approach was also used for a solid phase supported synthesis of 2a (Scheme 16) [36]. In this modification, the RCM served to disconnect substrate 80 from the solid support by a cyclorelease process.
As an alternative to the RCM, Yamaguchi macrolactonization was also used for the synthesis of 1a, 1c (Scheme 17) [34]. Thus, Wittig reaction of phosphonium salt 71 with aldehyde 67 gave a 9:1-Z/E-mixture of olefins 81. Conversion into aldehyde 82 followed by aldol addition with keto acid 59 led to a 1:1 diastereomeric mixture of aldol adducts 83, which were separated, deprotected and cyclized to 84 by Yamaguchi lactonization.
A related protocol was also applied to the synthesis of 1b, 2b (Scheme 18) [35]. The only modification was the unselective Wittig reaction of 71 with ketone 68 to give 85 as a 1:1-E/Z-mixture which was converted to aldehyde 86.
To improve the stereocontrol of this sequence [37,38] aldehyde 86 was prepared as a pure (Z)-olefin (Scheme 10) via an E-selective Wittig olefination of aldehyde 70 with phosphorane 90.
Aldol addition of (Z)-86 with ketone 60 (Scheme 20) furnished a 3:1 diastereomeric mixture of 97 in favor of the desired 6R,7S-diastereomer. Without separation, the 7-OH group was silylated, the primary alcohol at C1 was deprotected selectively and oxidized to the acid 99. Selective deprotection of the 15-OTBS group followed by Yamaguchi lactonization gave 88 which was separated into the diastereomers. Deprotection gave 2b and epoxidation led to 1b eventually.
5.3 Schinzer synthesis
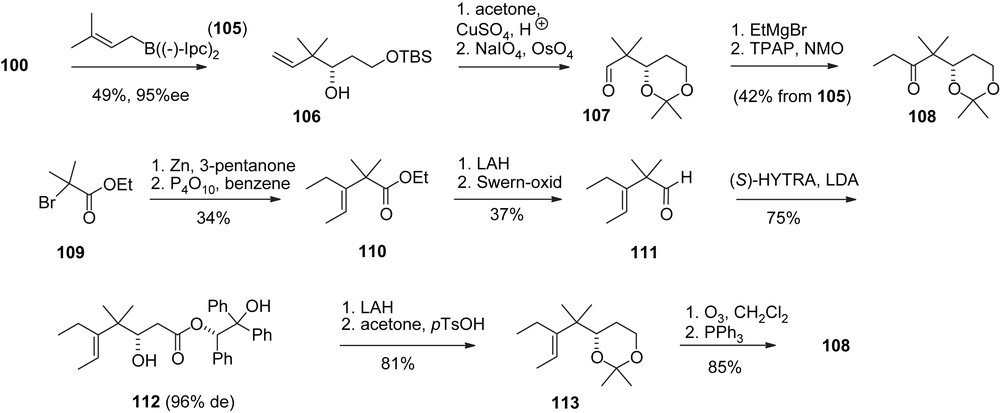
Schinzer's group was chronologically third to complete syntheses of both 1a, 1b and 2a, 2b. The preparation of the key fragments was different from the Danishefsky and Nicolaou approaches [39]. For instance the C21–C13-fragment 6 was synthesized from aldehyde 100 (Scheme 21). The C1–C5-acetonide 108 was available from 100 via Brown allylation or from a Reformatsky addition of bromoester 109 to 3-pentanone (Scheme 22).
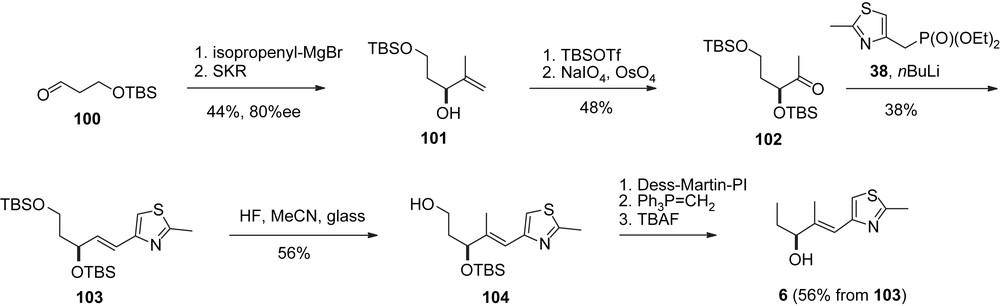
Schinzer's synthesis of epothilone A, part I.
Schinzer's synthesis of epothilone A, part II.
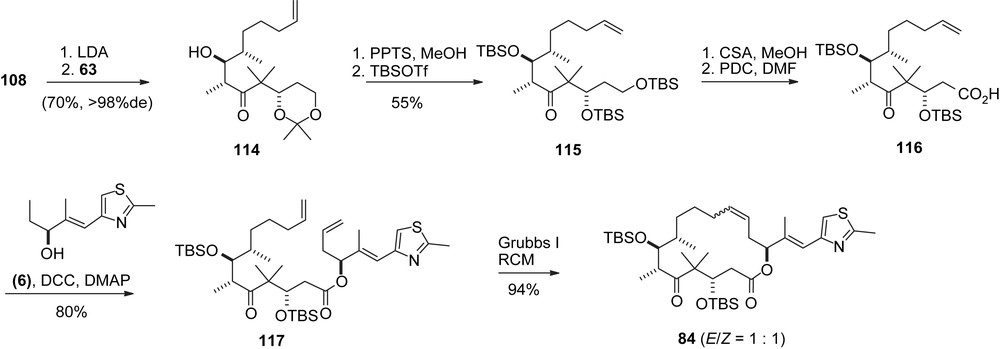
A major innovation was the acetonide moiety in 108 which turned out to be beneficial for the aldol addition to aldehyde 63 (Scheme 23). In contrast to Nicolaou's procedure a high selectivity in favor of adduct 114 was achieved. The rest of the synthesis [40] closely resembles Nicolaou's RCM approach.
Schinzer's synthesis of epothilone A, part III.
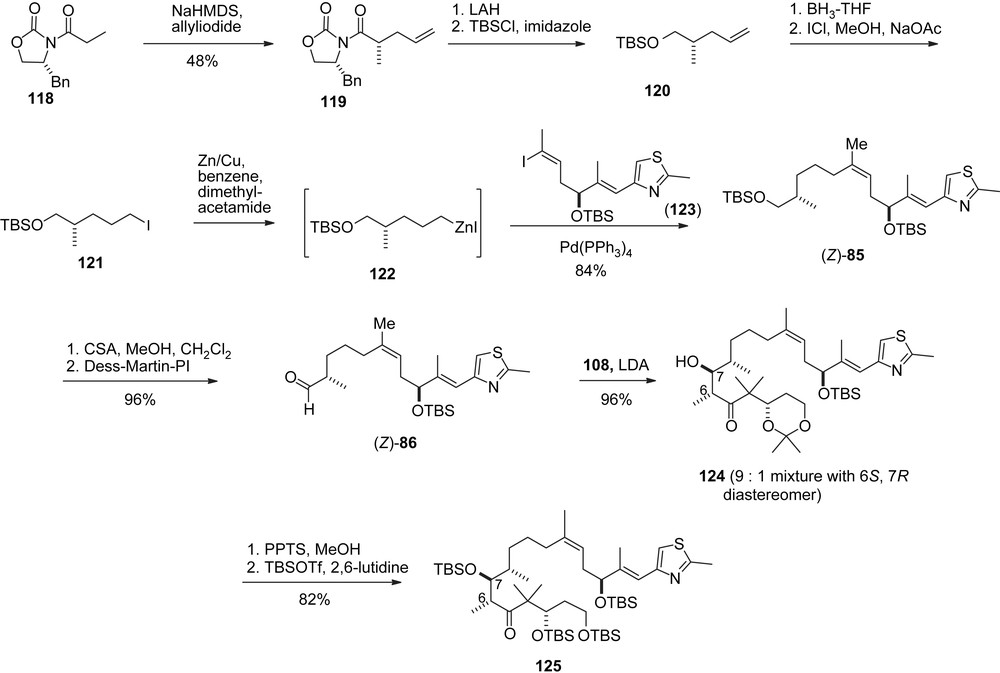
Schinzer's synthesis of 1b/2b (Scheme 24) [41] was based on a Zn-modification of Danishefsky's B-alkyl-Suzuki coupling to prepare pure (Z)-aldehyde 86. The aldol addition of (Z)-86 to ketone 108 led to adduct 124 with a d.r. of 9:1. Acetonide hydrolysis followed by global O-silylation delivered Nicolaou's intermediate 125.
Schinzer's synthesis of epothilone B.
5.4 Fürstner's alkyne RCM [42]
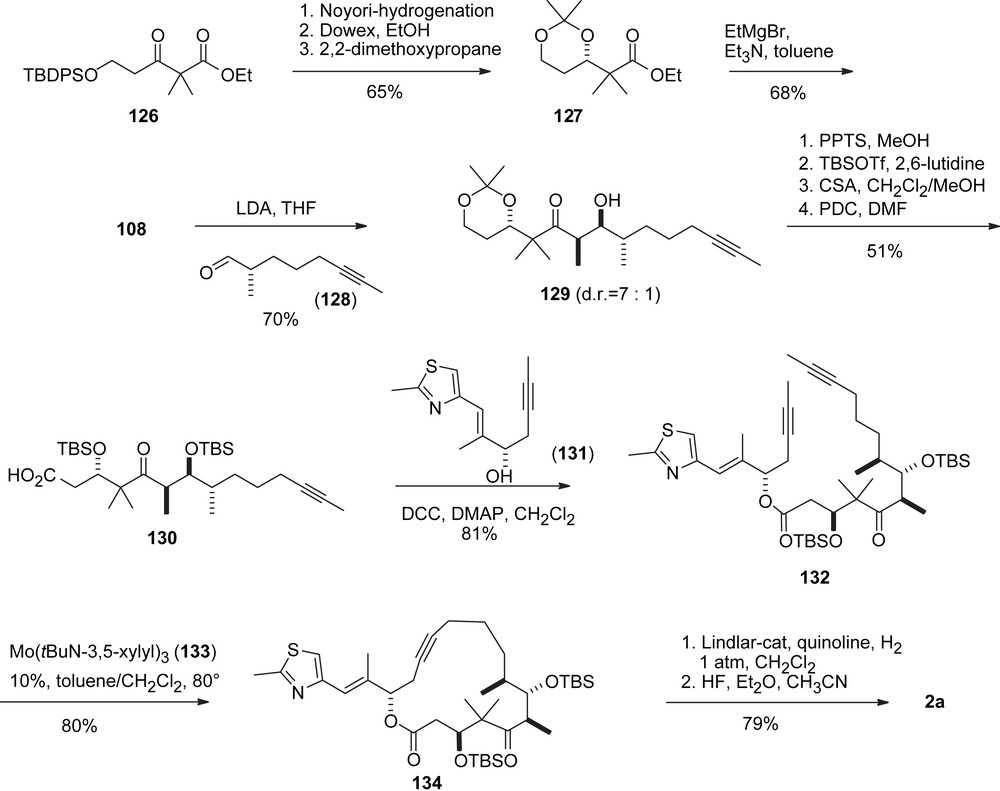
The early RCM approaches to 1 by Danishefsky, Nicolaou and Schinzer were all flawed by the missing E/Z-selectivity. In their synthesis of 2a, Fürstner et al. (Scheme 25) applied an alkyne RCM approach to generate macrolide 134 which was converted into the (Z)-olefin exclusively via Lindlar hydrogenation. The synthesis started with Schinzer's ketone 108, which was used for an aldol addition with aldehyde 128, to give adduct 129 with good selectivity. Esterification of acid 130 with alcohol 131, prepared from Nicolaou's intermediate 6 via Corey–Fuchs homologation to introduce the alkyne moiety, gave seco-intermediate 132. The RCM was achieved in good yield with Mo–catalyst 133.
Fürstner's alkyne metathesis approach to epothilone C.
5.5 Mulzer syntheses
The Mulzer group has published three different approaches to 1b, 2b. The first one (Scheme 26) [43], although it was carried through to the final target, is a formal one and aims for an easier access to Nicolaou's compounds 60 and (Z)-86 (Scheme 19). Mulzer's contribution lies in a facile introduction of the stereogenic center at C3 in ketone 60 via a Kiyooka type aldol addition [44], and a stereocontrolled synthesis of the (Z)-C12,13-olefin moiety in aldehyde (Z)-86. Thus, commercially available hydroxyl lactone 135 via hemiketal 136 and Wittig olefination with 137 gave alcohol 138. These steps have subsequently also been used by Ley [45] (Scheme 43). For the introduction of the (Z)-olefin a Still-Gennari olefination was employed to give enoate 139 which was converted to iodide 140. Sulfone 143 was obtained in good yield from Roche's ester 141 via tosylate 142. Alkylation of 143 with iodide 140 furnished 144 which was desulfonylated and converted to (Z)-86, in which both stereogenic centers at C15 and C8 have thus been derived from the chiral carbon pool.
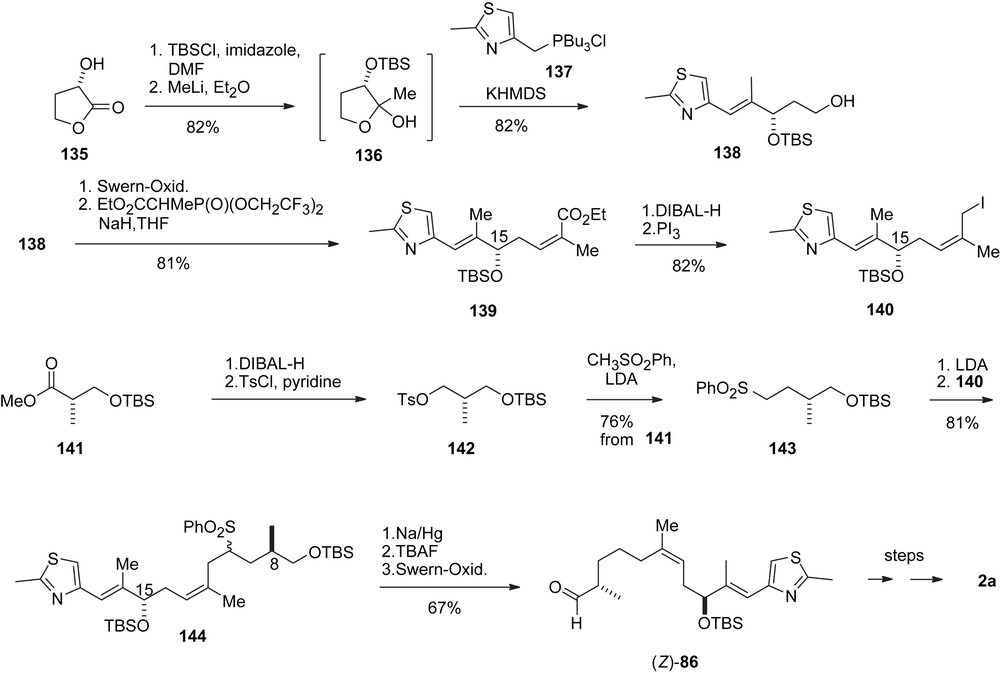
Mulzer's synthesis of epothilone D.
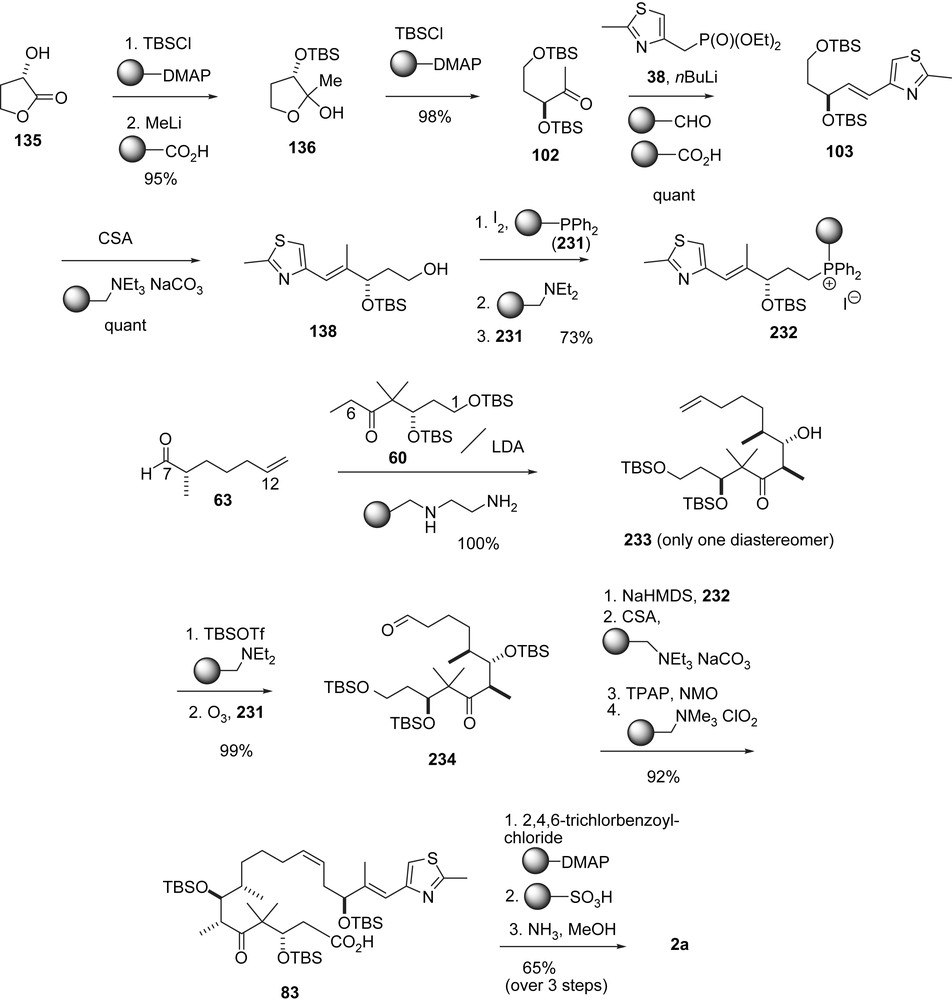
Ley's synthesis of epothilones A and C using immobilized reagent and scavengers, part II.
In an alternative approach to (Z)-86 [46] a silicon tethered RCM reaction of di-olefin 146, easily available from known alcohol 145 was employed (Scheme 27). Cycloolefin 147 was obtained as an easily separable 5:1-Z/E-mixture which was converted to aldehyde (Z)-86 as shown.
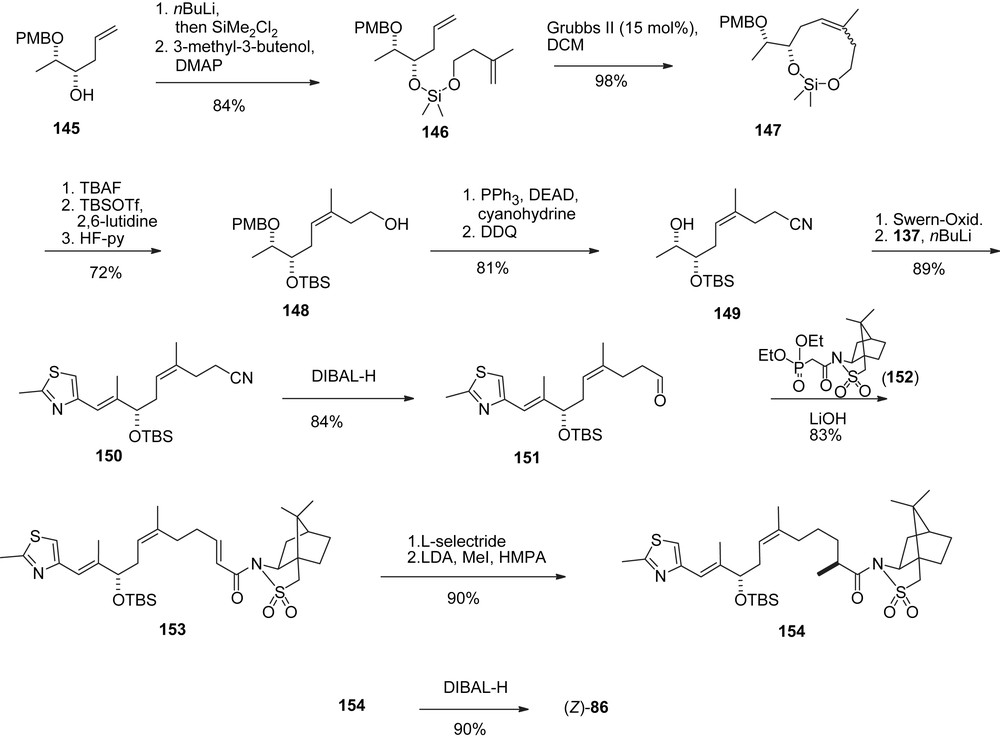
Mulzer's silicon tethered RCM approach to aldehyde (Z)-86.
Whereas these two approaches led to 2b first, the third approach furnished 1b directly (early epoxide approach, Scheme 28) [47]. By this concept, which was later adopted in Carreira's synthesis, the low-yielding nonselective and potentially hazardous epoxidation of 2b with peroxides was avoided. The synthesis started with the chain elongation of intermediate 155 to 156, and its conversion into ketone 157 which originally was obtained as an epimeric mixture. Base catalyzed epimerization gave diastereomerically pure 157. A chelate-Cram induced Grignard addition, followed by PMB deprotection and oxidation furnished tertiary alcohol 158 selectively from which mesylate 159 was easily available. Base induced cyclization gave the desired epoxide from which aldehyde 160 was obtained after ozonolysis. The conversion into 1b follows the established aldol addition–macrolactonization strategy.
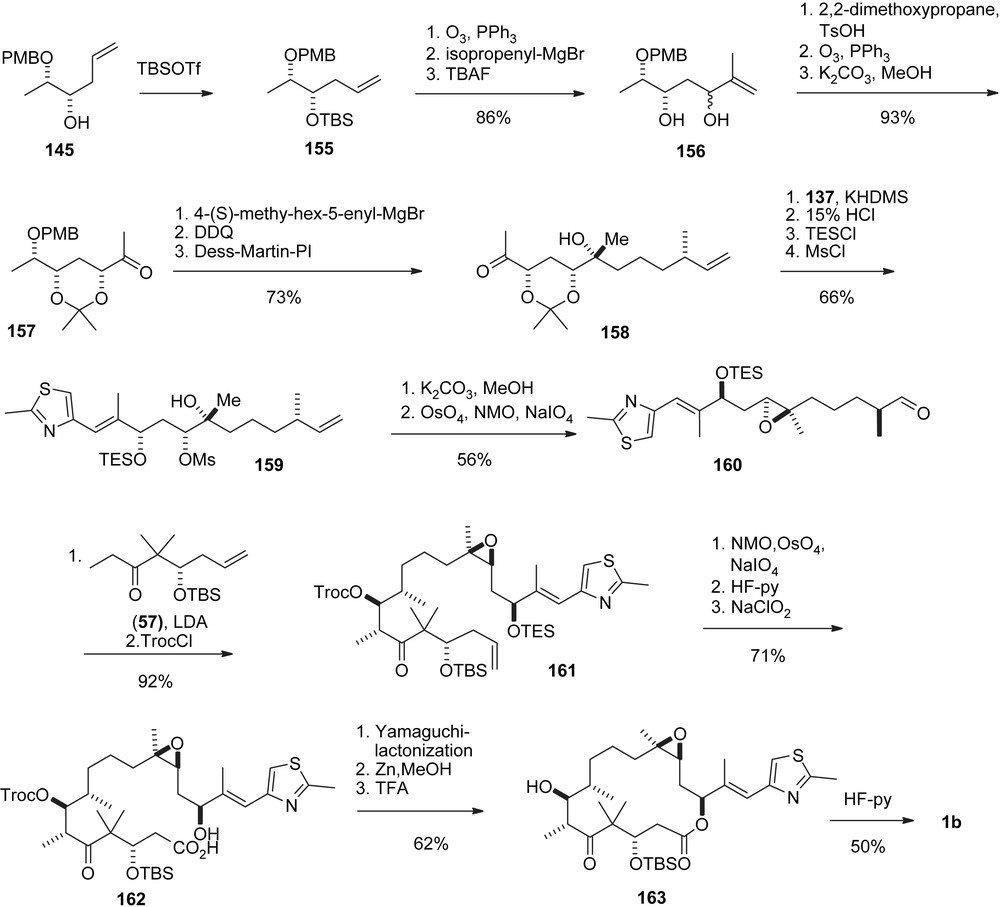
Mulzer's early epoxide approach to epothilone B.
5.6 Carreira's synthesis [48,49]
The Carreira group has also opted for Mulzer's “early epoxide” approach [47]. Moreover, they developed a highly innovative nitrile–oxide–olefin cycloaddition to establish the C12–C15 section in both the epothilone A and B series. Thus (Scheme 29), olefin 164 was oxidized to the aldehyde and treated with 3-methylbutyn-3-ol under asymmetric catalysis to give 165 with high selectivity. Additions of this type have been developed earlier in the group. Removal of the terminal tertiary alcohol and reduction gave the vinyl alcohol 166 which was subjected to a stereocontrolled 1,3-dipolar cycloaddition with nitriloxide 168, prepared in situ from oxime 167. The resulting isoxazoline 169 was olefinated with aldehyde 4 to give the C21–C7-epothilone A fragment 170a.
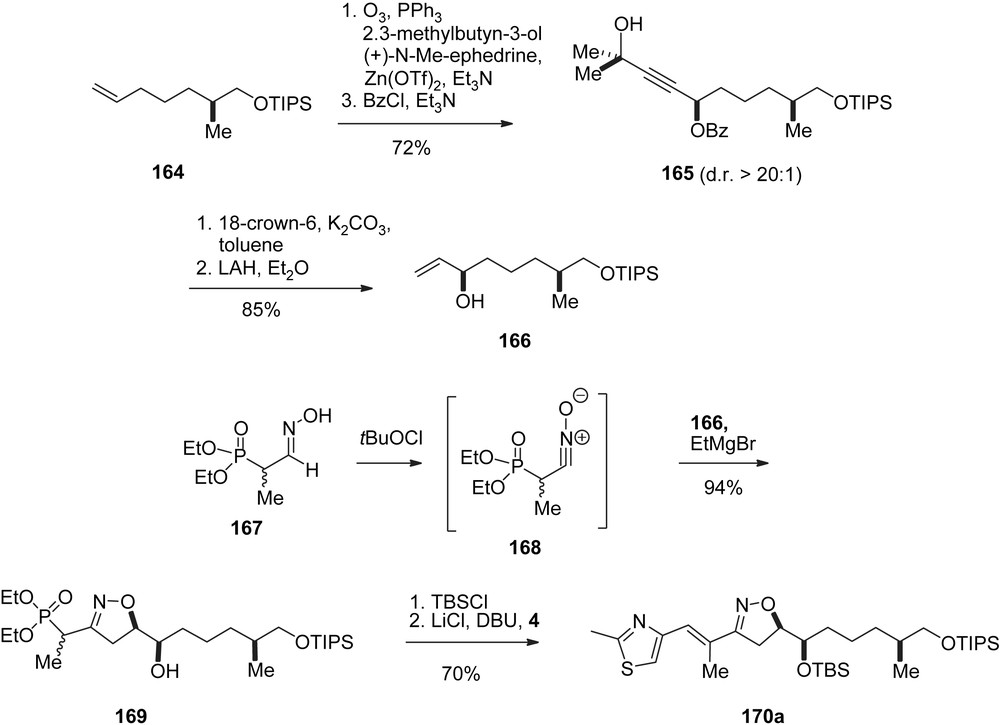
Carreira's synthesis of epothilones A and B, part I.
The corresponding epothilone B intermediate 170b (Scheme 30) was obtained starting from the addition of 168 to (R)-3-buten-2-ol. Adduct 171 was converted to ketone 173. Chelate-Cram induced addition of Grignard derivative 174 gave 170b selectively.
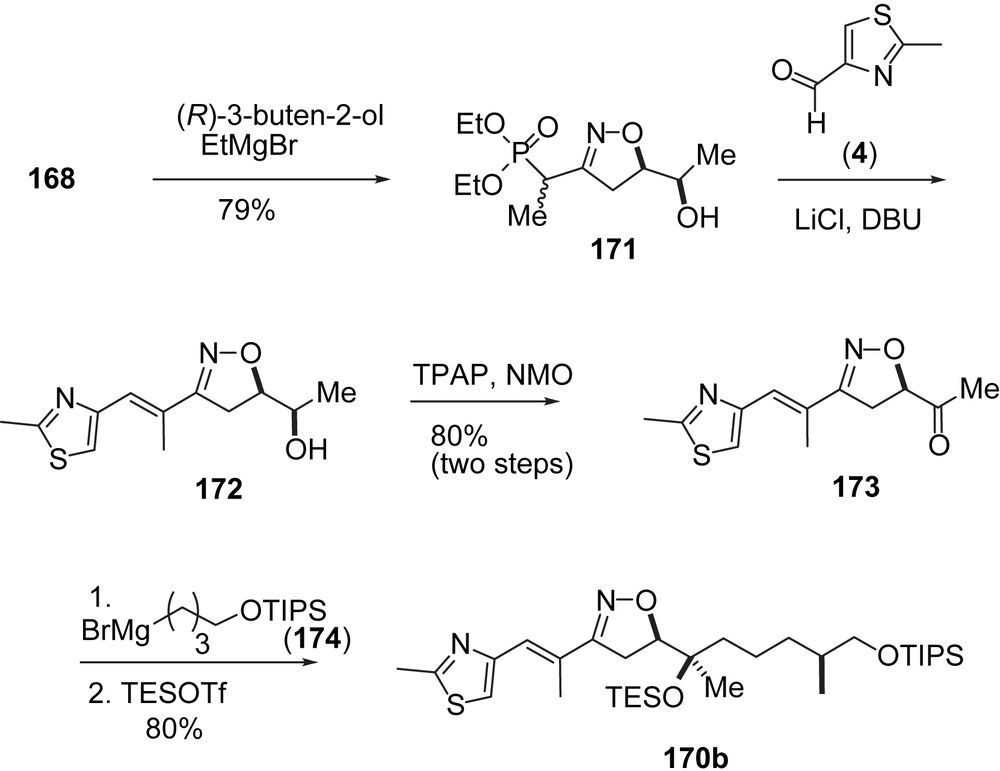
Carreira's synthesis of epothilone B, part II.
In parallel sequences (Schemes 31 and 32) intermediates 170a, 170b were processed towards the formation of 1a, 1b. Specifically, the isoxazoline ring was cleaved reductively to give triol 171, which was converted into epoxide 173 via the cyclic sulfite 172. Ditesylation followed by selective mono-detesylation and oxidation of the primary OH-function gave aldehyde 160, which underwent a highly selective aldol addition with ketone 57 to give aldol adduct 161 after Troc-protection of the resulting C7-alcohol function. The endgame was modelled after Mulzer's synthesis (Scheme 28) [47] to provide 163 via seco-acid 162 and macrolactone 174.
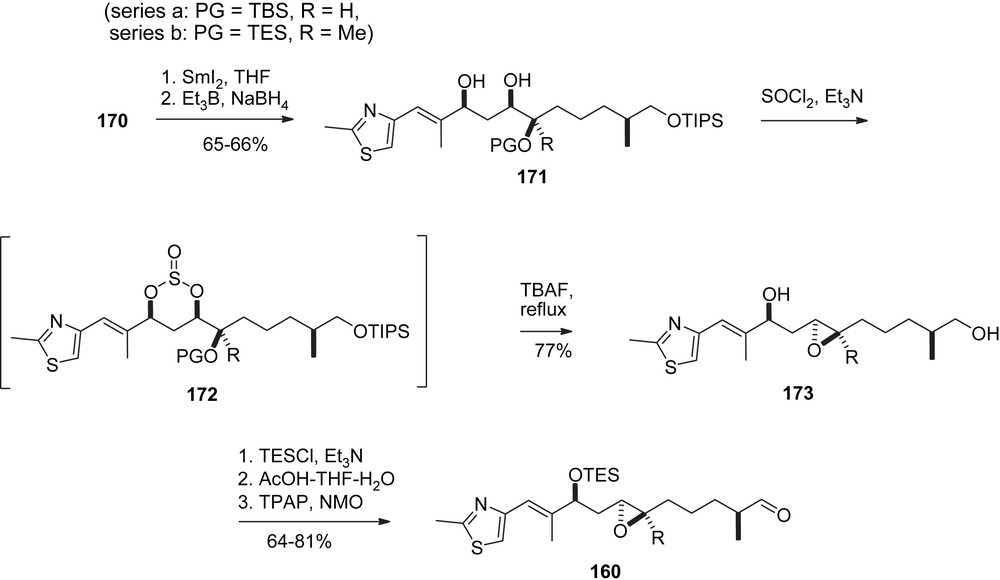
Carreira' synthesis of epothilones A and B, part III.
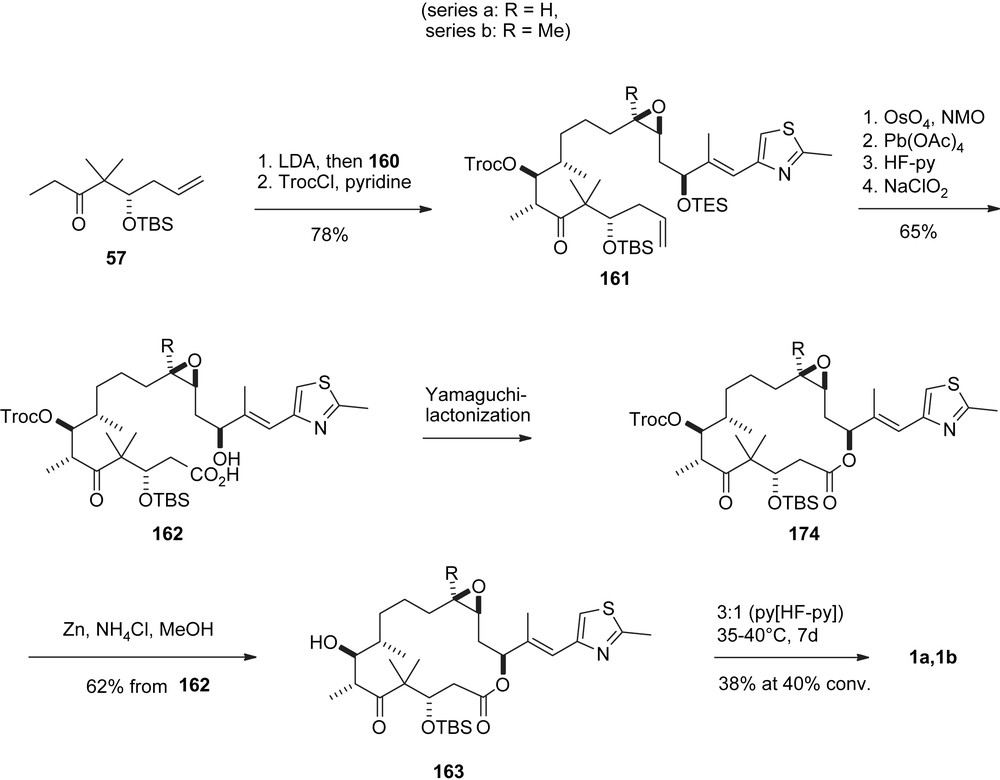
Carreira's synthesis of epothilones A and B, part IV.
5.7 Sinha syntheses [50–53]
In their synthesis of 2a, which incorporates modifications of both Nicolaou's RCM and Danishefsky's macrolactonization approach, the Sinha group has made use of “in-house” catalytic antibody methodology (Schemes 33 and 34) [50]. Thus, a racemic mixture of aldol adducts 176a, 176b was prepared and subjected to retro-aldol cleavage catalyzed by antibody AB 38C2. Enantiomer 176a was obtained in 96% ee. Conversely, aldehyde 7 was converted into adduct 177 via antibody catalyzed aldol addition with acetone, however, in low conversion and only 75% ee. Compound 177a was converted into the C1–C10-segment of 1 via catalytic hydrogenation to give a separable mixture of diastereomers 178, 179. Pure 178, after O-silylation and methylation, was subjected to an aldol addition to give after silylation and diastereomer separation intermediate 180 in unreported yield. The phenol ring was oxidized to the carboxylic acid and then converted into C1–C10-aldehyde 181 (Scheme 33).
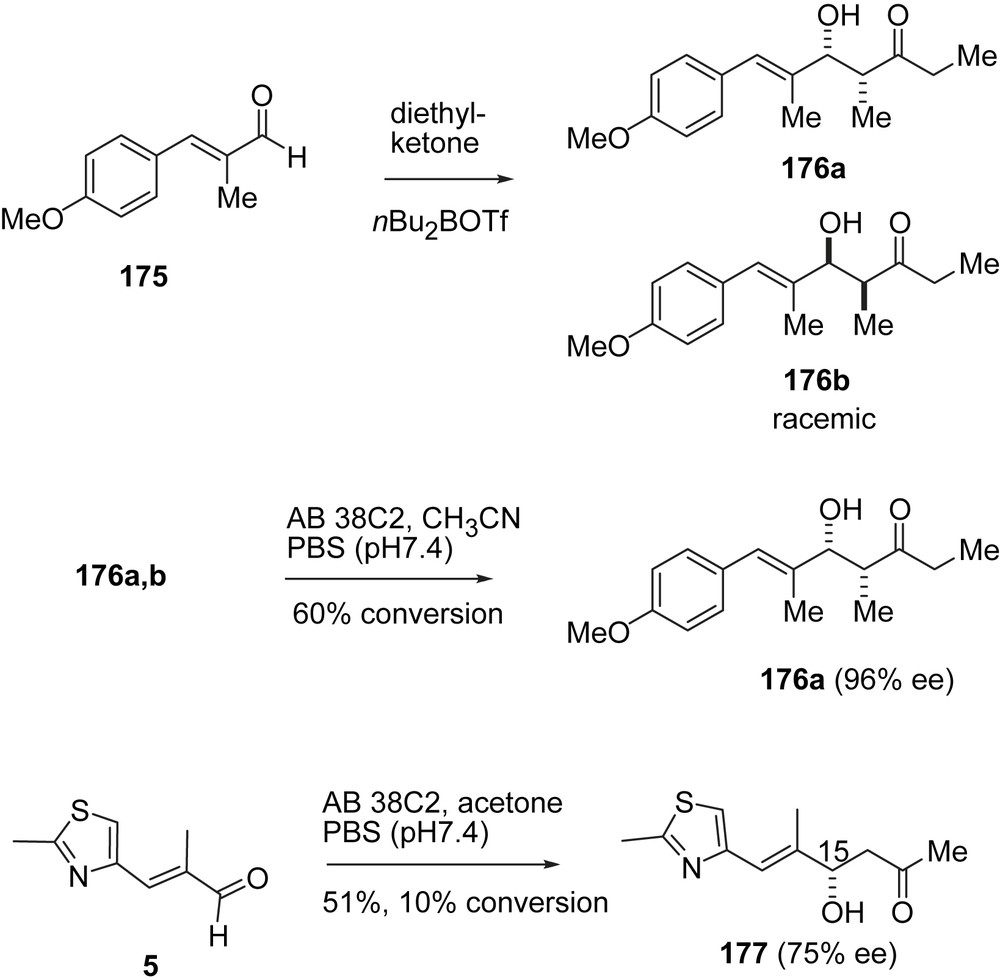
Sinha's synthesis of epothilone B, part I.
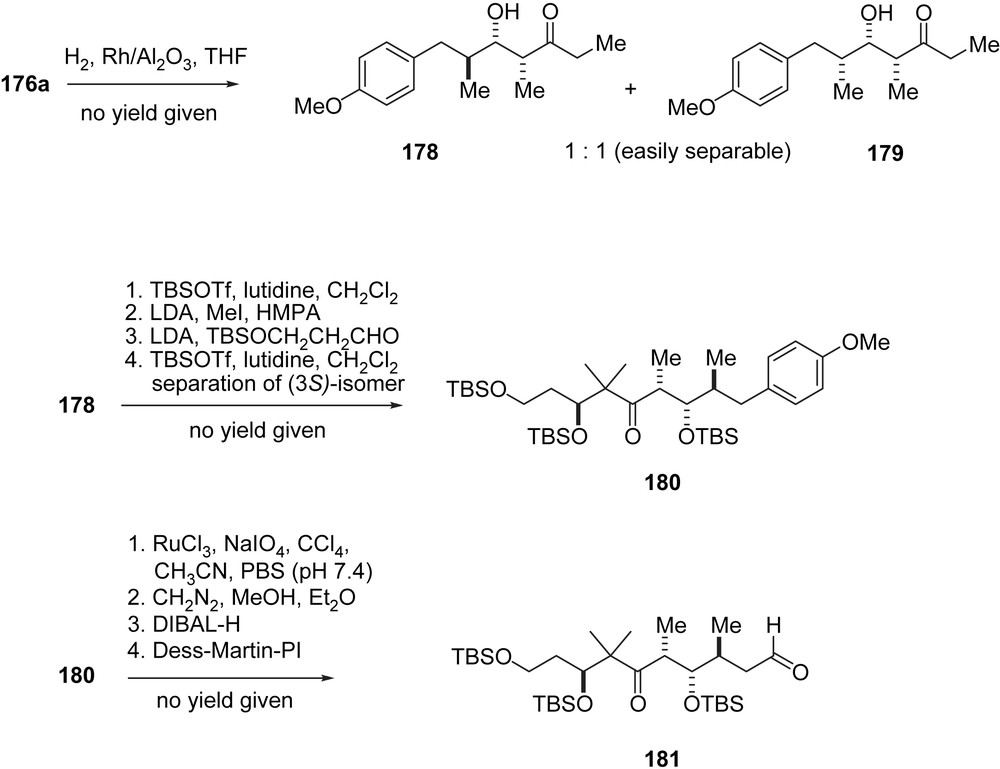
Sinha's synthesis of epothilone B, part II.
Via Horner olefination and hydrogenation 181 was transformed into ester 182, which was then elaborated via 183 into the olefinic carboxylic acid 184 (Scheme 35). Aldehyde 6 was prepared from 177 via enol ether 185 and hydroxyl ketone 186 and olefinated to alcohol 69. Esterification with acid 184 followed by RCM and desilylation led to 2a, in analogy to Nicolaou's earlier approach. Additionally, the Sinha group reported a synthesis of 1b along an “early epoxide approach” [53].
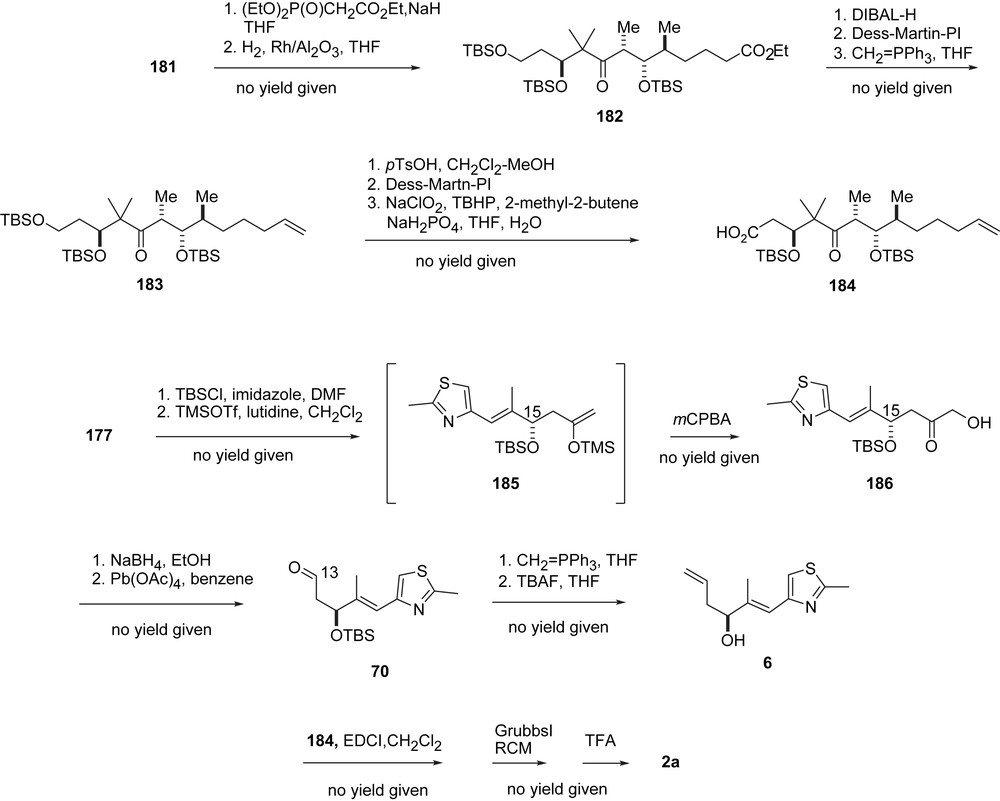
Sinha's synthesis of epothilone B, part III.
5.8 Shibasaki approach [54–57]
The Shibasaki group has developed a variety of chiral multifunctional catalysts (e.g. 187–189, Fig. 4) which they apply to natural product synthesis [54]. Their epothilone A/B synthesis (Schemes 36–39) is an adaptation of Danishefsky's B-alkyl-Suzuki reaction of vinyl iodides 9, 10 and terminal olefin 207 [55]. Thus (Scheme 36), aldehyde 5 underwent a highly enantioselective cyanosilylation with catalyst 187 [56] to form 190 which was converted into aldehyde 191 and then alkyne 193. Hydromagnesiation–iodination of 193 was used to generate Danishefsky's epothilone A vinyl iodide 9 stereoselectively. The preparation of the epothilone B vinyl iodide 10 [57] involves a homologation of aldehyde 191 to Schinzer's aldehyde 70 which was then elaborated into 10 following the Danishefsky/Schinzer precedence.
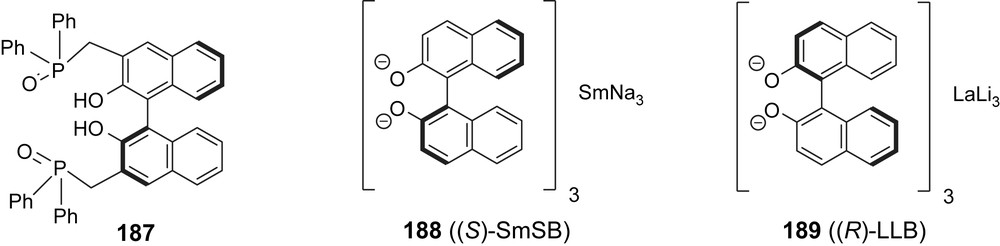
Asymmetric catalysts from the Shibasaki group.
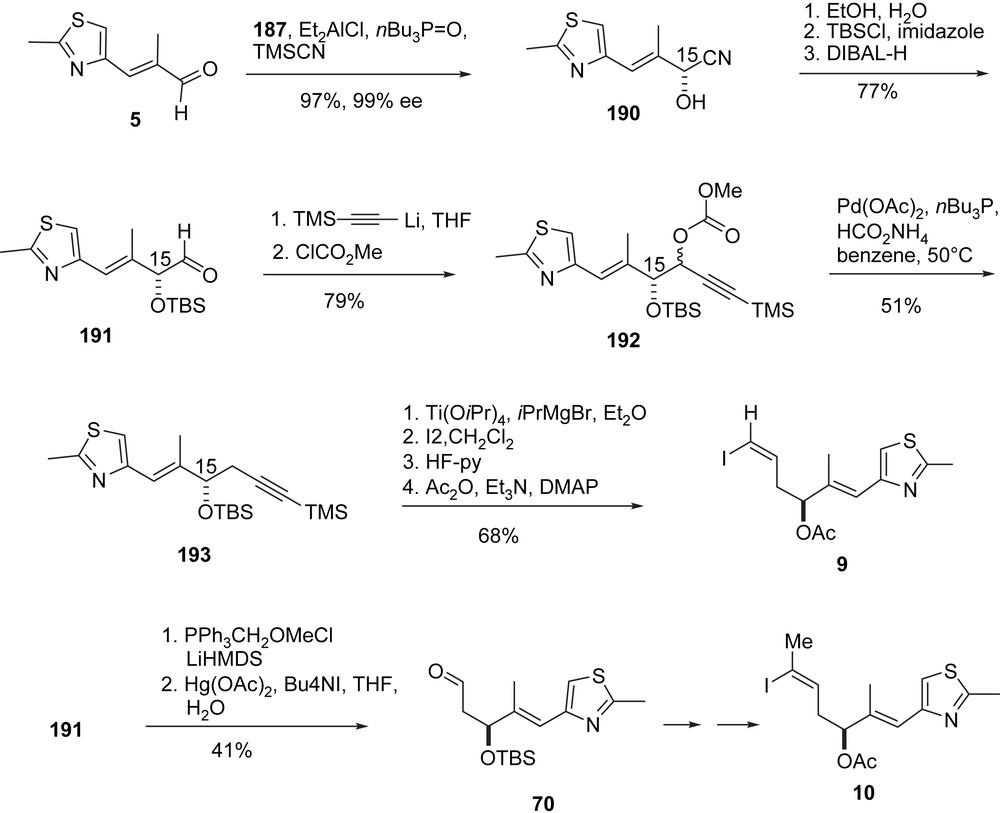
Shibasaki's synthesis of epothilones C and D, part I.
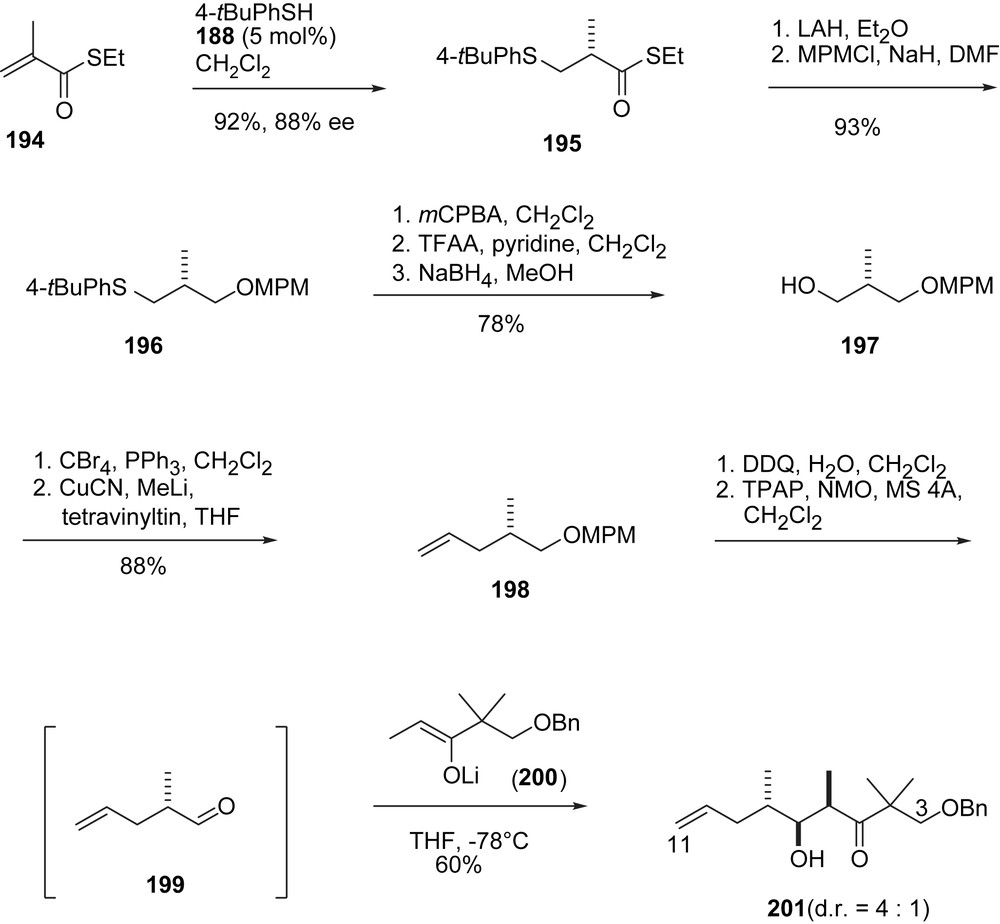
Shibasaki's synthesis of epothilones C and D, part II.
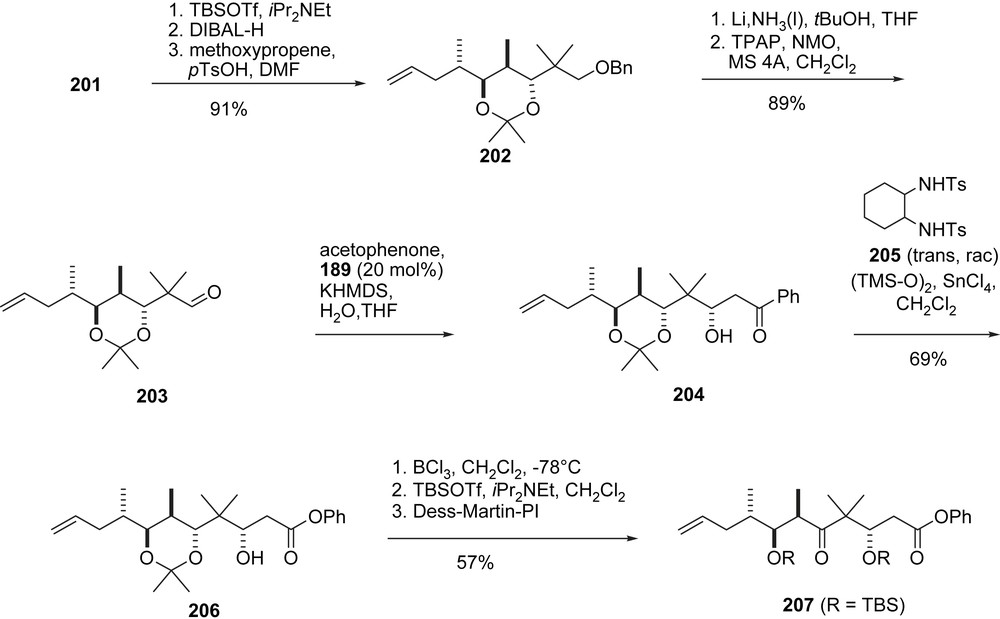
Shibasaki's synthesis of epothilones C and D, part III.

Shibasaki's synthesis of epothilones C and D, part IV.
The synthesis of 207 (Schemes 37 and 38) started with a 1,4-addition of 4-t-BuPh–SH to thiol ester 194 under the enantiocatalysis of 188 to give 195. Reduction of the thiol ester and O-protection gave thioether 196, which was converted into 197 via Pummerer oxidation–reduction. Chain elongation to olefin 198 was followed by MPM deprotection and oxidation to the labile aldehyde 199 which was added to enolate 200 to give the epothilone A/B C3–C11-fragment 201 (Scheme 37). Standard methodology was used to convert 201 into acetonide 202 and then aldehyde 203 (Scheme 38). Aldol addition with acetophenone under the catalysis of 189 gave phenyl ketone 204, which was converted to phenyl ester 206 via a novel Baeyer–Villiger oxidation, catalyzed by diamide 205. In three additional steps ketone 207 was prepared, which was the coupling partner of 9 and 10 to give the 2a-precursor 208 and the 2b-precursor 209, respectively (Scheme 39).
5.9 Synthesis by E.J. Thomas [58]
Thomas' contribution lies in a novel stereoselective approach to the (Z)-12,13-double bond (Schemes 40 and 41). The sequence started with an alkylation of sulfone 211 with iodide 210 to give 212 as a mixture of diastereomers. Deprotection and reprotection via 213 followed by an SR2′ addition of tributylstannane gave 214, which was added to aldehyde 215 under twofold allylic Thomas rearrangement to give the homoallylic alcohol 216 as a diastereomeric mixture, however, with purely (Z)-olefin geometry. From 216, the superfluous OH-function was removed in a Barton–McCombie sequence to furnish 217, which was converted into alcohol 218 and then ketone 219 by conventional methodology. Usual Horner olefination with 38 followed by deprotection and oxidation gave aldehyde 220, which is identical with Nicolaou's intermediate (Z)-86 except for the 15-OSEM protecting group. Aldol addition with ketone 221, prepared from pantolactone analogously to Schering's synthesis of sagopilone (Schemes 44 and 45) gave adduct 222 with high diastereoselectivity. The rest of the synthesis is essentially identical to Nicolaou's precedence (Scheme 18).
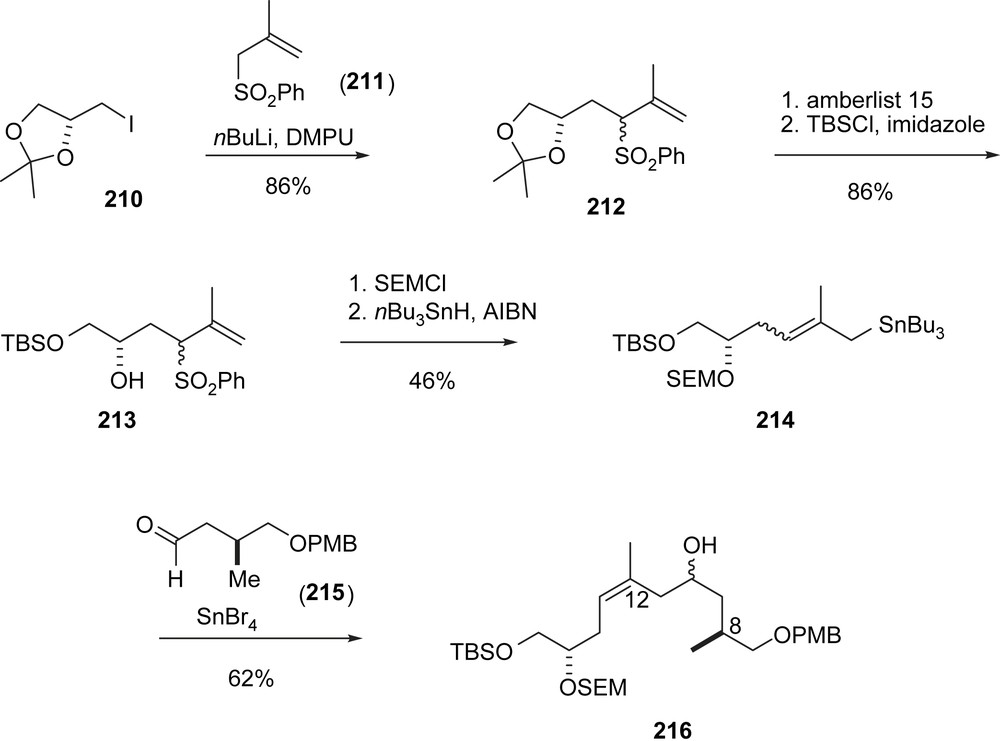
E.J. Thomas's synthesis of epothilone D, part I.
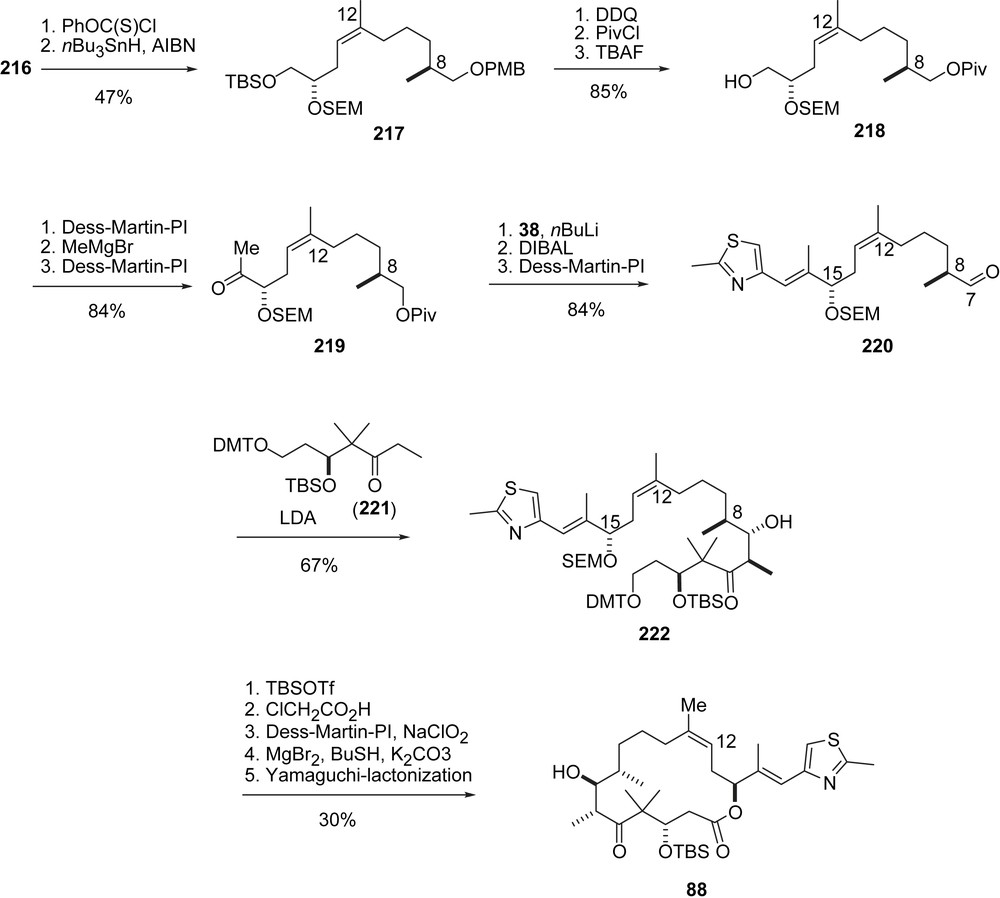
E.J. Thomas's synthesis of epothilone D, part II.
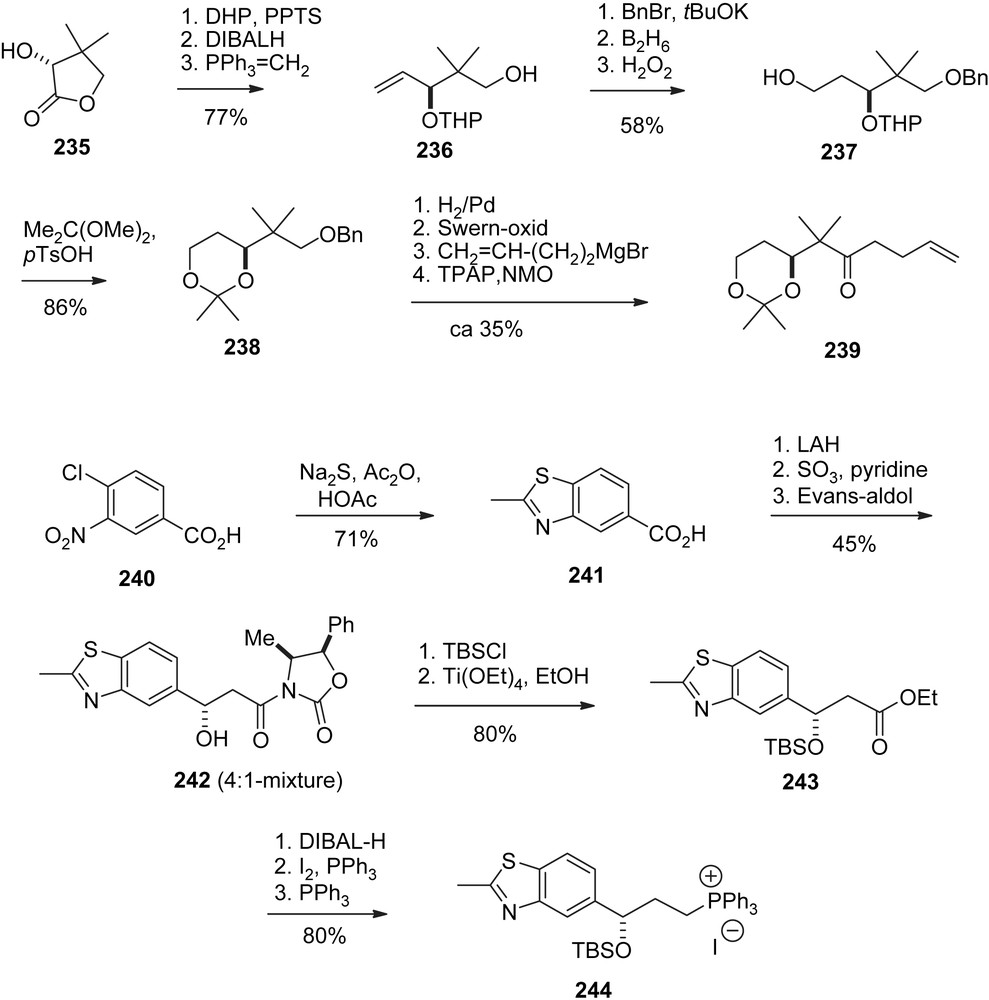
Schering synthesis of ZK-Epo (sagopilone), part I.

Schering synthesis of ZK-Epo (sagopilone), part II.
5.10 Ley's approach [45]
In pioneering studies to improve large-scale preparations of complex natural products, the Ley group focused on the use of immobilized reagents and scavengers. This philosophy is reflected in an approach to 1a in which (Schemes 42 and 43) the key steps are the aldol addition of ketone 60 to aldehyde 63 to form 233 and the (Z)-selective Wittig reaction of aldehyde 234 and the ylide generated from the immobilized phosphonium salt 232. The preparation of ketone 60 and fragment 103 is adapted from Mulzer's synthesis of 1b [43] and the Wittig reaction between 232 and 234 stems from Nicolaou's synthesis of 1a (Scheme 17). Ley's achievement lies in the fact that the acids, bases and the diphenyl phosphine previously required for catalyzing reactions, destroying intermediates and isolating reaction products are fixed to a solid support. In this way, aqueous workup, filtration or chromatography are largely avoided and the yields are extremely high, in most steps nearly quantitative.
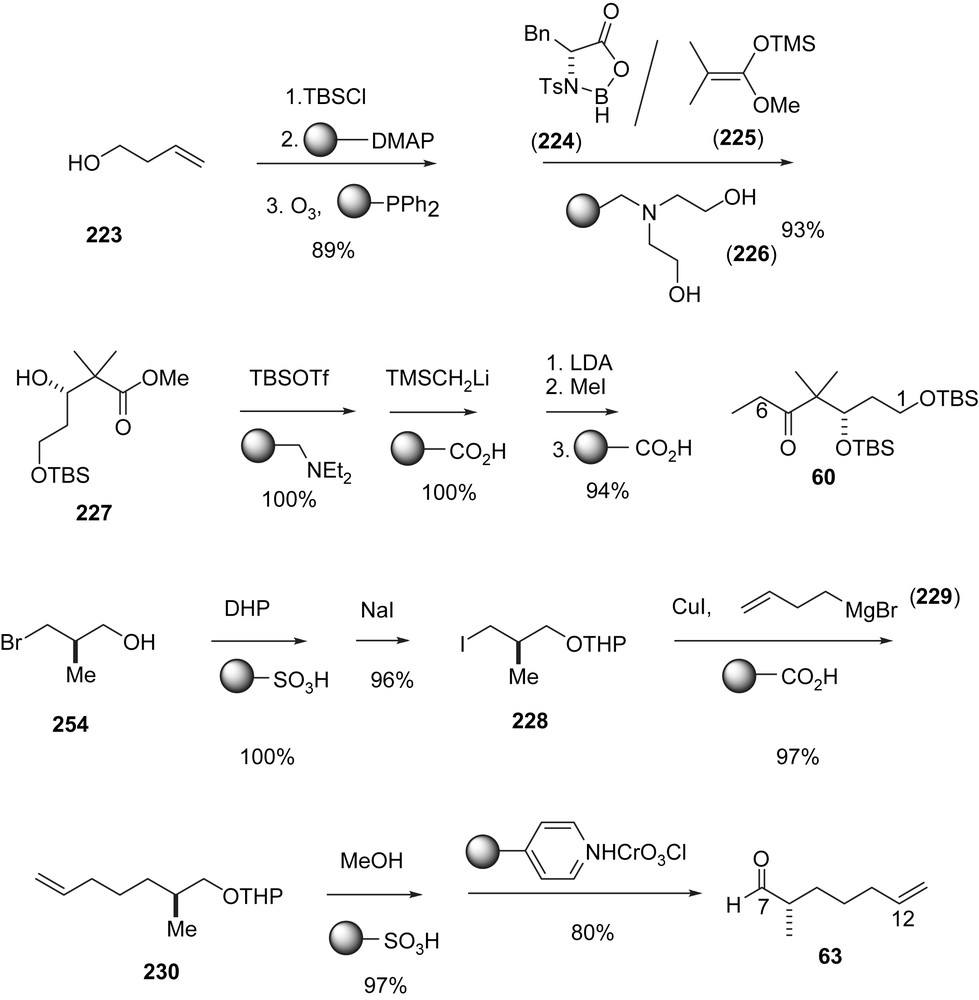
Ley's synthesis of epothilones A and C using immobilized reagent and scavengers, part I.
5.11 Synthesis of epothilone analogs
For SAR studies (see Section 6) a broad variety of epothilone derivatives were desirable. From early on, the Nicolaou group, together with the Danishefsky group, applied their synthetic approach for structural variations in essentially all sections of the epothilone skeleton. From the methodological viewpoint basically little new was developed. Thus it may suffice to mention two representative examples, one from the Nicolaou group [59] which was used to generate highly active epothilone B analogs [60], and one from the Danishefsky manifold [61] (trifluorodehydroEpoB, fludelone).
5.12 Conclusion and an industrial application (ZK-Epo (sagopilone)) [62]
The total synthesis of the epothilones has undoubtedly been one of the largest projects ever in the history of organic chemistry. At first sight relatively little new methodology has emanated from these huge worldwide efforts. However, several useful novelties may be gleaned: application of RCM reactions to a rather complex substrate, all kinds of stereoselective approaches to generate a (Z)-tri-substituted double bond, long range effects on stereoselective aldol additions that connect oxygenated fragments, the robustness and generality of Yamaguchi macrolactonization, and last but not least stereo- and regioselective epoxidation with DMDO and the stability of epoxides towards a variety of aggressive reagents.
In fact, many of the academic approaches have been transferred to the kilogram scale industrial synthesis of ZK-Epo (253, sagopilone) [62], a highly potent unnatural analog of 2a, which is in Phase II clinical trial (see Section 7.6) (Schemes 44 and 45). The synthesis essentially is a hybrid of Schinzer and Nicolaou components based on three major fragments 239, 244, and 248. The Wittig olefination of ketone 250 and phosphonium salt 244 furnishes a 1:1-E/Z-mixture of 249. After chromatographic separation the undesired E-olefin is recycled by photochemical cis–trans-equilibration. The aldol addition of Schinzer's ketone 239 and aldehyde 250 gives adduct 251 with “good selectivity”, yet only 64% yield. The endgame follows the routine Yamaguchi lactonization, desilylation, and DMDO epoxidation (selectivity 7:1) protocol. The overall sequence consists of no less than 44 steps, all told. In this respect, the synthesis is an impressive achievement and emphatically demonstrates that multistep synthesis is not a purely academic playground, but can be performed on an industrial scale if so desired.
6 Semisynthetic derivatives
For biologically active structurally complex natural products the chemical derivatization of material isolated from natural sources often represents the only feasible means to explore structure–activity relationships (SARs) and to produce analogs with more favorable pharmacokinetic and pharmacological properties [63–65]. Typically, six out of seven epothilones (see Section 7) that have entered clinical evaluation in humans so far (including the natural product epothilone B (1b)) are made by derivatization, and only one is produced by total chemical synthesis (253, sagopilone; see previous section).
Lactam-based epothilone analogs are metabolically more stable than the parent natural macrolactones [66,67]. Therefore, the BMS (Bristol–Myers–Sqibb) group has developed a route capitalizing on the reaction of an allylic Pd–π complex with a nitrogen nucleophile. Thus, the treatment of 1a, 1b with NaN3 in the presence of Pd(PPh3)4 leads to azido acids 255a and 255b, respectively, with full retention of configuration at C15 (Scheme 46).
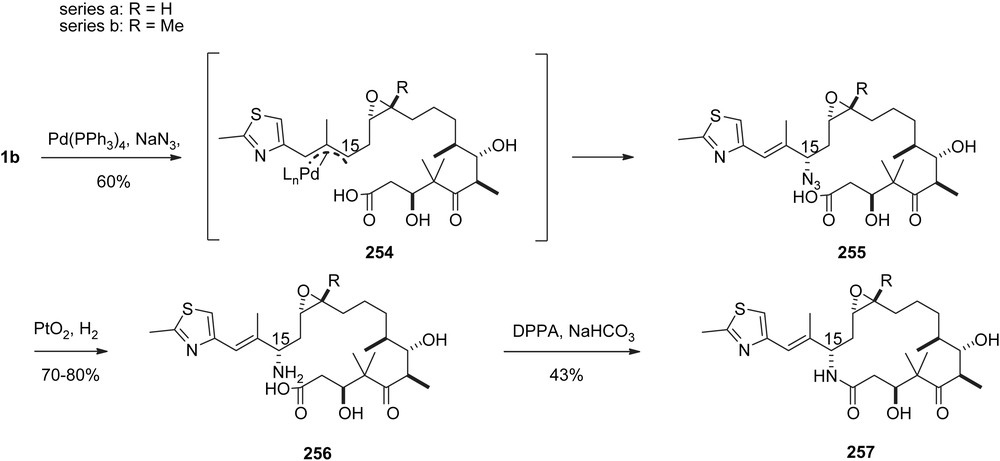
BMS synthesis of Aza-EpoB.
Reduction of the azide group followed by cyclization furnished the lactam analogs of EpoA and B, 257a and 257b.
A second modification refers to the C21 position through Polonovski rearrangement [68] of N-oxide 258 and further transformation of the ensuing C21-trifluoroacetoxymethyl group [69,70] to 259 and 260 (Scheme 47).
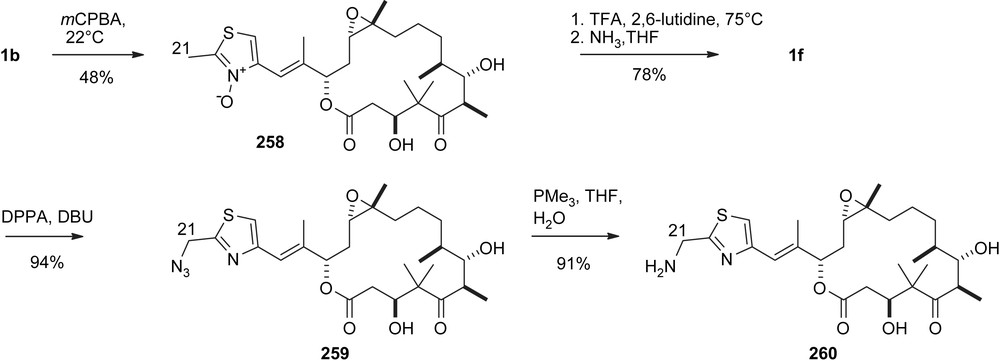
Modification of C21.
7 Clinical data
For seven epothilone-based drugs (Scheme 48) clinical trials have been performed [71–73].

Epothilones in clinical evaluation.
7.1 Patupilone (EPO906, 1b)
Patupilone was the first epothilone to enter clinical trials [74]. In Phase II the most promising data have been obtained in prostate, ovarian, and NSCL (non-small-cell lung cancer) patients. In particular, good results were observed for platinum- and taxane-resistant ovarian carcinomas and platinum-pretreated NSCLC [75].
7.2 Ixabepilone (261)
The BMS–epothilone B-lactam 261, now called ixabepilone, received FDA approval for the treatment of metastatic or advanced breast cancer on October 16, 2007 [76]. Tumor types that have been investigated with 261 include breast, prostate, colorectal, non-small-cell lung, gastric, hepatobiliary, gynaecological, and pancreatic cancers. In addition, studies have been conducted for the treatment of sarcoma, melanoma, and non-Hodgkin's lymphoma [77].
7.3 KOS-862 (EpoD, 2b)
Several Phase I trials have been reported for KOS-862 (R1492, 2d, deoxyepothilone B) [78,79]. Although tumor shrinkage was observed in these studies in two patients with large cell and mediastinal B-cell lymphomat the clinical development has been terminated [80]. Lately, Phase II trials have been reported [81].
7.4 BMS-310705 (262)
As a back-up for 261, two Phase I studies with BMS-310705, which is more water-soluble than 261, have been reported in abstract form [82–84]; no Phase II data are (publicly) available for the compound at this point. Overall, the toxicities associated with BMS-310705 treatment are similar to those observed for 261 [85,86].
7.5 KOS-1584 (263)
Interim results of ongoing Phase I trials with KOS-1584 (R1645) have been reported [87–89]. No recommended Phase II dose has yet been defined.
7.6 Sagopilone (ZK-Epo (sagopilone), 253)
Sagopilone, from Bayer–Schering, is the only fully synthetic epothilone analog so far in Phase I/II clinical development [90,91]. As communicated by the company, 253 is currently evaluated “in an extensive program of clinical Phase II studies in various oncological indications”… At the time of reporting 63 patients had entered the study, with data being available on 30 patients. The trial was still ongoing at the time of reporting.
8 Conclusions
As yet, data from more than 20 Phase II trials with four different epothilones (patupilone, ixabepilone, KOS-862, sagopilone) have been reported. Measurable antitumor activity has been observed in metastatic breast cancer, ovarian cancer, and hormone-resistant prostate cancer, with sporadic activity in colon cancer. So far, however, only ixabepilone has received FDA approval for the treatment of metastatic or advanced breast cancer.
1 Crystallographic data of the structure have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no CCDC-241333 and CCDC-241334. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44)1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).