

1 Introduction
It is now well-recognized that natural products have directly or indirectly contributed to the discovery and development of as much as 75% of our current treatments for cancer and infectious diseases [1]. In addition, Lipitor, a single example of the statin class of antilipidemic drugs was not only the top-selling drug in U.S. Markets, but it was also derived from a natural product, compactin. Despite this impressive track record, natural products are no longer an in-house discovery vehicle for major pharmaceutical companies. One rationale for this could be an apparent desire to find lead compounds for more diverse indications. However, pharmaceutical companies have themselves articulated a strong belief that “promising leads come faster and more frequently out of combinatorial chemistry and synthetic techniques” [2]. Unfortunately, the importance of chemical diversity in the discovery of new therapeutic agents cannot be underestimated and natural products have been shown to fill a significantly greater percentage of chemical diversity space than synthetic libraries [3].
Material supply issues can significantly hamper rapid therapeutic development of natural product lead compounds. The development of Taxol® is an excellent example [4]. However, despite large investments of funds in search of a synthetic solution, total synthesis efforts resulted in little more than an academic exercise. This is in stark contrast to recent efforts in the evaluation of the sponge-derived polyketide discodermolide. When it became clear that isolation of natural material was not an adequate source of material, Novartis produced 50 g of synthetic discodermolide [5]. The structural differences between Taxol® and discodermolide were the key determinant of the result of each effort. Methods for the production of synthetic fragments related to polyketides, such as discodermolide, have evolved to the point where even the most complex of these structures can be prepared on a scale necessary for full biological evaluation. Moreover, advancement in the characterization and genetic engineering of polyketide synthase gene clusters can provide an alternative, manufacturing scale source of material through heterologous expression or analogues through precursor-directed and combinatorial biosynthesis [6–8].
Peloruside A is an excellent example of a marine derived polyketide with significant therapeutic potential. It was isolated from a marine sponge, Mycale hentscheli, found off the coast of New Zealand [9]. Peloruside A showed potent cytotoxicity (18 nM) against P388 murine leukemia cell line. In contrast to many stereochemically rich polyketides, peloruside A 1 is highly oxygenated with limited sites of methyl substitution, Fig. 1. While solution NMR studies provided the relative stereochemistry of the compound's 10 stereogenic centers, the absolute stereochemistry remained unknown until a recent total synthesis of the ent-peloruside A by the De Brabander group at the UTSW Medical Center [10]. The first total synthesis of the naturally occurring enantiomer appeared shortly after [11].
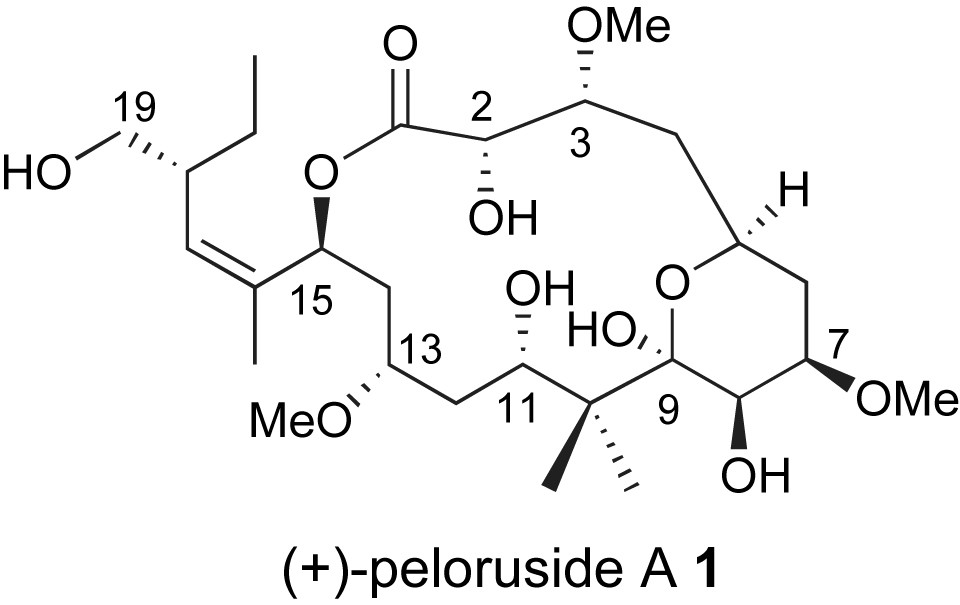
Structure of peloruside A.
In 2002, Miller and coworkers reported a study which established peloruside A as a potent cytotoxic agent with paclitaxel-like microtubule-stabilizing activity [12]. Recently, it has been reported that peloruside A appears not to bind to the Taxol®-binding site on β-tubulin [13]. In fact, laulimalide was able to displace peloruside A and thus these compounds appear to have similar binding properties. In fact, synergistic effects have been observed with taxoid site drugs including Taxol®, epothilone, discodermolide, eleuthorobin, and others [14]. Transfer-NOE experiments in the presence of microtubules have provided insight into the bound-conformation of peloruside A and docking experiments identified a potential binding site on α-tubulin [15]. However, recent experimental efforts using hydrogen–deuterium exchange mass spectrometry supported an alternative binding site [16]. Based on the significant in vivo activity, Reata Pharmaceuticals has initiated advanced preclinical development [17].
Page and coworkers have investigated the environmental influences contributing to the metabolite production of Mycale hentscheli [18]. Their efforts concluded that an aquaculture program designed to harvest compounds such as peloruside A must carefully control a number of biological factors which trigger metabolite production. Thus, prior to the identification and characterization of the gene cluster responsible for peloruside A production, total synthesis may play an important role in meeting the material supply requirements of early clinical evaluation. This review will cover synthetic efforts towards the synthesis of peloruside A with an emphasis on overall synthetic strategy. De Brabander's [10] and Taylor's [11] efforts have been previously included in a detailed review of recent total syntheses of marine natural products and will only be summarized herein [19].
2 Total synthesis
Late-stage macrolactonization is a common element in the synthetic strategies towards naturally occurring macrolides. Both total synthesis efforts successfully exploited this process through cyclization of highly functionalized seco-acids thus taking full advantage of conformational preorganization. In the De Brabander lab, after saponification of C1-methyl ester 2, exposure of allylic alcohol to classic Mitsunobu conditions [20] followed by acidic deprotection of the C2-methoxymethyl, C8- and C19-silyl ethers provided ent-peloruside A 3 through a clean inversion process, Scheme 1 [10]. Interestingly, related cyclizations of the diastereomers resulting from epimeric pairs at C13 and C15, uncovered mechanistic differences including an ionization pathway due to the allylic nature of the C15-activated intermediate [21]. These issues offer additional support for the influence of stereochemistry and conformation on the efficiency of biomimetic lactonizations [22].
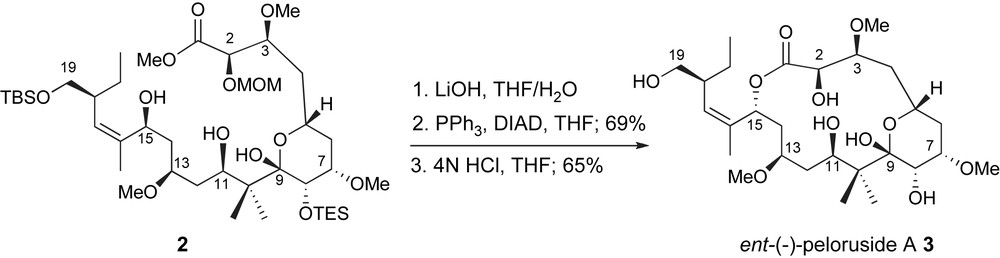
De Brabander macrolactonization to ent-peloruside A.
The generation of the C5–C9-pyran represented a critical milestone of the De Brabander's route to peloruside A, Scheme 2. Fragment coupling between the enolate of methyl ketone 5 and aldehyde 4 followed by oxidation provided β-diketone 6 in excellent yield for the two steps. Cyclization to 4-pyrone 7 proceeded under the acidic conditions necessary for deprotection of the C5-OTES group. A stereoselective Luche reduction [23] at low temperature provided the C7-allylic alcohol, which directed a selective epoxidation of the enol ether. In situ methanolysis of the intermediate glycal epoxide provided the methyl pyranoside in high yield as a single diastereomer. Selective methylation of the presumably less sterically encumbered equatorial alcohol completed the structural features of the peloruside pyran system and provided key intermediate 8 after silylation.
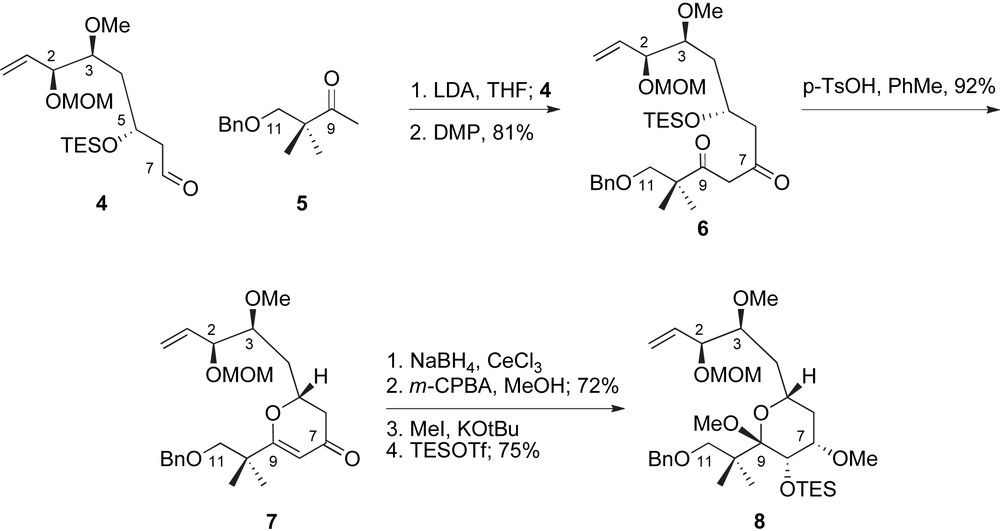
De Brabander's synthetic sequence for the preparation of the C1–C11 fragment.
De Brabander and coworkers completed the carbon skeleton of peloruside A by a fragment coupling between the C1–C13 and C14–C19 structural units, Scheme 3. An aldol reaction between the enolboronate derived from methyl ketone 10 and aldehyde 9, while efficient, unfortunately proceeded with limited diastereoselectivity. The two C13-isomers were readily separable and taken on independently to the macrolactonization. Selective methyl ether formation was accompanied by acetal hydrolysis and isolation of C15-ketone 11. Reduction of the carbonyl did not require a specific stereochemical outcome as the concomitant cyclization could be accomplished with inversion or retention of stereochemistry depending on the choice of reaction conditions. However, stereoselective reduction of C15 was ultimately accomplished by a reagent-controlled method [24]. Exposure of ketone 11 to (S)- or (R)-B-Me-CBS-oxazaborolidine and BH3·DMS provided the diastereomeric allylic alcohols including 2 in 80–94% yield with high diastereoselectivity.
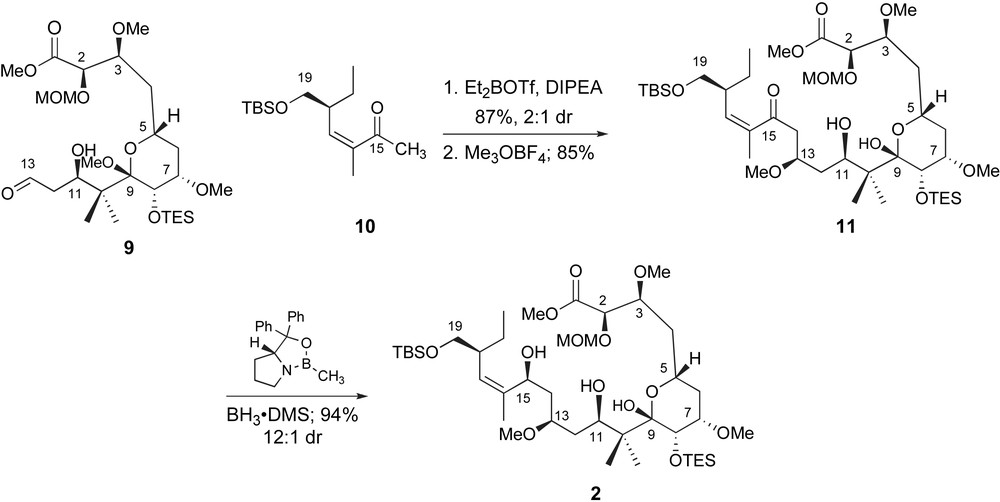
De Brabander's completion of the seco-acid with a late-stage fragment coupling and ketone reduction.
Jin and Taylor [11] utilized an activated ester intermediate, a Yamaguchi macrolactonization [25], to effect cyclization after saponification of a C1-oxazolidine 12, Scheme 4. The chiral auxiliary, necessary for the selective generation of the C2,C3-vicinal stereogenic centers, was carried through several steps as a masked carboxylic acid. Completion of the total synthesis was accomplished through functionalization of the 4-pyrone of lactone 13. Interestingly, epoxidation of the intermediate glycal resulted in selective deprotection of the C11-methoxymethyl group. The authors proposed that the MOM-group participated in the fragmentation of the glycal epoxide via anchimeric assistance. Hydrolysis of the intermediate carbonium ion then provided the methyl pyranoside and a C11-hydroxyl group. Completion of the total synthesis was accomplished by deprotection of the C2- and C19-ethers.
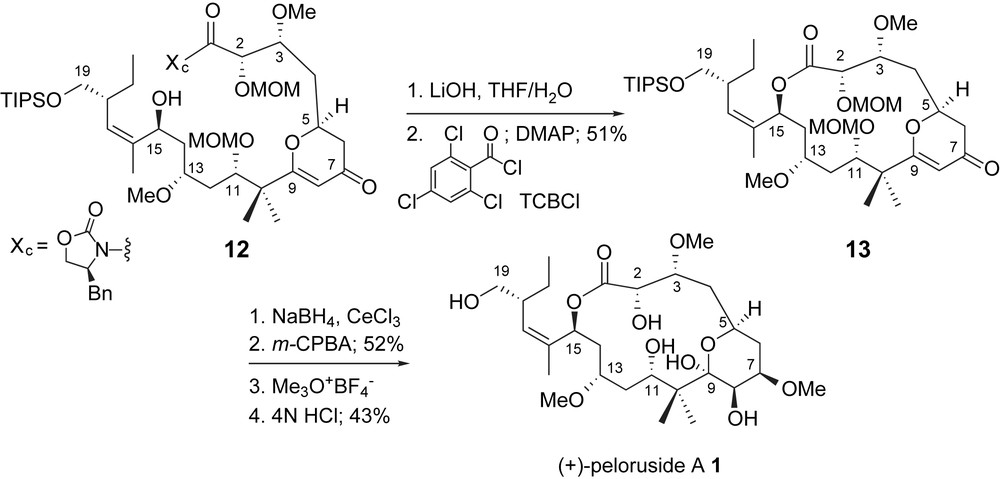
Taylor's macrolactonization and completion of peloruside A.
The Taylor group's approach to the C9–C17 fragment was a linear strategy from aldehyde 14 as shown in Scheme 5. Allylation with Brown's allylborane reagents [26] resulted in the formation of the expected homoallylic alcohol with high diastereoselectivity. Unfortunately, reaction by-products resulting from the chiral reagent hampered isolation on a large scale. It was then found that an unselective allylation followed by separation and interconversion of the undesired isomer provided high yields of the desired intermediate in a more practical fashion. The C15 stereogenic center was then utilized for the generation of the C13-stereogenic center through a Barlett–Smith iodo-carboxylation sequence to provide carbonate 15 [27]. Transesterification with methanol provided the intermediate epoxyalcohol which after single carbon homologation with dithiane, methyl ether formation, and hydrolysis provided aldehyde 16. The C11-stereogenic center, as alcohol 17, was produced by an 1,3-anti-selective Mukaiyama aldol reaction [28]. In preparation for the macrolactonization, the complete carbon skeleton of peloruside A was produced through 4-pyrone formation in a manner related to the De Brabander's route. However, the production of this intermediate through a more convergent route resulted in an overall shorter synthetic sequence of 29 steps.

Taylor's stereoselective route to the C9–C17 fragment.
3 Towards the synthesis of peloruside A
Several additional research labs have communicated preliminary results describing their synthetic efforts towards a total synthesis of this interesting marine polyketide. Paterson has presented a triply convergent strategy through sequential aldol reaction via methyl ketones 18 and 20 with structural unit 19 serving as latent “dialdehyde” linchpin, Fig. 2 [29]. The stereoselectivity of each fragment coupling would rely on the established preference for 1,5-anti-stereochemical relationships as previously demonstrated by Paterson [30].
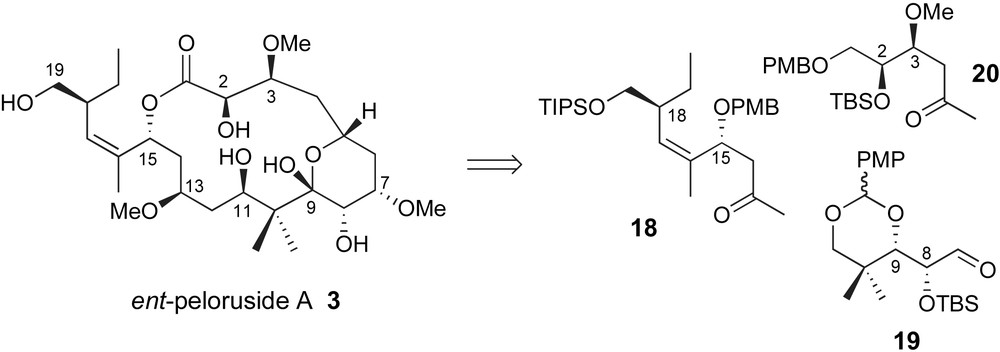
Paterson's “Di-aldehyde Linchpin” strategy.
A sequential pair of stereoselective aldol reactions, shown in Scheme 6, provided access to methyl ketone 18. The C18 stereogenic center was created in high yield by the condensation of formaldehyde with boron enolate of chiral ketone 21 derived from lactate. The Z-trisubstituted olefin was generated through a Still–Gennari olefination [31] and a reduction–oxidation sequence provided aldehyde ent-14. The C15 stereogenic center was then produced by an aldol reaction with acetone. Utilization of a chiral boron enolate [32] allowed the production of the desired C15-diastereomer with good stereoselectivity. Finally, generation of the PMB ether produced ketone 18.

Paterson's route to the C12–C19 fragment.
With methyl ketone 18 in hand, Paterson explored the first aldol fragment coupling utilizing a model linchpin, aldehyde 23 (Scheme 7). The c-Hex2B-enolate of 18 was generated in the usual fashion. Coupling with aldehyde 23 provided C11,C15-anti-aldol adduct 24 in excellent yield and selectivity. Generation of the desired C11-configuration was supported by Mosher ester analysis [33]. The C11-alcohol was then exploited for stereoselective reduction of the C13-ketone through an Evans–Tishchenko reduction [34] with SmI2 and propionaldehyde to provide the C9–C19 fragment 25.
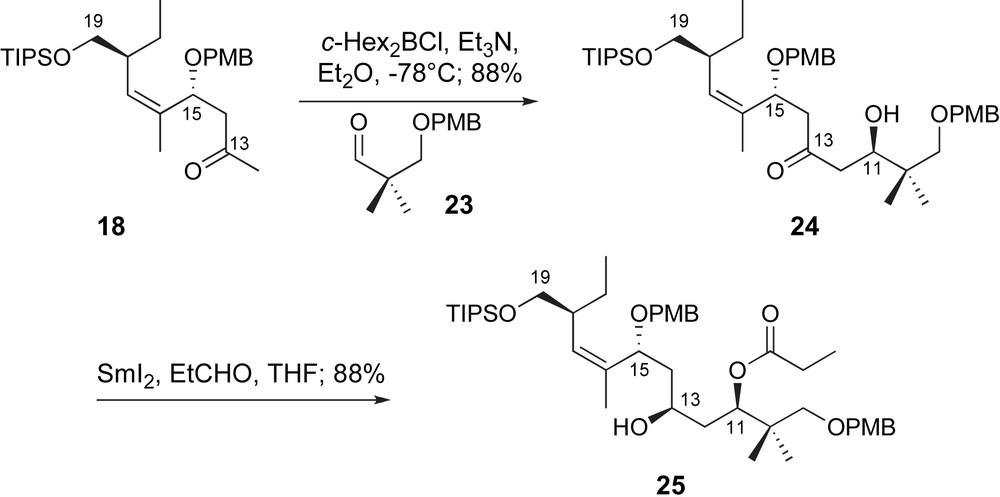
A 1,5-anti-aldol and Evans–Tishchenko reduction.
In early 2004, Zhou and Liu reported a highly convergent synthesis of the C1–C16 backbone of peloruside A [35]. The key fragment coupling for the creation of the C10–C11 bond was an aldol reaction between a highly substituted enolate derived from ketone 27 and aldehyde 26 Fig. 3. Both fragments were generated in highly stereoselective manners exploiting iterative use of Brown chiral allylation technology [26].
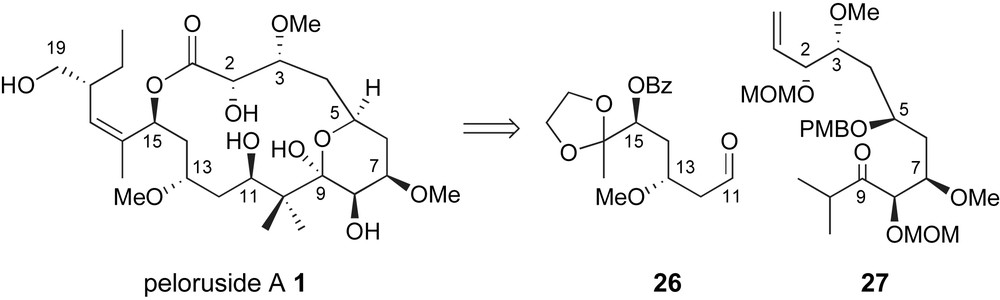
Zhou's stereoselective aldol strategy for generating the C10–C11 bond.
Once both fragments were available, Zhou and Liu explored a variety of aldol coupling conditions. Unfortunately, the highly substituted nature of the necessary enolate derived from ketone 27 hindered clean reactivity. No reactivity was observed under Mukaiyama aldol conditions, initiated by Lewis acids such as BF3·OEt2 or TiCl4. Moreover, generation of intermediate titanium or boron enolates also met with disappointment. Fortunately, however, some modest reactivity was observed with a lithium enolate generated at low temperatures with lithium diisopropyl amide (LDA). As shown in Scheme 8, condensation of the lithium enolate of ketone 26 and aldehyde 27 generated the C1–C16 backbone of peloruside A with fair stereoselectivity for the desired configuration at C11.
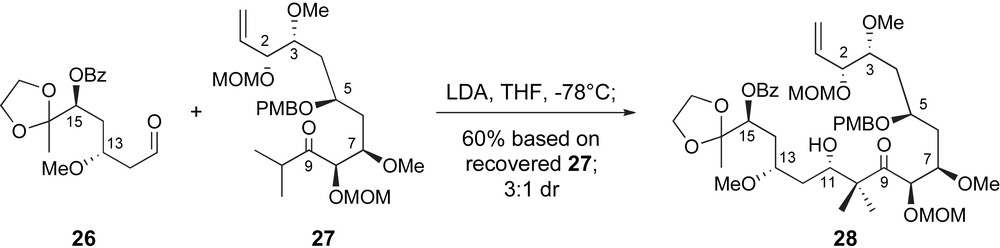
Key fragment coupling in Zhou's synthetic route to the C1–C16 segment.
Roush and Owen have proposed a highly convergent strategy for the preparation of the carbon skeleton of peloruside A [36] which utilizes two applications of the double allylboration reaction previously developed in their lab [37]. The bis-nucleophile 31 serves as a linchpin between terminal fragments 14 and 30 and dialdehyde equivalent 29, Fig. 4. With this bold strategy, essentially all of the chirality associated with this highly oxygenated natural product would be controlled by the chiral allylborane reagent.
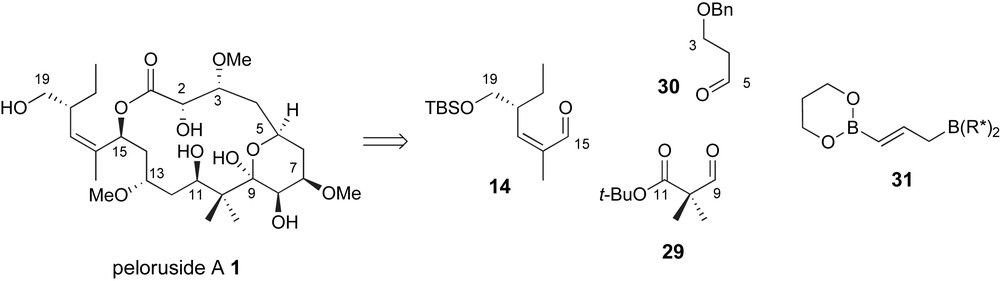
Roush's common linchpin strategy using a double allylboration reagent.
Successful demonstration of this concept is presented in the preparation of the C3–C11 fragment 35, as detailed in Scheme 9. Sequential exposure of the bis-nucleophile 31 to aldehydes 29 and 30 resulted in the isolation of diol 33 in 36% yield as a single diastereomer and geometric isomer. The diastereoselectivity observed in the product supports selective formation of the intermediate 8,9-anti-boronate 32 and a highly organized cyclic transition state. The enantioselectivity observed in the product 33 and thus intermediate 32, as determined by Mosher analysis, was >95%. Completion of the C3–C11 fragment then required generation of the C7,C8-anti-vicinal diol unit. After selective silyl protection of the homoallylic C5-alcohol 33 [38], the E-olefin was epoxidized from the α-face with high facial selectivity presumably directed by the allylic alcohol [39]. Further protection of C9 as a p-methoxybenzyl ether was followed by Zn(II)-mediated epoxide fragmentation and isolation of lactone 34. Subsequent methylation and hydride reduction provided the C3–C11 fragment of peloruside A, 35.
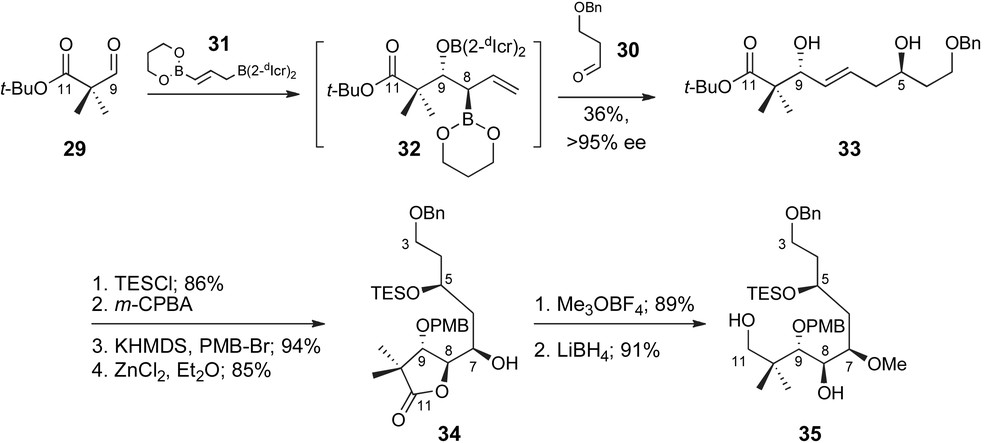
Roush's synthesis of the C3–C11 fragment.
From a complementary perspective, Pagenkopf and coworkers have proposed the use of a C8–C12 “bis-enolate” linchpin 37 for generation of the carbon skeleton of peloruside A, Fig. 5 [40]. The key fragment coupling step would require a glycolate anti-aldol with aldehyde related to 38 and generate the C7,C8-vicinal diol unit.
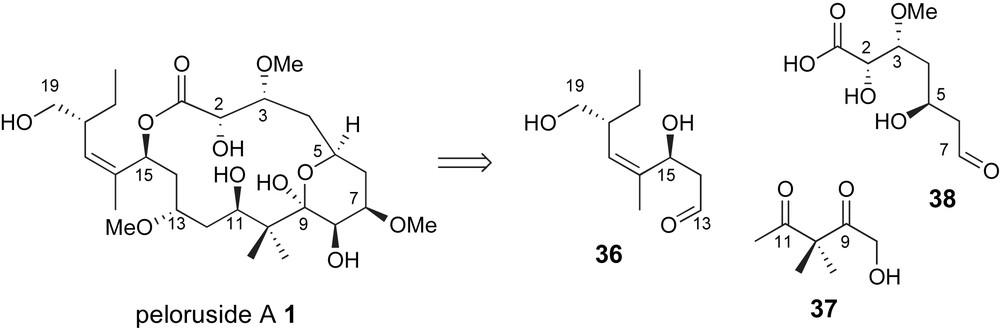
Pagenkopf's glycolate aldol strategy for generation of the C7–C8 bond.
Pagenkopf generated the C1–C7 fragment rapidly and in a practical fashion, as aldehyde 40, through manipulation of readily available triacetal glucal. While a variety of aldol conditions were explored for generation of the C7–C8 bond, classic Mukaiyama conditions proved the most effective. As shown in Scheme 10, trimethylsilyl enol ether 39 underwent coupling with aldehyde 40 promoted by BF3·OEt2. The desired 5,7-anti-7,8-anti-adduct 41 was generated in stereochemical excess (3.5:1) in good overall yield. Unfortunately, exploration of alternative Lewis acids failed to improve upon this ratio. At this point, both termini underwent oxidative cleavage with ozone and, after selective oxidation of the C1-aldehyde, the resulting carboxylic acid was protected as a methyl ester. Despite the modest stereoselectivity observed for the initial fragment coupling, the relative ease in preparation of each fragment within this strategy supports its potential to access useful quantities of peloruside A.
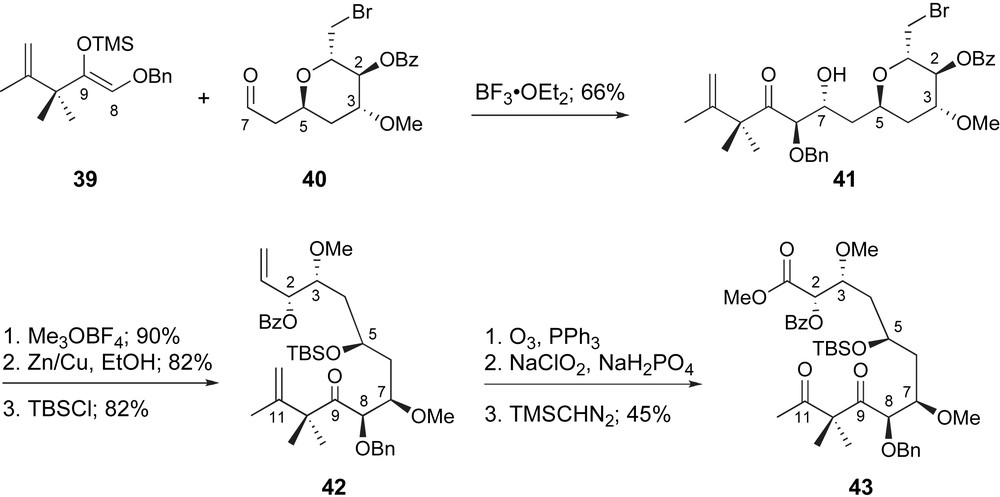
Pagenkopf's glycolate aldol coupling and generation of the C1–C12 fragment.
Hoye and Ryba have presented a creative bi-directional approach to a C8-epimer of the C1–C9 region of peloruside A via terminal group differentiation, Scheme 11 [41]. Ozonolytic cleavage of the terminal alkenes in 44 produced a pseudo-C2-symmetric dialdehyde intermediate 45. Selective formation of lactone 46 was presumably a result of kinetic oxidation of an equilibrating mixture of diastereomeric hemi-acetal intermediates. A related investigation of competitive lactonization reactions of bis-methyl esters provided additional insight into the controlling factors of this mechanistically interesting selectivity.
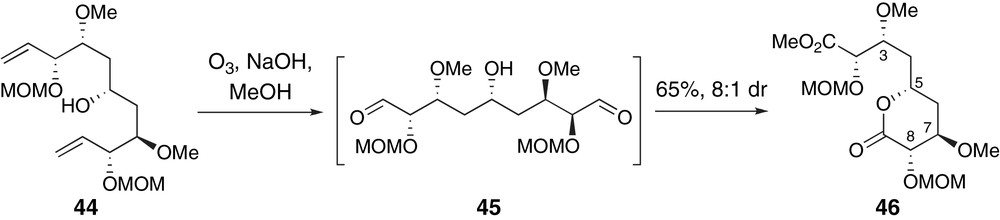
Hoye's kinetic lactonization to the C8-epimer of the C1–C9 fragment of peloruside A.
Ghosh and Kim have proposed a fragment coupling strategy complementary to Zhou's (Fig. 3). An aldol coupling to form the C9–C10 bond would also necessitate a highly substituted enolate of a C11 ketone and an α-oxygenated C9-aldehyde. In 2003 they presented a synthetic route to the C1–C9 region of ent-peloruside A [42a]. The vicinal diol functionality found at C2,C3 and C7,C8 was each incorporated by a Sharpless asymmetric dihydroxylation reaction [43] (Scheme 12). The route began with α,β-unsaturated ester 47 available via a three-step sequence from 1,3-propan-diol [44]. Asymmetric dihydroxylation with AD-mix-α proceeded in high yield and 90% ee. Functional group manipulation provided 48. After a two-step conversion to β,γ-unsaturated ketone 50, hydride reduction in the presence of LiI provided 1,3-syn-product 51 in 87% yield. The stereoselectivity was rationalized by a chelated transition state 50 and axial addition of hydride. Acylation and ring-closing metathesis [45] provided the δ-lactone 52 which enabled the selective generation of the Z-alkene at C7,C8. Saponification of the lactone, protection of the secondary alcohol and esterification followed by dihydroxylation with AD-mix-β afforded the desired diol 53, in 72% yield as a 91:9 mixture of diastereomers.
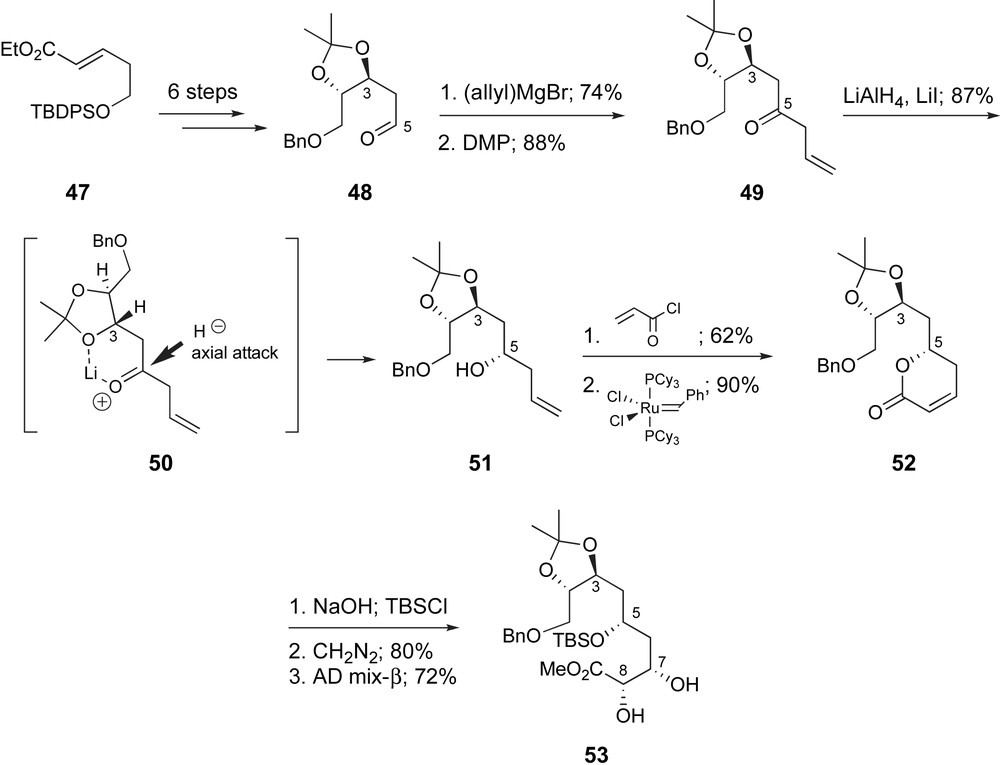
Ghosh's asymmetric dihydroxylation approach to the C1–C9 fragment of ent-peloruside A.
Later in 2003, Ghosh and Kim published their synthetic route to their C1–C19 fragment, ketone 57, Scheme 13 [42b] The C15-homoallylic alcohol was prepared as previously presented. Homologation of the terminal alkene was accomplished by the generation of an intermediate acrylate and ring-closing metathesis to provide the δ-lactone 55. The C13-stereogenic center was generated by a two-step sequence; stereoselective epoxidation and reductive ring opening. Protection of the secondary hydroxyl as a t-butyldiphenylsilyl ether provided lactone 56. The lactone was then converted to ketone 57 by standard methods.
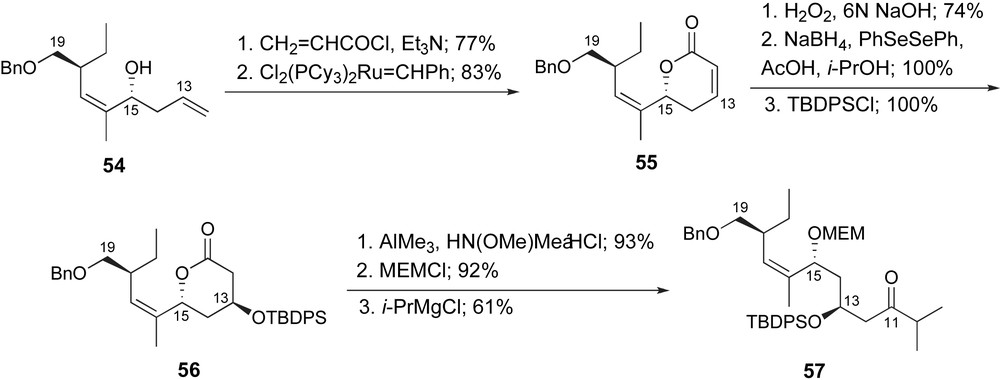
Ghosh's synthesis of the C10–C19 fragment of ent-peloruside A.
Shortly after Ghosh's communications, Gurjar and coworkers reported a related route to C1–C11 region of peloruside A [46]. As shown in Scheme 14, dihydroxylation of α,β-unsaturated lactone 59 stereoselectively generated the C7,C8-vicinal diol functionality. The lactone was formed in a fashion similar to Ghosh exploiting a ring-closing metathesis reaction. However, in contrast, this substrate underwent osmylation with high facial selectivity without the need for chiral ligands. Moreover, the C2,C3-stereogenic centers were provided by a practical choice of starting material, d-glucose.

Gurjar's chiral pool approach to the C1–C11 fragment of peloruside A.
Finally, Roulland and Ermolenko reported an efficient synthetic route to methyl ketone 67 which represents the C12–C19 region of peloruside A [47]. As detailed in Scheme 15, non-racemic, homoallylic alcohol 64, prepared in one step from (S)-propylene oxide, underwent diastereoselective epoxidation catalyzed by VO(acac)2. After protection of the secondary alcohol as a PMB ether, the epoxide was converted to the isomeric allylic alcohol in high yield. Acylation with crotonyl chloride provided the β,γ-unsaturated ester 66, which after deprotonation with LDA, underwent α-alkylation with ethyl iodide to provide a mixture of diastereomers (18R:18S, 2:3). Ring-closing metathesis conditions provided exclusively the 18R-lactone 68 and unreacted 18S-67. Lactone 68 was isolated in 36% yield which is 95% based on the starting material, 18R-67. Moreover, the authors demonstrated that unreacted ester 18S-64 can be epimerized with LDA, AcOH and recycled into the sequence. Lithium aluminum hydride reduction of lactone 68 produced an intermediate diol, which was selectively protected to provide 69. Manipulation of protecting groups followed by oxidation of C13 provided the C12–C19 fragment as methyl ketone 70. The overall sequence is fairly efficient and highlighted by a useful, diastereo-differentiating RCM reaction.
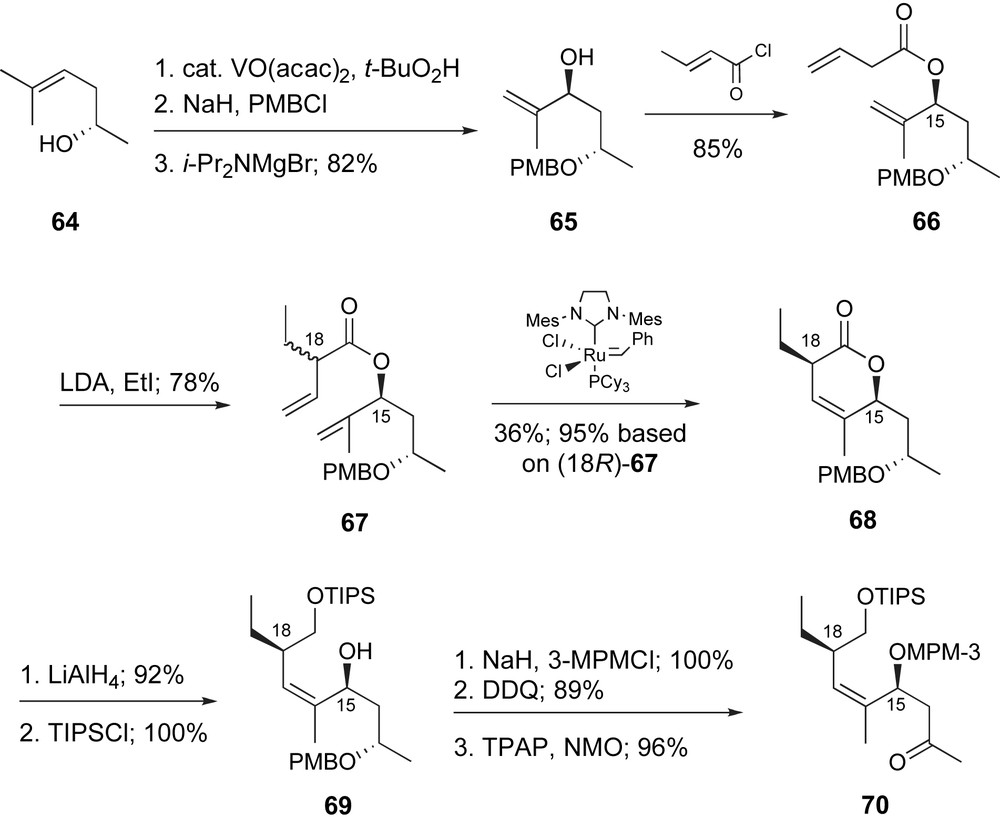
Ermolenko's ring-closing metathesis approach to the C12–C19 fragment.
4 Addendum
During the review of this manuscript a third total synthesis of peloruside A was reported by Ghosh and coworkers, Fig. 6 [48]. Their second generation approach relied on a complex aldol fragment coupling to generate the C10–C11 bond that had previously been investigated by Zhou, Fig. 3. This report included optimized routes to aldehyde fragment 71 and ketone 72. Moreover, the successful approach contained several findings that significantly complement previously presented efforts.
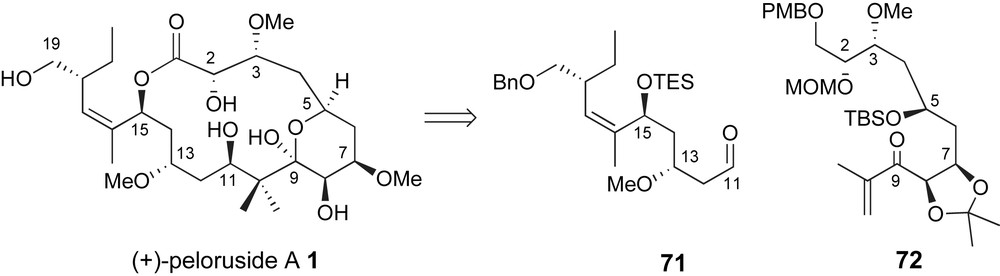
Ghosh's aldol fragment coupling strategy for generating the C10–C11 bond.
As discussed above, Zhou and coworkers had previously explored an aldol coupling strategy to form the C10–C11 bond. Due to the sterically hindered enolate intermediate, derived from ketone 27, Scheme 8, it is not surprising that optimized conditions only provided moderate yields and stereoselectivity. In contrast, Ghosh creatively generated the necessary lithium enolate, not through typical LDA deprotonation, but through a conjugate reduction of enone 72 with l-selectride, Scheme 16. Reaction of this enolate with aldehyde 71 at −78 °C provided the aldol adduct in 92% yield with 4:1 diastereoselectivity for the desired stereoisomer 73. After deprotection of the TES and PMB ethers, the C1-primary alcohol was oxidized to the carboxylic acid in two steps. Yamaguchi lactonization then provided the macrocyclic lactone 74 in 64% yield. A unique aspect of this route is the fact that the dihydropyran unit was not established prior to lactonization due to the protected C5-alcohol. One would assume that the conformation of 74 is quite different from the natural system. However, it is not clear if the presence of the C7,C8-dioxolane provided some entropic advantage. Completion of the total synthesis required sequential protecting group removal steps. Mild acid simultaneously deprotected the C5-TBS ether and the isopropylidene resulting in an equilibrium mixture of dihydropyran and open hydroxy–ketone isomers. Despite this complication, selective methyl ether formation at C7 proceeded well. Hydrogenolysis of the benzyl group followed by aqueous acid removal of the C2-methoxymethyl group provided synthetic peloruside A, identical to reported data for natural material.
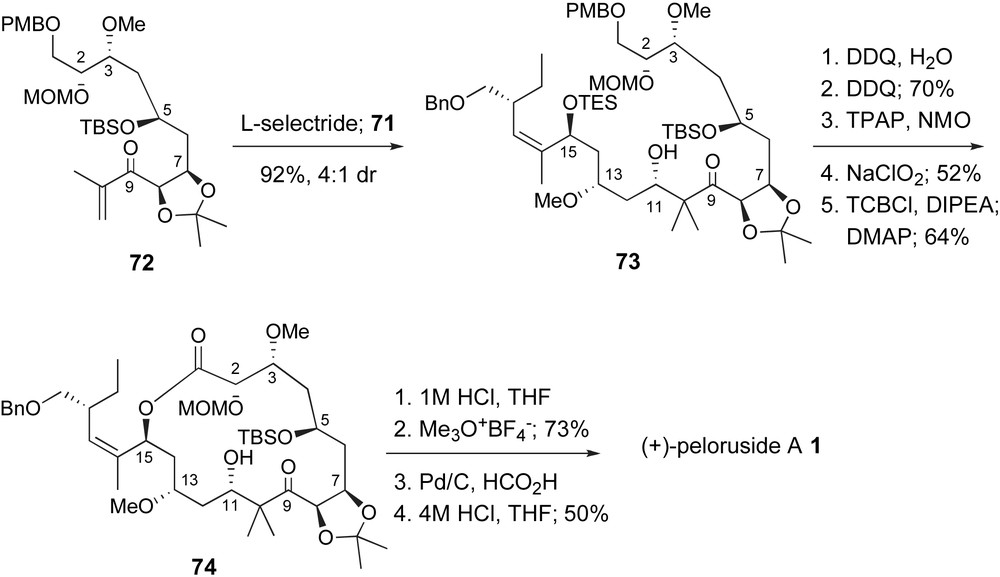
Fragment coupling and end game in the Ghosh's synthesis of peloruside A.
5 Summary and prospective
The previous presentation represents a snapshot of the synthetic progress made towards peloruside A as of April 2008. While only three groups have completed total syntheses to this challenging polyketide target, several additional groups have made significant progress. Moreover, these efforts have shown remarkable diversity in synthetic strategy and in the development of broadly applicable new tactics. At this point, it is not clear whether any of these individual approaches can provide reliable quantities of material required for preclinical and, if necessary, early clinical development. However, potential success may reside with a creative combination of a subset of these efforts.
As has been true with other compilations of natural product synthetic efforts, it is clear that chemists can prepare any naturally occurring compound. Unfortunately, we have yet to routinely demonstrate an ability to prepare compounds of this complexity at a practical level necessary for fully exploiting their chemotherapeutic potential. Thus, synthetic chemistry is far from a mature field when one carefully considers the ever-evolving needs of society and the resulting practical constraints.