

1 Introduction
The intense contemporary interest in the design and synthesis of active site models of iron–iron hydrogenases derives from their ability to catalyse reduction of protons to hydrogen [1–3]. Much work has been performed in the past years on understanding the reaction mechanism for H–H bond formation in order to improve the efficiency of electrochemical hydrogen production catalyzed by active site model systems [4–6]. To convert the solar energy into a fuel, we are interested in light-driven proton reduction (hydrogen production) in bio-inspired chemical systems, such as FeFe-hydrogenase active site models as catalysts [7]. One way to realize this idea is the direct connection of a photosensitizer to a 2Fe2S complex, making the hydrogen production catalyst photoactive. Although several photoactive catalysts have been reported during the past years [8,9], they are based on noble metal ions, such as Pt or Pd, with one exception where cobalt is used [9]. Very few reports of photocatalysts based on hydrogenase active site model complexes have been published. However, we have earlier synthesized three systems where photosensitizers were covalently linked to Fe2S2 catalytic complexes (Chart 1) [7,10–12].
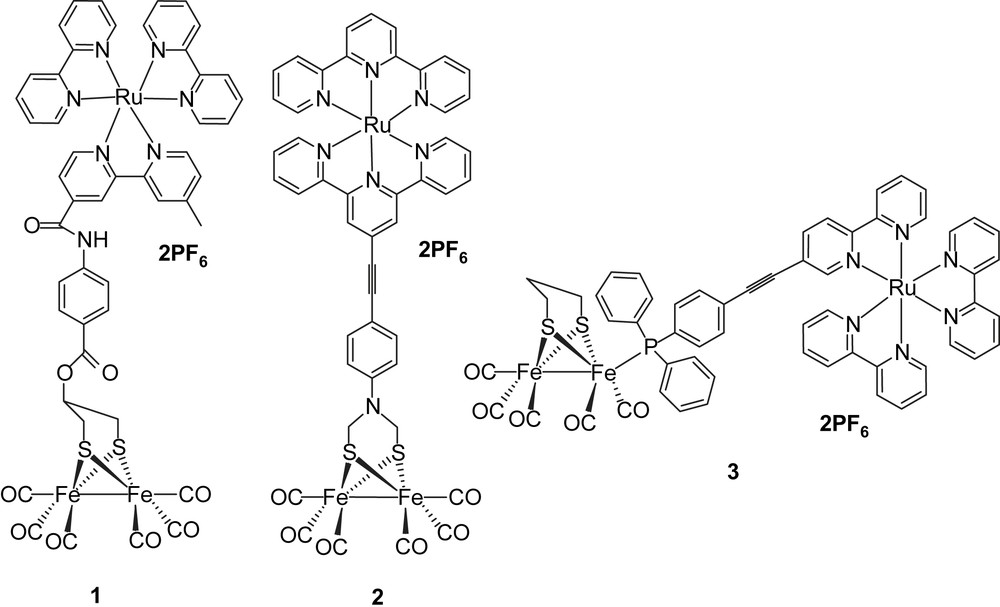
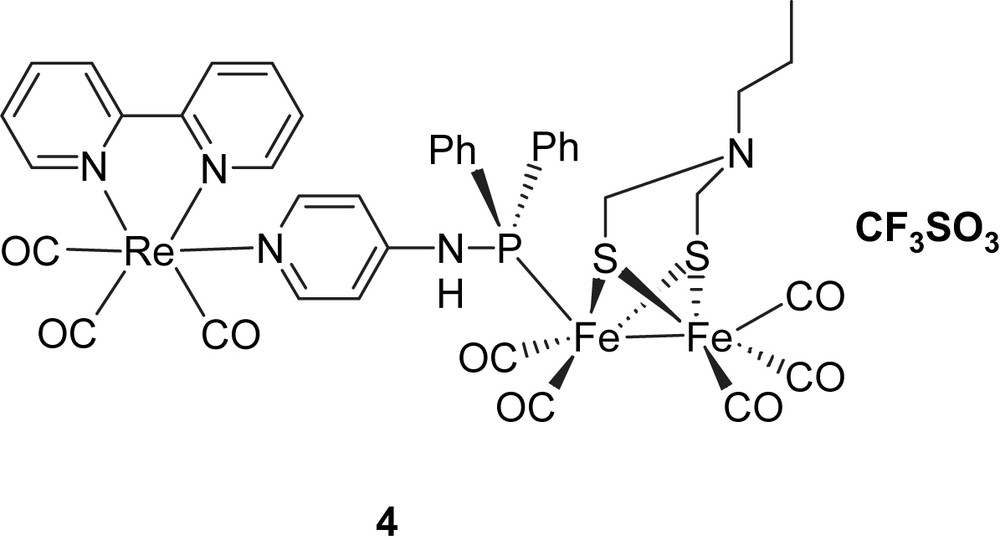
In one case (1), a Ru(bpy)3 photosensitizer was linked via an ester group to the bridge head of a propanedithiolate (PDT)-bridged diiron catalyst. Photophysical measurements showed that the excited state of Ru(bpy)3 in system 1 was quenched by the Fe2S2 catalyst. By studying the electrochemistry of system 1, it was found that the one-electron reduction of the Fe2S2 catalyst was too negative to be reached by the excited state of Ru(bpy)3. In the second case (2), a Ru bis(terpyridine) complex as the light-harvesting component and an azadithiolate Fe2S2 complex were coupled through a rigid link. From the value of the reduction potential of the excited state of [Ru(terpy)2]2+ (ca. −0.8 V vs SCE) and relevant electrochemical data of the 2Fe2S subunit (ca. −1.09 V vs SCE), it was apparent that the driving force for an electron transfer from the photo-generated *[Ru]2+ excited state to the diiron complex was uphill by about 0.59 eV, as calculated with the Rehm–Weller equation, and therefore, thermodynamically unfavorable [11]. In the third case (3), the Ru(bpy)3 photosensitizer was attached to a PDT-bridged diiron catalyst via a phosphine through ligand exchange with CO [12]. However, the same conclusion was drawn after transient absorption measurements: only reductive quenching of the excited state of Ru-polypyridine photosensiziers by Fe2S2 catalysts occurred independently of the nature of the link. To overcome this reductive quenching problem, one way is to decrease the reduction potential of the Fe2S2 catalyst, making the desired electron transfer from the excited state of photosensitizer to the Fe2S2 catalyst possible. An alternative way is to increase the energy level of the excited state of photosensitizer. Since it is known that the excited states of ReI(CO)3(bpy)(py) complexes have considerably higher redox potentials [11] than the RuII(bpy)2 complexes, we decided to study Re complexes. We have, therefore, prepared complex 6, which from the energy of the excited state and the ReI/ReII redox potential has a calculated reduction potential in the excited state of ca. −2 V. This should in principle be sufficient for photo-induced electron transfer to an Fe2S2 catalyst. In addition, the use of a phosphine to coordinate to the Fe2S2 catalyst should permit protonation of the catalyst by shifting the redox potential towards more positive values. Based on these considerations, we have designed a new type of molecular dyad 4, featuring a rhenium-containing subunit and an Fe2S2 cluster bridged with 4-(diphenylphosphinoamino)pyridine, with which it should be possible to realize the light-driven proton reduction. Because of the electronic interaction between iron and rhenium, complex 4 may also behave as a mimic of the cysteine-linked [Fe4S4] cluster, which is part of the electron-transfer chain to and from the active site of Fe-only hydrogenases. In this paper, we report the synthesis and electrochemistry of the rhenium-containing Fe2S2 molecular dyad 4 (Chart 2).
2 Results and discussion
2.1 Synthesis and spectral characterizations
Prior to this research, no similar complexes have been reported, although many substitution reactions of phosphorus-containing ligands of the type PR3 for CO have been studied [13]. The synthetic route for the preparation of target molecule 4 is depicted in Scheme 1. The commercially available Re(CO)5Cl was treated with 2,2′-bipyridine (bpy) in refluxing toluene to give fac-(bpy)Re(CO)3Cl (5). This was then coordinated to the pyridine in refluxing ethanol in the presence of CF3SO3H to afford novel complex 6 [14]. In principle, Re can coordinate to either N or P of the ligand, but N is evidently preferred in our case. By strict control of the reaction time to ca. 2 h, the coordination is selective and we could obtain complex 6 with a free phosphine ligand.
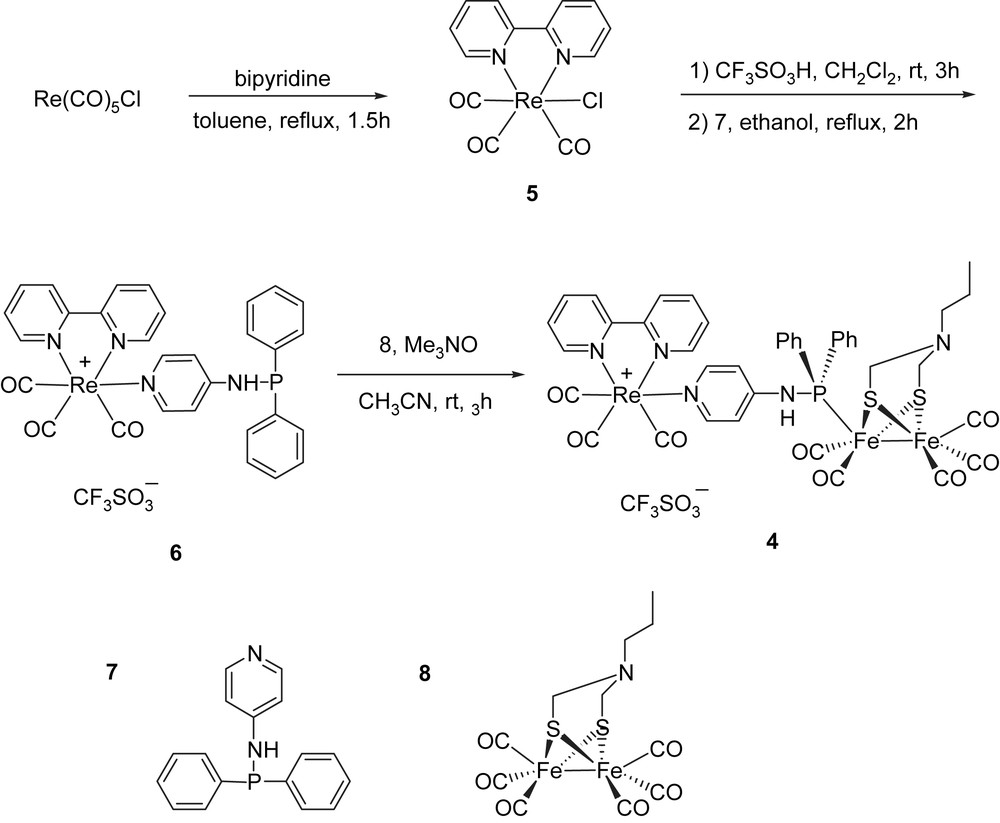
The synthetic procedure of complex 4.
The final ligand exchange reaction of CO in 8 by the phosphine in 6 was achieved in CH3CN with the assistance of the decarbonylation reagent Me3NO. The carbonyl substitution reaction was accomplished in high yield with good selectivity for mono-substitution at the Fe2S2 unit. The trialkyl phosphine complexes are well known, while those containing NH–PR2 ligands are rare.
With this red-colored complex 4 in hand, we have made great efforts to obtain the single crystals, but in vain. Therefore, compound 4 was identified via a combination of spectroscopic methods (1H, 13C, 31P NMR, HRMS and UV–vis). The characteristic signal for methylene protons on the 2Fe2S subunit (doublet at δ 2.84 and 2.94 ppm) is obvious. The protons at very low field (δ, 7.93, 8.46, 8.76 and 9.37 ppm) consist of the expected signals of bipyridine subunits. It also unambiguously shows the corresponding resonance at δ 7.53 and 7.79 ppm for phenyl protons. The most important spectroscopic feature of 4 is the 31P NMR spectrum, in which the resonance signal is found at 95.5 ppm. The most intense peak observed in the mass spectrum at m/z 1105.8 corresponds to [M–CF3SO3−]+ (positive mode). Fig. 1 shows the IR spectra for complexes 6 and 4 in the carbonyl region absorptions between 1910 and 2050 cm−1. By comparison, the peaks at 1914, 1926 and 2030 cm−1 correspond to the CO vibrations of the rhenium moiety, while those at 1956, 1978, 1986 and 2049 cm−1 correspond to the CO vibrations of the Fe2S2 moiety.

The IR spectra for complexes 4 and 6.
The UV–vis absorption spectra of complex 4 are shown in Fig. 2 with 6 and 8 for comparison. Both 6 and 8 have intense absorptions at 282 nm (for 6) attributed to the ligand centered transition (πL → πL∗), a broad band at ca. 380 nm (for 6) due to MLCT, and 329 nm (for 8) assigned to a d(Fe) → d∗(Fe) transition. These assignments were based on comparison with data from [fac-(bpy)Re(CO)3Cl] and [(bpy)Re(CO)3(py-PTZ)]+ [15]. For complex 4, however, only weak absorptions are observed, at 274 and 358 nm. The strong donor capacity of the –PPh2 ligand increases the electron density of iron atoms and contributes to the dπ(Fe) → π∗(PPh2) MLCT (metal-to-ligand charge transfer) transition which appears at around 358 nm. The decrease of the intensity of the absorption at band 282 nm for complex 6 after coordination with 8 to form 4 may be due to decreased interaction between rhenium and the ligands (pyridine and bipyridine). The strong absorption around 329 nm disappears on the formation of 4. Instead, an observable dπ(Fe) → π∗(PPh2) MLCT transition appears at 358 nm. We could thus see that the absorption in the near-UV region depends greatly on the coordination sphere around the iron nuclei.
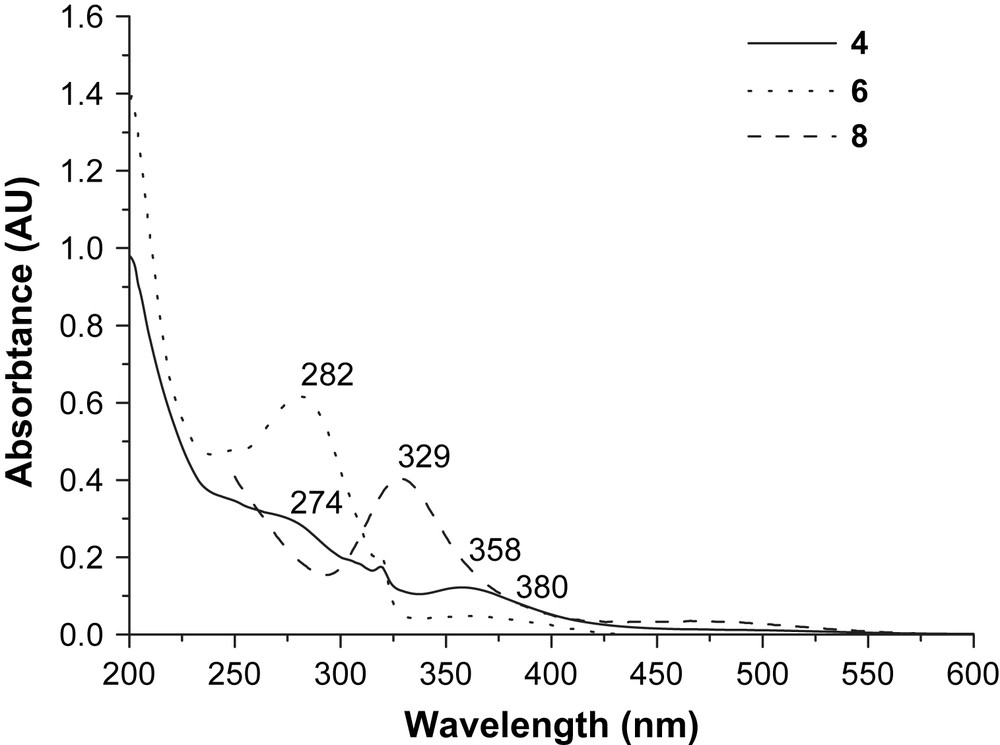
The UV–vis spectra for 4, 6 and 8 (0.01 mM) in CH3CN.
2.2 Electrochemistry
The redox potentials of complex 4 were determined by cyclic voltammetry (CV) in MeCN, using tetrabutylammonium hexafluorophosphate as the supporting electrolyte. Reduction and oxidation potentials were given vs internal standard Ag/AgNO3 and compared to those of the reference compound 6 (Table 1), which facilitates the assignment of the individual redox waves (Fig. 3).
Electrochemical data of complexes 4 and 6 (V vs Ag/Ag+)
| Complex | Epsemiox ReI/ReII | Epirrox FeIFeI/FeIIFeI | Epc FeIFeI/Fe0FeI | Epc Fe0FeI/Fe0Fe0 | Epc bpy/bpy− | Epc bpy−/bpy2− |
| 6 | +1.43 | – | – | – | −1.54 | −1.84 |
| 4 | ca. +1.30 | +0.43 | −1.58 | −1.96 | ca. −1.58 | −1.80 |
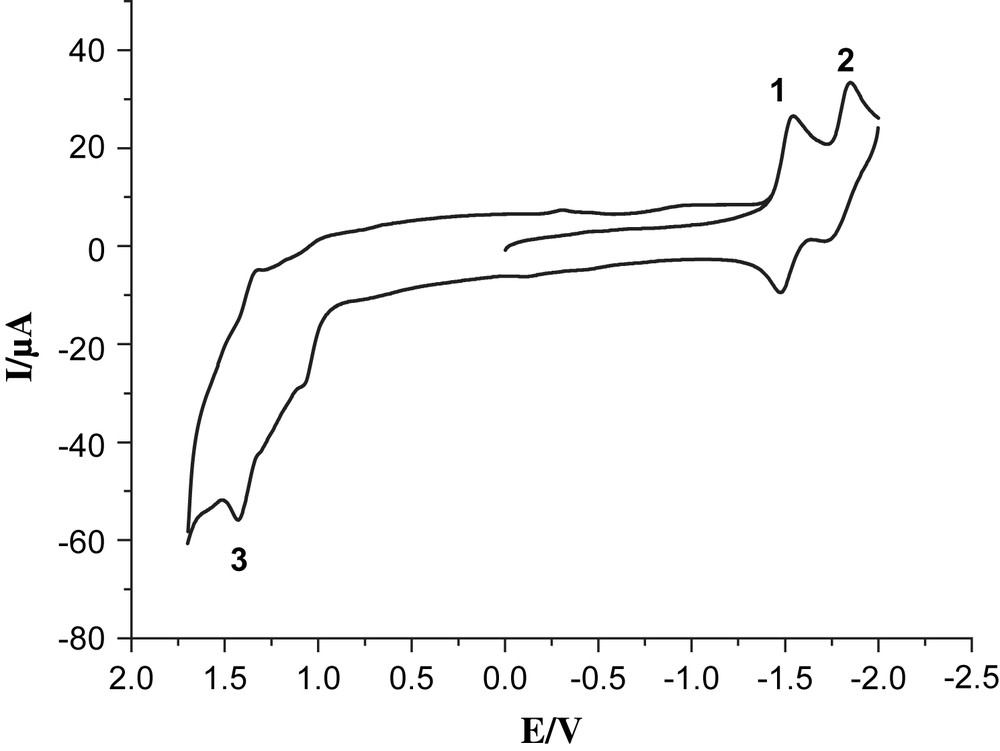
Cyclic voltammogram of 6, 1.0 mM in 0.05 M n-Bu4NPF6/CH3CN at a scan rate of 100 mV/s with Ag/Ag+ as a reference electrode.
Complex 6 exhibited clearly two reversible reduction couples at −1.54 and −1.84 V vs Ag/AgNO3 (labeled by 1 and 2) and one semi-reversible oxidation couple at 1.43 V (labeled by 3). Also oxidation of the phosphine in complex 6 is observed as a shoulder at 1.14 V, which may be compared with the readily observed oxidation at 1 V of the ligand itself (compound 7). In addition, oxidation of the solvent contributes as a strong background current. While the solvent oxidation in the absence of the Re-catalyst does not start until ca. 1.7 V (Fig. 3), the presence of the rhenium complexes 4 and 6 results in sharply increasing solvent oxidation already at 1.3–1.5 V, making the assignment of the exact value of ReI/ReII redox couple difficult. The two reduction waves observed are described as one-electron processes for bpy/bpy− and bpy−/bpy2−, respectively [16]. In the case of the trinuclear complex 4, the first reversible reduction peak at −1.58 V (labeled by 1′) is ascribed to a mixture of the two overlapping processes FeIFeI/Fe0FeI and bpy/bpy− (Fig. 4). The reduction couples at −1.80 and −1.96 V (labeled by 2′ and 3′) are assigned as bpy−/bpy2− and Fe0FeI/Fe0Fe0, respectively. The irreversible oxidation peak at 0.43 V (labeled by 4′) is assigned to FeIFeI/FeIIFeI and the shoulder at ca. 1.3 V (labeled by 5′) is assigned to ReI/ReII. These data follow the general trend discussed earlier for our reported Fe2S2 complexes [17,18]. Because of –PPh2 donor ligand, complex 4 exhibits an irreversible oxidation at a potential which is lower by ca. 300 mV compared to those of all-carbonyl analogues [11].
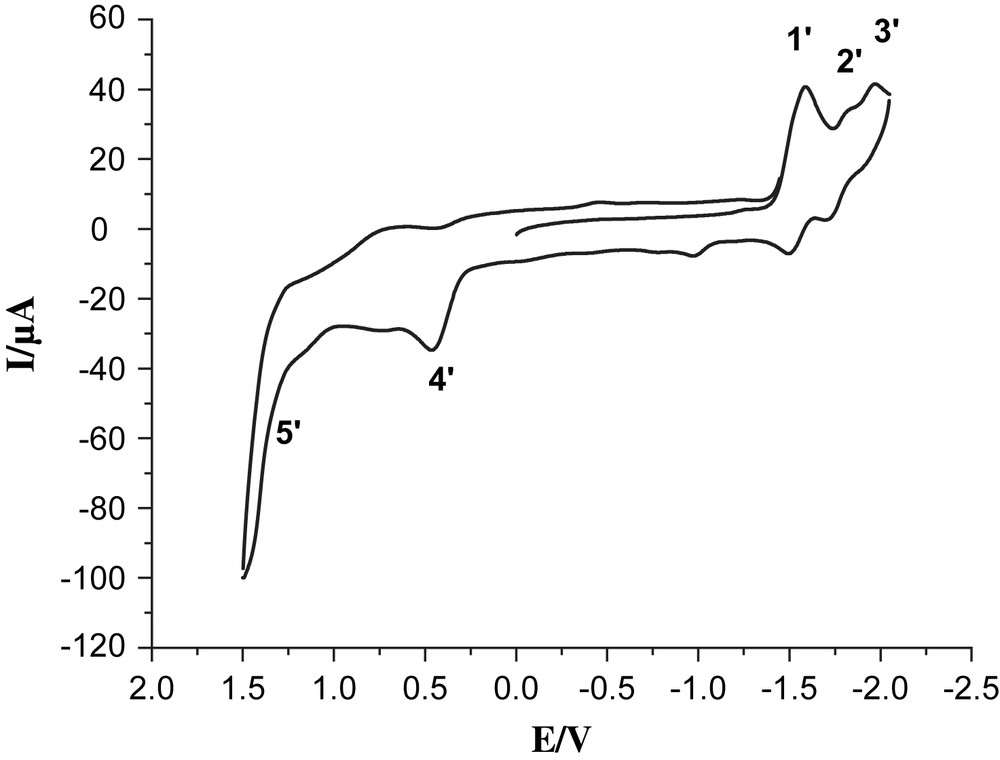
Cyclic voltammogram of 4, 1.0 mM in 0.05 M n-Bu4NPF6/CH3CN at a scan rate of 100 mV/s with Ag/Ag+ as a reference electrode.
Proton reduction catalyzed by complex 4 was studied by CV in the presence of a strong acid, CF3SO3H in CH3CN. A new reduction peak appeared at about −1.23 V when 1 equiv of CF3SO3H was added (Fig. 5). As the acid concentration was increased, all the reduction peaks grew and moved to more negative potentials. These features are indicative of catalytic proton reduction. Obviously, the anodic shift on protonation (ca. 350 mV) makes the electron transfer from the excited state of Re moiety to the Fe2S2 unit thermodynamically feasible.
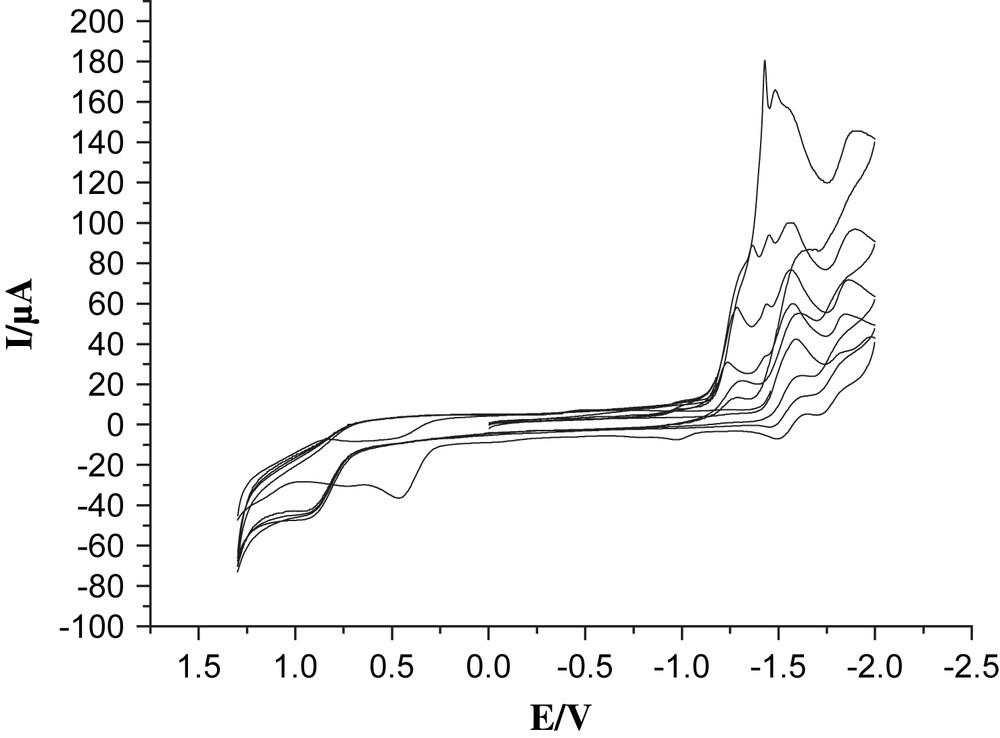
Cyclic voltammograms of 4 (1.0 mM) with CF3SO3H (0, 1, 2, 3, 4 mM upwards) in 0.05 M n-Bu4NPF6/CH3CN at a scan rate of 100 mV/s with Ag/Ag+ as a reference electrode.
3 Experimental section
3.1 Reagents and instruments
All reactions and operations related to organometallic complexes were carried out under a dry, oxygen-free nitrogen atmosphere with standard Schlenk techniques. All solvents were dried and distilled according to the standard methods prior to use. Commercially available chemicals, including n-propyl amine, 4-aminopyridine, paraformaldehyde, diphenylchlorophosphine and Re(CO)5Cl were used without further purification. The reagents LiEt3BH and triflic acid were purchased from Aldrich.
Infrared spectra were recorded on a JASCO FT/IR 430 spectrophotometer. 1H and 13C NMR (mainly CDCl3 solution) spectra were collected on a Varian INOVA 400 NMR spectrometer. Mass spectra were recorded on an HP1100 MSD instrument, and HRMS on an HPLC-Q-Tof MS (Micro) system.
Acetonitrile (Aldrich, spectroscopy grade), which was used for the electrochemistry, was dried with molecular sieve (4 Å) and then freshly distilled from CaH2 under N2. A solution of 0.05 M n-Bu4NPF6 (Fluka, electrochemical grade) in CH3CN was used as electrolyte. Electrochemical measurements were recorded using a BAS-100 W electrochemical potentiostat. The electrolyte solution was degassed by bubbling with dry argon for 10 min before measurement. Cyclic voltammograms were obtained in a three-electrode cell under argon. The working electrode was a glassy carbon disc (diameter 3 mm) successively polished with 3 and 1 μm diamond pastes and sonicated in ion-free water for 10 min. The reference electrode was a non-aqueous Ag/Ag+ electrode (1.0 mM AgNO3 in CH3CN) and the auxiliary electrode was a platinum wire.
3.2 Procedures
3.2.1 4-Diphenylphosphinoaminopyridine (7)
A stirred solution of 4-aminopyridine (4.7 g, 50 mmol) in dry CH2Cl2 (40 mL) was cooled to 0 °C under an atmosphere of N2 before diphenylchlorophosphine (4.5 mL, 25 mmol) was added over a period of 30 min. After warming the mixture to room temperature, the aminopyridine hydrochloride salt was filtered off and the solvent was removed from the filtrate to yield crude 4-(diphenylphosphinoamino) pyridine, which was recrystallized from hexane-dichloromethane to give white pure product (5.2 g, 75%). 1H NMR (CDCl3): δ (ppm) 6.92 (d, J = 12.0 Hz, 2H), 7.37–7.47 (m, 10H), 8.19 (d, J = 8.0, 2H). MS (API-ES): m/z 279.3 [M + H]+. 31P NMR (CDCl3): δ (ppm) 19.4 (s).
3.2.2 [(bpy)Re(CO)3-4-diphenylphosphinoaminopyridine] (CF3SO3) (6)
Complex 5 (185 mg, 0.4 mmol) and CF3SO3H (0.7 mL, 8 mmol) were stirred in CH2Cl2 (20 mL) for 3 h. Then the solvent was removed and the residue was washed with anhydrous diethyl ether (40 mL) before filtration. A solution of 7 (111 mg, 0.4 mmol) in EtOH (40 mL) was added to the filtrate and refluxed for 2 h. Finally, the solvent was removed on a rotary evaporator and the crude product was purified by column chromatography with dichloromethane/acetone (8:1) as an eluent to give complex 6 as a yellow solid (170 mg, 50%). 1H NMR (CD3COCD3): δ (ppm) 7.07 (d, J = 6.0 Hz, 1H, bpy), 7.26 (t, J = 9.2 Hz, 2H, py), 7.40–7.47 (m, 6H, ph), 7.57 (t, J = 7.2 Hz, 2H, ph), 7.63 (d, J = 6.4 Hz, 1H, bpy), 7.75–7.80 (m, 2H, ph), 7.97–8.01 (m, 2H, py), 8.31 (t, J = 8.2 Hz, 1H, bpy), 8.46 (t, J = 8.0 Hz, 1H, bpy), 8.52 (d, J = 8.4 Hz, 1H, bpy), 8.76 (d, J = 8.0 Hz, 1H, bpy), 8.99 (d, J = 4.4 Hz, 1H, bpy), 9.42 (d, J = 4.4 Hz, 1H, bpy). 13C NMR (CD3COCD3): δ (ppm) 120.67, 123.87, 125.39, 125.80, 128.95, 129.64, 130.07, 130.65, 131.26, 132.26, 133.23, 138.42, 141.22, 142.21, 152.44, 154.86, 156.68, 158.07. 31P NMR (CD3COCD3): δ (ppm) 111.31 (s). IR (CH2Cl2): ν(CO) 2029, 1925 and 1914 cm−1; MS (API-ES): m/z 705.3 [M − CF3SO3−]+. HRMS (ES) m/z calcd for C30H23N4O3PRe 705.1065, found: 705.1055.
3.2.3 [(μ-SCH2NnPrCH2S)Fe2(CO)5{(bpy)Re(CO)34-diphenylphosphinoaminopyridine}] (CF3SO3) (4)
A solution of 8 (94 mg, 0.22 mmol) [18] and Me3NO·2H2O (24 mg, 0.22 mmol) in CH3CN (40 mL) was stirred for 10 min at room temperature and 6 (187 mg, 0.22 mmol) in CH3CN (5 mL) was added. After 3 h, the solvent was removed and the residue was washed with CH2Cl2/acetone (3:1) as eluent to give 4 (220 mg, 80%). 1H NMR (CD3COCD3): δ (ppm) 0.67 (t, J = 7.4 Hz, 3H), 1.14–1.18 (m, 2H), 2.26 (t, J = 7.8 Hz, 2H), 2.84 (d, J = 13.6 Hz, 2H), 2.94 (d, J = 11.6 Hz, 2H), 6.69 (d, J = 6.8 Hz, 2H, py), 7.53 (s, 6H, ph), 7.79 (t, J = 8.8 Hz, 4H, ph), 7.93 (t, J = 3.4 Hz, 2H, bpy), 7.98 (d, J = 6.4 Hz, 2H, py) 8.46 (t, J = 7.6 Hz, 2H, bpy), 8.76 (d, J = 8.4 Hz, 2H, bpy), 9.37 (d, J = 5.2 Hz, 2H, bpy). 13C NMR (CD3COCD3): δ (ppm) 11.74, 20.02, 51.44, 61.29, 114.67, 128.46, 128.79, 129.68, 130.69, 131.94, 135.55, 136.41, 136.86, 142.71, 150.41, 151.18, 211.09, 213.04. 31P NMR (CD3COCD3): δ (ppm) 95.49 (s). IR (CH2Cl2): ν(CO) 2049, 2032, 1986, 1978, 1956, 1927, and 1914 cm−1; MS (API-ES): m/z 1105.8 [M − CF3SO3−]+. HRMS (ES) m/z calcd for C42H34Fe2N5O8PReS2 1105.9843, found: 1105.9879. Elemental analysis (%) calcd for C41H34F3Fe2N5O11PReS3 CH3COCH3: C, 40.25; H, 3.07; N, 5.33. Found: C, 40.42; H, 3.13; N, 4.91.
4 Conclusion
In conclusion, a novel trimetallic Re–Fe complex 4 has been synthesized through the coordination of –PPh2 of a rhenium photosensitizer to a diiron catalyst with the assistance of the decarbonylation reagent Me3NO. Spectral data (IR, UV–vis) and electrochemistry indicate that there exists some interactions between Re and Fe moieties of complex 4. The rhenium moiety might act as a model for the cysteine-linked [Fe4S4] cluster, which is part of the electron-transfer chain to and from the active site of Fe-only hydrogenases, and it also gives a new model for light-driven proton reduction. Further investigations on excited-state properties and light-induced hydrogen production of system 4 are in progress.
Acknowledgements
We are grateful to the Ministry of Science and Technology of China (Grant No. 2001CCA02500) and the Chinese National Natural Science Foundation (Grant No. 20633020) for financial supports of this work. We also thank the Swedish Energy Agency, K & A Wallenberg Foundation and the Swedish Research Council for financial support of this work. This work was also supported by Program for Changjiang Scholars and Innovative Research Team in University (IRT0711).