

1 Introduction
Hydrogenases are enzymes adept at performing the reversible interconversion between proton and dihydrogen. Among two major kinds of metal-containing hydrogenases, i.e., FeFe-hydrogenase and NiFe-hydrogenase [1–4], the first type is extensively studied because of its high efficiency in H2 production and thus the possibility of achieving H2 production technologies of practical use [5]. Since X-ray crystallographic studies on FeFe-hydrogenase revealed the active site to be composed of a “butterfly” diiron unit which is linked to a typical Fe4S4 cuboidal subcluster [6–8,10], great efforts have been made to synthesize diiron models incorporating some structural or functional features of the active site. Due to closely resembling the diiron unit of the FeFe-hydrogenase, complexes of general formula [Fe2(CO)6(μ-SR)2], the so-called Reihlen type, attract interest of chemists renewedly [1,9], with the result that many ADT (1,3-azadithiolate) or PDT (1,3-dithiolate) bridged model complexes combining various terminal ligands, such as phosphine, cyanide, pyridine, carbine, have been synthesized [1,10,11]. Although a few of the model complexes have been designed to promote the reduction of protons to dihydrogen at relatively high potentials [12], there are still many problems to be addressed, such as overpotential and dioxygen sensitivity [13]. Theoretical studies pointed out that “asymmetric substitution of strong donor ligands is the most viable method of making better synthetic diiron complexes that will serve as both structural and functional models” [13], and in the meantime we find that the influence of the bridge between two Fe atoms is also a key factor [14], for which our work has focused on both the “bridge” and asymmetric substitution.
As a potential alternative to electrocatalysis, photosensitizers are good candidates to be linked to the bridge with the possibility of driving H2 activation by light [12]; diiron models with bridging aromatic ring, which could combine various ligands, including photosensitizers, are excellent substrates for this task [15]. Somewhat surprisingly, aromatic-thiolate-functionalized diiron models are paid little attention in the past decades. In the context of our interest in photochemical and to investigate the effects of aromatic rings on 2Fe2S cores, we turned to study mono-S-aromatic ring bridged biomimetic complexes obtained by oxidative addition reaction of iron(0) carbonyl compounds with thiols [9]. Here we describe some preliminary results of our studies in this field.
2 Experimental
2.1 Material and instruments
All reactions were carried out under dry, oxygen-free nitrogen using standard Schlenk techniques. Analytical-grade reactants, such as Fe3(CO)12, aromatic thiols and PMe3, were commercially available and used as-received. Solvents were dried and distilled prior to use according to standard methods. Elemental analysis was carried out on a Vario EL III Elemental Analyser. IR spectra were taken on a SpectrumOne FT-IR spectrophotometer using KBr pellets in the range of 4000–400 cm−1. 1H NMR spectra were collected on a Varian Unity 500NMR spectrometer. Mass spectra were recorded on a DECAX-3000 LCQ Deca XP instrument.
2.2 Synthesis of hexacarbonyldithiolatodiiron complexes (1–4)
Thiophenol (2 equiv, 0.6 ml, 6 mmol) was added to a solution of Fe3(CO)12 (1.5 g, 3 mmol) in 40 ml THF under a nitrogen atmosphere. The mixture was refluxed for 4 h at 70 °C, until the color turned from dark green to dark red. After cooling to room temperature, the solvent, i.e. THF, was evaporated under vacuum. The remaining solid was dissolved in a minimum amount of n-hexane, followed by filtration through silica gel. A red fraction was collected by elution with n-hexane. Recrystallization of the crude product from freshly distilled hexane at −18 °C gave compound 1 (Scheme 1).

Synthesis of complexes 1–4.
Compound 1. Yield 1.13 g, 76%, red solid (found: C, 43.21; H, 2.33; Calc. for C18H10Fe2O6S2: C, 43.41; H, 2.02%); δH (500 MHz, CDCl3, Me4Si): 7.20 (10H, 2C6H5); m/z 499.5 (M − H+).
Complexes 2–4 were prepared by procedures similar to those of 1, with differences arising from the corresponding aromatic thiols used as reactants, o-CH3O–C6H4–SH for 2, p-CH3O–C6H4–SH for 3 and p-Cl–C6H4–SH for 4. Crystals of 2 and 3 suitable for X-ray analysis were obtained from concentrated solution of hexane at −18 °C, respectively, and 4 from the concentrated solution of hexane layered with Et2O at room temperature.
Compound 2. Yield 1.20 g, 72%, red solid (found: C, 42.85; H, 2.20; Calc. for C20H14Fe2O8S2: C, 43.04; H, 2.53%); δH (500 MHz, CDCl3, Me4Si): 7.33–7.42 (2H, 12-H, 19-H), 7.20–7.29 (2H, 10-H, 17-H), 6.91–7.00 (2H, 11-H, 18-H), 6.81–6.83 (2H, 9-H, 16H), 3.92 (6H, 2OCH3); m/z 559.2 (M − H+).
Compound 3. Yield 1.10 g, 66%, red solid (found: C, 42.58; H, 2.1; Calc. for C20H14Fe2O8S2: C, 43.04; H, 2.53%); δH (500 MHz, CDCl3, Me4Si): 7.24–7.31 (4H, 8-H, 12-H, 15-H, 19-H), 6.80–6.81 (4H, 9-H, 11-H, 16-H, 18-H), 3.75 (6H, 2OCH3); m/z 559.2 (M − H+).
Compound 4. Yield 1.19 g, 70%, red solid (found: C, 39.00; H, 1.73; Calc. for C18H8Fe2O6S2Cl2: C, 38.13; H, 1.42%); δH (500 MHz, CDCl3, Me4Si): 7.25–7.37 (8H, 2C6H4); m/z 568.8 (M − H+).
2.3 Synthesis of PMe3-disustituted complex (5)
PMe3 (2 mM, 2 ml) was added to a solution of 2 (0.279 g, 0.5 mmol) in 10 ml of freshly distilled hexane under a nitrogen atmosphere. After 12 h of stirring, the color changed from red to purple, and some purple solid was deposited. The solvent and unreacted PMe3 were removed under vacuum. The resulting brown residue was dissolved in CH2Cl2 and filtered through silica gel by flash chromatogram. The purple-red fraction was collected. Recrystallization in distilled CH2Cl2 gave black crystals suitable for X-ray analysis (Scheme 2).
Synthesis of complex 5.
Compound 5. Yield 0.29 g, 90%, red solid (found: C, 44.5; H, 4.8; Calc. for C24H32Fe2O6S2P2: C, 44.04; H, 4.93%); δH (500 MHz, CDCl3, Me4Si): 7.548 (1H, 14-H, 21-H), 7.246–7.149 (4H, 13-H, 15-H, 20-H, 22-H), 6.721 (2H, 16-H, 23-H), 3.825 (6H, 2OCH3), 1.524 (18H, 2PMe3); δP (202 MHz, CDCl3, Me4Si): 21.138 (PMe3); m/z 655.5 (M − H+).
2.4 Electrochemistry
Acetonitrile (spectroscopy grade) was used as the solvent for the electrochemistry with a solution of 0.1 M n-Bu4NPF6 in MeCN being used as the electrolyte. Before measurements, the electrolyte solution was degassed by bubbling Ar through it for 15 min. All results were obtained at a scan rate of 100 mV/s by using a CHI630A potentiostat and a three-electrode cell under argon. The working electrode was a glassy carbon disc (diameter 3 mm) polished with 0.5-μm diamond pastes and washed with MeCN before use. The reference electrode was an Ag/AgCl electrode (3 M KCl in H2O) and the auxiliary electrode was a platinum wire. All potential data are quoted against the Fc/Fc+ potential in CH3CN.
2.5 X-ray structure determination of complexes 3 and 5
Single crystals of complexes 3 and 5 were performed on a Mercury-CCD diffractometer equipped with a graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). All data were collected at 293(2) K by using a ω–2θ scanning mode. An empirical absorption correction was made by multi-scan type. These structures were solved by direct methods and refined by full-matrix least-squares techniques with SHELX-97 program [16,17]. Anisotropic displacement parameters were refined for all non-hydrogen atoms. The hydrogen atoms were added in the riding model and not refined. Crystallographic data are outlined in Table 1.
Crystallographic data summary for complexes 3 and 5
| 3 | 5 | |
| Empirical formula | C20H14Fe2O8S2 | C24H32Fe2O6P2S2 |
| Formula weight | 558.13 | 654.26 |
| Space group | P2(1)2(1)2(1) | P2(1)/n |
| a [Å] | 8.9277(6) | 10.111(6) |
| b [Å] | 12.7546(8) | 15.363(6) |
| c [Å] | 20.2760(13) | 19.629(8) |
| α [°] | 90 | 90 |
| β [°] | 90 | 98.20(2) |
| γ [°] | 90 | 90 |
| V [Å3] | 2308.8(3) | 3018(2) |
| Z | 4 | 4 |
| ρcalcd [g/cm3] | 1.606 | 1.440 |
| λ (Mo Kα) [Å] | 0.71073 | 0.71073 |
| T [K] | 293(2) | 293(2) |
| μ [mm−1] | 1.480 | 1.240 |
| Ra | 0.0499 | 0.0719 |
| wRb | 0.0853 | 0.1885 |
a R = ∑||Fo| − |Fc||/∑|Fo|.
b wR = [∑w(|Fo| − |Fc|)2/∑wFo2]1/2.
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (CCDC #667910 and #667911), and can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the address mentioned above; fax: (internat.) +44-1223/336-033; e-mail: deposit@ccdc.cam.ac.uk].
3 Results and discussion
3.1 Molecular structures of 1–5
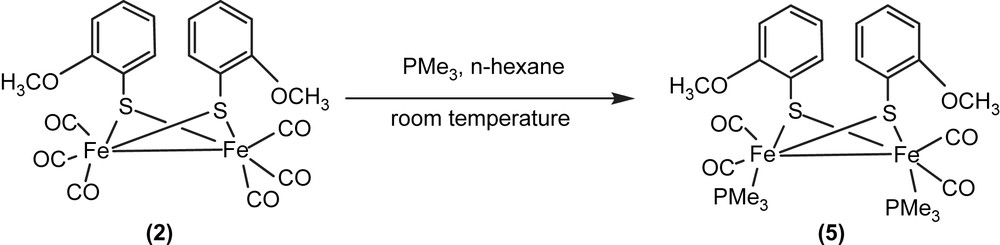
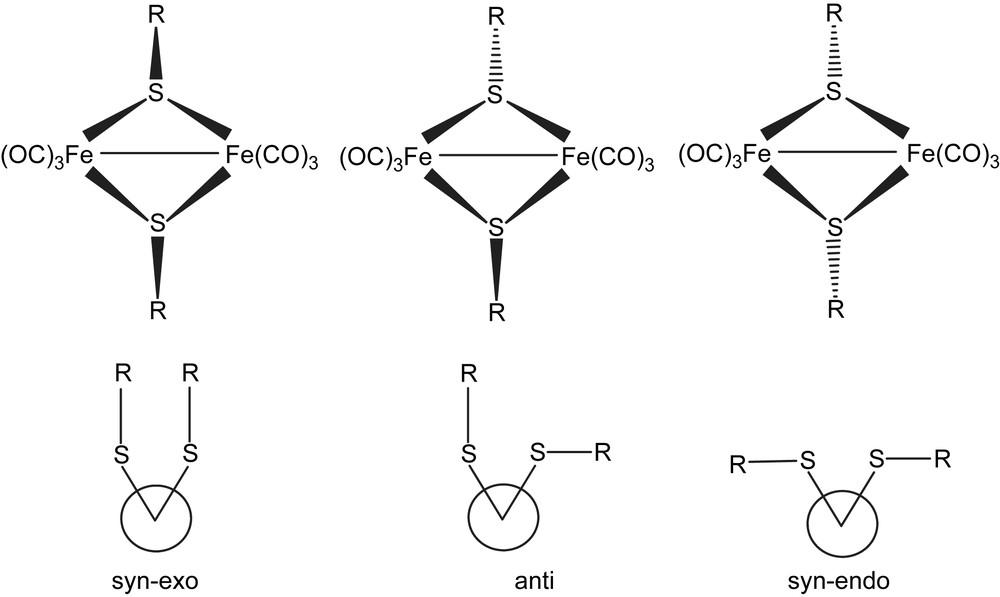
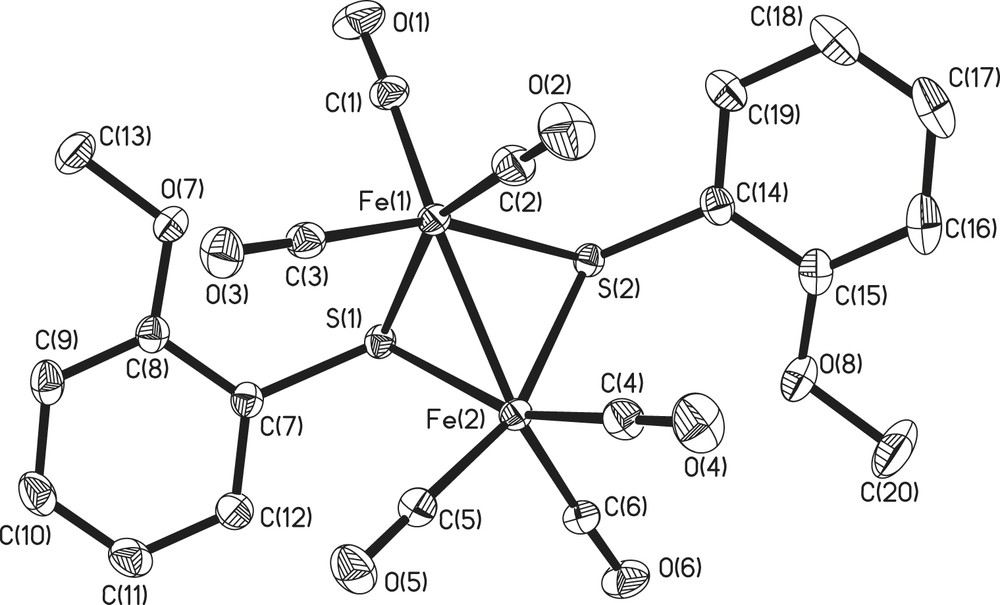
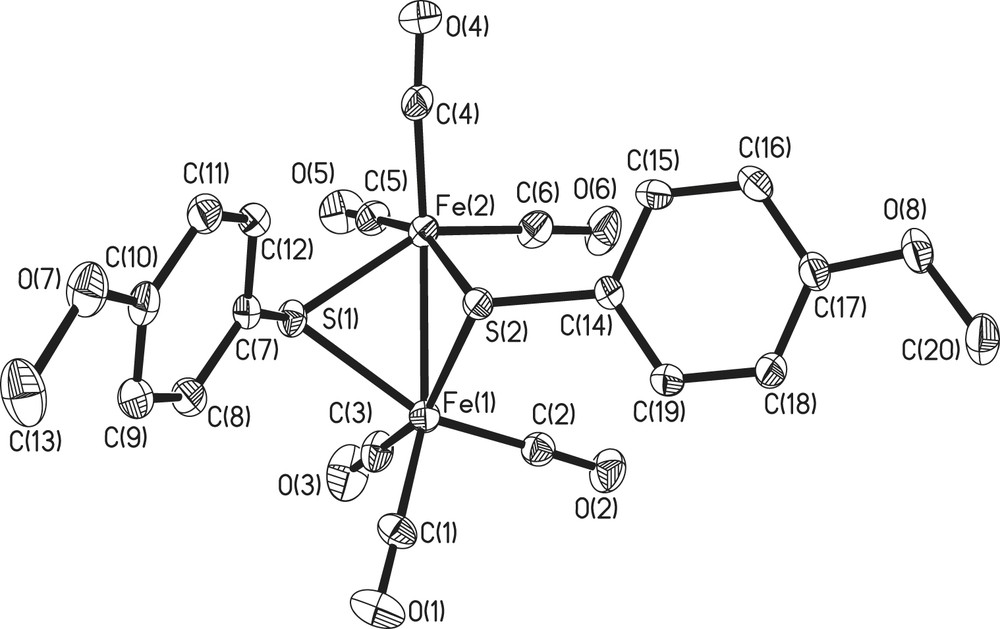
All the five complexes have 2Fe2S cores of butterfly conformation. There are three possible stereoisomers for 2Fe2S core, as shown in Fig. 1, and only two types have been observed, with 1, 3–5 belonging to anti-type and 2 syn–endo-type (Figs. 2–4). Compounds 2 and 4 have wider Fe(1)–Fe(2)–S(1)–Fe(1)–Fe(2)–S(2) dihedral angle (93, 99.1°, respectively) than the others (78–80°). The Fe–Fe bond lengths of all-carbonyl species (1–4) are similar (ca. 2.50 Å), implying that the substitutes on the phenyl ring have little effects on the Fe–Fe bond, while that of 5 is significantly longer (2.5540 Å), which is caused by the introduction of PMe3. All the values described above are in agreement with the results observed from ADT or PDT bridged analogues, Fe2(CO)6(μ-SCH2)2–N–C5H9 (6), Fe2(CO)4(PMe3)2(μ-SCH2)2–N–C5H9 (7), Fe2(CO)6(SCH2CH2CH2S) (8), (Table 2) [14,15]. It is notable that in 2 the two methoxyl ligands are on both sides of the C(7)–S(1)–S(2) plane, while in 5 two methoxyl ligands are at the same side of C(18)–S(2)–S(1) plane, which indicates fluxionality of the phenyl ring in solution. In 5, two phosphine ligands feature an apical/basal configuration, which is sterically less crowded and controlled by electronic effects (Figs. 2 and 4).
Three stereoisomers of 2Fe2S core (top: wedge-and-dash structures, bottom: Newman projections down the Fe–Fe axis).
Crystal structure of complex 2.
Crystal structure of complex 3.
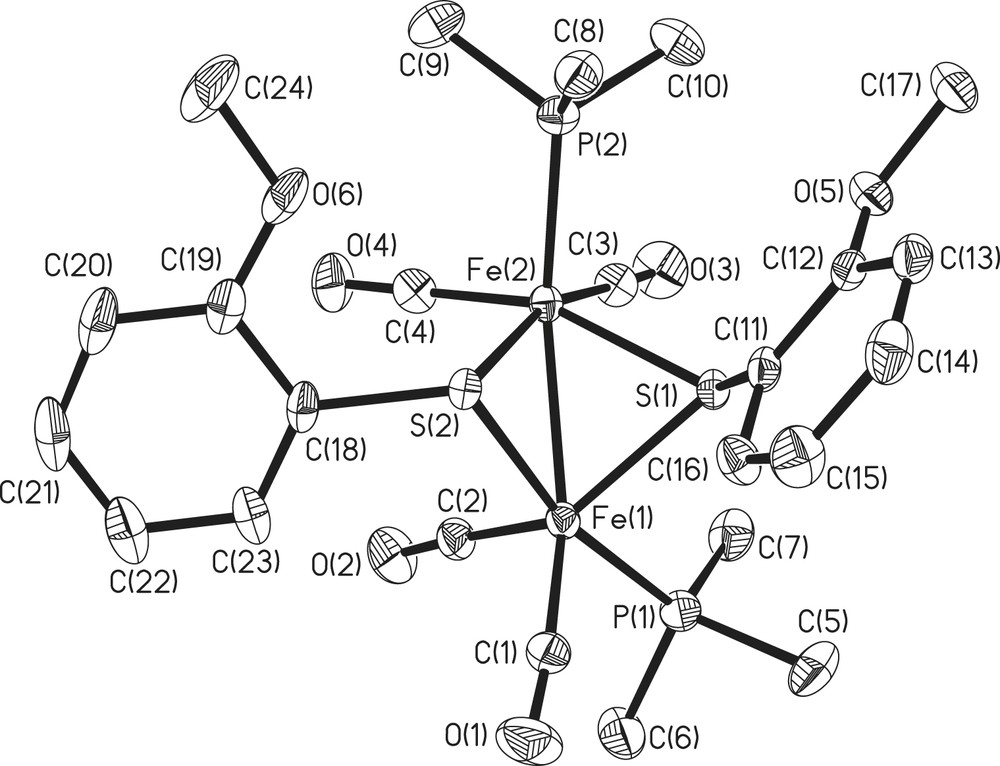
Crystal structure of complex 5.
Selected crystal structure data of complexes 1–8
| Complex | Fe–Fe bond length [Å] | Fe(1)–Fe(2)–S(1)–Fe(1)–Fe(2)–S(2) dihedral angle [°] |
| 1 | 2.5092(9) | 79 |
| 2 | 2.4979(5) | 93 |
| 3 | 2.5081(9) | 79.8 |
| 4 | 2.5029(11) | 99.1 |
| 5 | 2.5540(14) | 78 |
| 6 | 2.5025(7) | 72.3 |
| 7 | 2.5279(13) | 72.6 |
| 8 | 2.510 | 70.6 |
3.2 IR spectroscopic characterization of complexes 1–5
The ν(CO) is similar for each of the all-CO species (1–4), consistent with their similar Fe–Fe bond lengths. The average ν(CO) value of 1–4 is somewhat higher than that of cycloalkyl-ADT species, such as Fe2(CO)6(μ-SCH2)2N–C6H11 (9) [14], while lower than that of Fe2(CO)6(μ-PDT) (8) [18]. The average value of the PMe3 substituted complex (5) is about 100 cm−1 lower than that of 1–4, and is somewhat lower than that of Fe2(CO)4(PMe3)2(μ-pdt) (10) (Table 3) [18]. These observations reflect a relatively stronger electron-donating ability of cycloalkyls than that of aromatic rings, while the ability of PDT is relatively lower, because enhancement of electron accumulation on Fe atoms would lead to a stronger back-bonding from Fe atoms to CO and a weakening of the CO triple bonds.
Comparison of ν(CO) bands in complexes 1–5 and 8–10
| Complex | ν(CO) |
| 1 | 2072, 2037, 2005, 1992, 1982 |
| 2 | 2074, 2031, 2007, 1992, 1980 |
| 3 | 2071, 2037, 2002, 1994, 1979 |
| 4 | 2073, 2032, 1994, 1985, 1972 |
| 5 | 1978, 1921, 1883 |
| 8 | 2074, 2036, 1995 |
| 9 | 2071, 2023, 1979 |
| 10 | 1979, 1942, 1898 |
The 31P NMR spectra of 5 in solution show a singlet, indicating fast fluxionality of PMe3.
3.3 Cyclic voltammograms in the absence/presence of HOAc
Complexes 1–5 each display two reduction peaks. Complexes 2, 3 and 5 manifested two oxidation peaks, while only one was observed for 1 and 4. Because methoxyl is a better electron-donating substitute, the electron accumulations on Fe atoms of 2 and 3 are richer than those of 1 and 4, which led to the reduction processes of 2 and 3 occurring at more negative potentials. The CV data of 5 were significantly shifted in a cathodic direction compared to 1–4, again because of strong electron-donating ability of PMe3.
All the electrochemical data, together with those of 8, Fe2(CO)6(μ-S–Et)2 (11), Fe2(CO)6(1,2-bi-SCH2–C6H4) (12), Fe2(CO)4(PMe3)2(μ-S–Et)2 (13) and Fe2(CO)6(μ-SCH2)N(C6H4OMe-p) (14), are listed in Table 4 [18,19]. The data of 1–4 are similar to those of 12, while 11 shows its reduction peaks at much more negative potentials when compared with 1–4, resulting from the difference between ethyl and aromatic ring. Also, PMe3 substituted complex 5 is more readily reduced than 13. These results indicate that aromatic ring-functionalized 2Fe2S models were reduced relatively easily.
Electrochemical data of 1–5 and 8, 11–14 (V vs Fc+/Fc)
| Complex | Epc [V] | Epc [V] | Epa [V] | Epa [V] |
| 1 | −1.44 | −2.26 | 0.81 | |
| 2 | −1.55 | −2.29 | 0.73 | 1.08 |
| 3 | −1.51 | −2.42 | 0.93 | 1.06 |
| 4 | −1.35 | −2.11 | 0.79 | |
| 5 | −2.08 | −2.45 | −0.23 | 1.03 |
| 8 | −1.74 | −2.35 | 0.74 | |
| 11 | −1.71 | −2.34 | 0.78 | |
| 12 | −1.56 | −2.21 | 0.80 | |
| 13 | −2.31 | −0.13 | ||
| 14 | −1.61 | −2.10 | 0.48 | 0.81 |
The reduction behaviors of complexes 1–5 were further studied by cyclic voltammetric techniques in the presence of HOAc, as a proton source.
As shown in Fig. 5, the first electrochemically irreversible reduction peak of 1 grew slightly with added increments of acid, indicative of H+ not being attached to the model complex at this stage, while the second reduction event at −2.26 V increased linearly with the concentration of acid, implying that H+ was introduced and electrocatalysis occurred at this potential. It is also notable in Fig. 5 that a new reduction wave emerged at about −1.70 V. Complexes 2–4 had catalytic cycles similar to those of 1, all manifesting three reduction events.
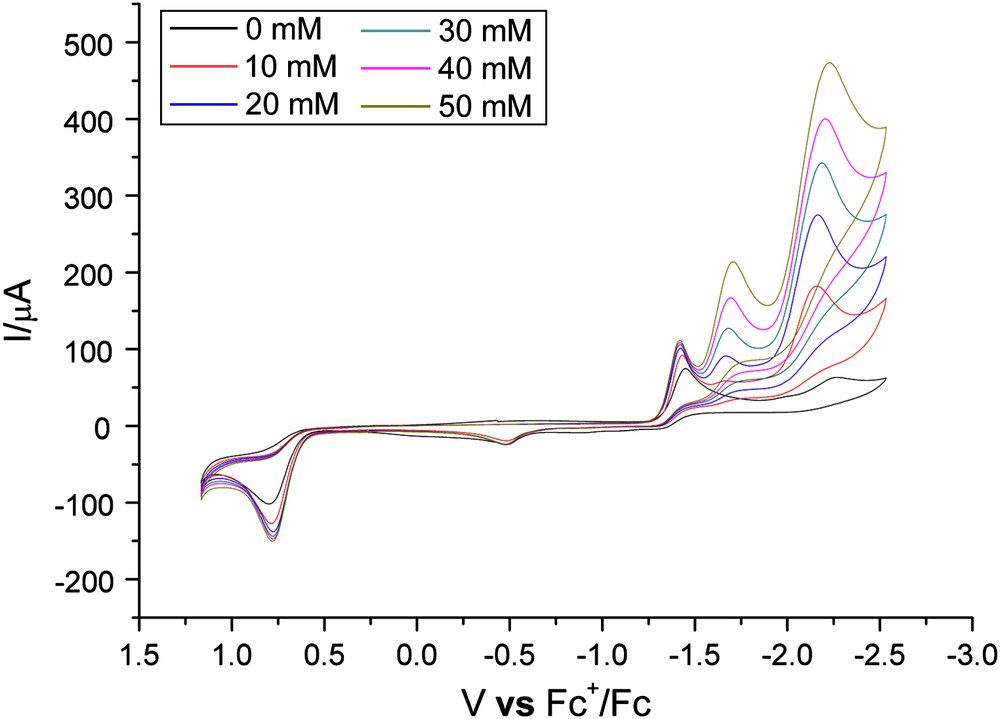
Cyclic voltammogram of 1 (2.0 mM) with HOAc.
A new case was found for 5 as indicated in Fig. 6. Both reduction peaks grew with the increment of acid, which is significantly different from the electrochemical behaviors of 13 [18]. After the sixth equivalent of HOAc was added, the two reduction peak heights no longer increased. Both reduction events of 5 are assumed to be H+-related.
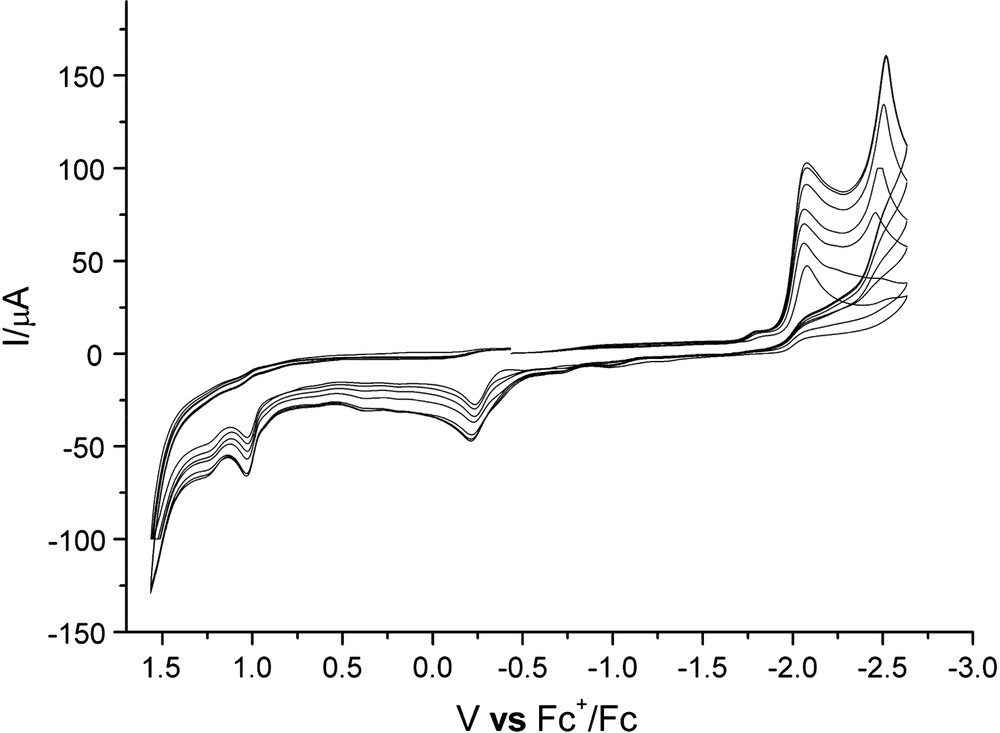
Cyclic voltammogram of 5 (2.0 mM) with HOAc (from 0 equiv to 6 equiv).
The catalytic mechanism of the catalytic processes by 1–5 will be the subject of another article.
4 Conclusion
Aromatic ring-functionalized 2Fe2S models incorporate key structural features of the diiron unit of FeFe-hydrogenase, possessing relative positive reduction potentials, for which they are promising models on the way of seeking dihydrogen catalysts. After being substituted, both reduction events of 5 are H+-related, which are different from those of 1–4. As a potential substrate to link photosensitizers, this species should be paid more attention and further studies are underway in our laboratory.
Acknowledgment
We are grateful to the National Key Foundation of China (No. 20633020), the Science & Technology Innovation Foundation of Fujian Province (No. 2007F3112), and the National Nature Science Foundation of China (No. 20471061) for financial supports of this work.