1 Introduction
Iron-carbonyl is a motif found at the active site of hydrogenases [1]. These metallo-enzymes catalyze efficiently the reduction of protons from water into dihydrogen. Their functional modeling could thus help finding catalysts alternative to platinum to be used in water electrolysers [2]. Hydrogenases are divided into two main classes, namely FeFe and NiFe, depending on the metal content of their active sites (Fig. 1); a third class, Fe hydrogenases, catalyzes the reversible dehydrogenation of methylene-tetrahydromethanopterin and also possesses an iron-carbonyl-based active site (Fig. 1) [3,4].
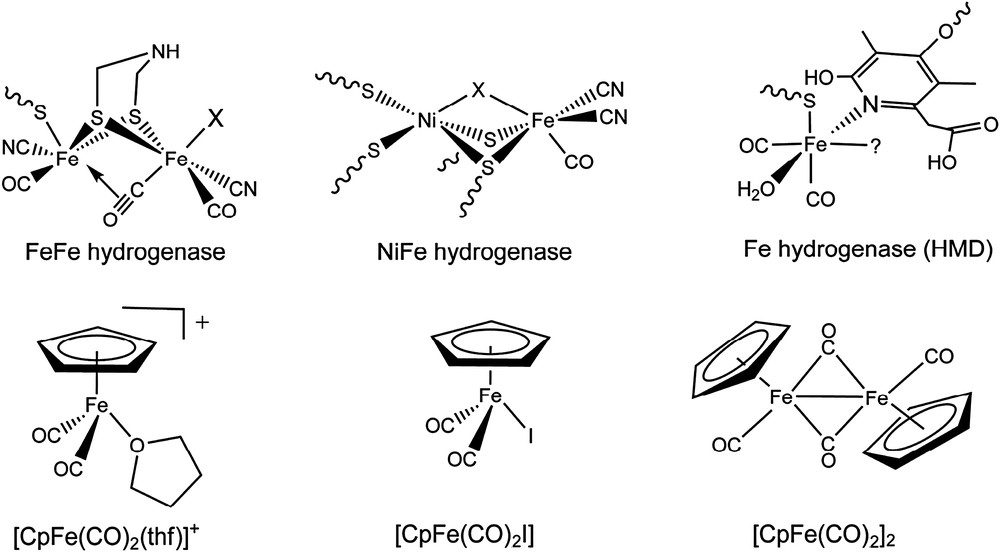
Molecular structure of the active sites of hydrogenases and model complexes considered in this study.
A great number of diiron complexes as both structural and functional mimics of FeFe hydrogenases are reported in the literature [5,6]. Regarding NiFe model compounds, the best functional mimics reported so far are nickel–ruthenium complexes [7,8]. Yet several dinuclear nickel–iron structural models have been synthesized, but either they do not display any catalytic activity or were not assayed for it [9].
Some of these structural models contain the {CpFe} moiety (Cp− = cyclopentadenyl anion) [10,11], but no catalytic hydrogenase activity has been reported in the literature for iron–cyclopentadienyl compounds. It has, however, been demonstrated by Darensbourg and coworkers that the Cp− ligand has electronic properties similar to the {Ni(SCys)2(X)} moiety found in the NiFe hydrogenase bimetallic cluster (Fig. 1) [12]. Furthermore, [CpFeII(CO)2Br] was used as a model by Böck and coworkers in the context of the biosynthesis of NiFe hydrogenase active site: this iron complex cleaves phenylthiocyanate in the presence of phenylthiolate and binds the cyanide reaction product [13].
The complex [CpFeII(CO)2(thf)]+ (Fig. 1) [14] appears as an attractive biomimetic candidate to be evaluated as a catalyst for hydrogen production for the following reasons: (i) it contains a labile ligand at the iron center, (ii) by analogy with [CpFeII(CO)2X] (X = Cl, Br, I) complexes, it should be reducible to the Fe0 state at relatively mild potentials [15], and (iii) the Fe0 species is highly nucleophilic and protonates easily to yield the quite stable and isolated hydride derivative [CpFeII(CO)2H] [16,17]. Moreover, derivatives of {CpFe(CO)2} are quite stable and straightforward inexpensive routes are available for their synthesis.
In the following, we describe the electrocatalytic properties of this complex for proton electro-reduction in DMF.
2 Experimental section
2.1 Materials
[CpFe(CO)2(thf)](BF4) was prepared according to the reported procedure [14]. The supporting electrolyte (n-Bu4N)BF4 was prepared from (n-Bu4N)HSO4 (Aldrich) and NaBF4 (Aldrich) and dried overnight at 80 °C under vacuum. [CpFe(CO)2I] (Aldrich), [CpFe(CO)2]2 (Ventron), triethylammonium chloride (Acros) and trichloroacetic acid (SDS) were used as received.
2.2 Methods and instrumentation
All electrochemical measurements were carried out in DMF under nitrogen at room temperature. Commercial DMF was used as received and degassed by bubbling nitrogen for 10 min. A standard three-electrode configuration was used consisting of a glassy carbon (3 mm in diameter, Radiometer) disk as the working electrode, an auxiliary platinum wire and an Ag/AgCl/aqueous AgClsat + KCl 3 mol L−1 (named Ag/AgCl through this text) reference electrode closed by a Vycor frit and directly dipped into the solution. In order to take into account the liquid junction potential between aqueous and non-aqueous solution, this electrode was calibrated with the internal reference system Fc+/Fc, which was found at 0.53 V vs Ag/AgCl in DMF. The Fc+/Fc couple (E0 = 0.400 V vs SHE)[53] can be used to quote potentials to SHE, when needed.
Cyclic voltammograms were recorded on an EG&G PAR 273A instrument. Solution concentrations were 1 mmol L−1 for the catalyst and 0.1 mol L−1 for the supporting electrolyte (n-Bu4N)BF4. Electrodes were polished on a MD-Nap polishing pad with a 1-μm monocrystalline diamond DP suspension and DP lubricant blue (Struers). Additions of trichloroacetic acid from a 0.1 mol L−1 solution in the electrolytic solution were made with the aid of a syringe.
Bulk electrolysis and coulometry were carried out on an EG&G PAR 273A instrument in DMF, using a 2-cm2 graphite cathode. The platinum-grid counter-electrode was placed in a separated compartment connected with a glass-frit and filled with a 0.1 mol L−1 solution of (n-Bu4N)BF4 in degassed DMF. A degassed DMF solution (10 mL) containing 0.1 mol L−1 (n-Bu4N)BF4 and 0.1 mol L−1 of TCA was first electrolyzed at −1.0 V vs Ag/AgCl until the current dropped below 500 μA. The catalyst was then added as a solid to reach a final {CpFe(CO)2} concentration of 1 mmol L−1 and electrolysis was then performed at the same potential for 2 h. Hydrogen was tested for purity using a Delsi Nermag DN200 GC chromatograph equipped with a thermal conductivity detector (TCD) detector.
3 Results
3.1 Cyclic voltammetry
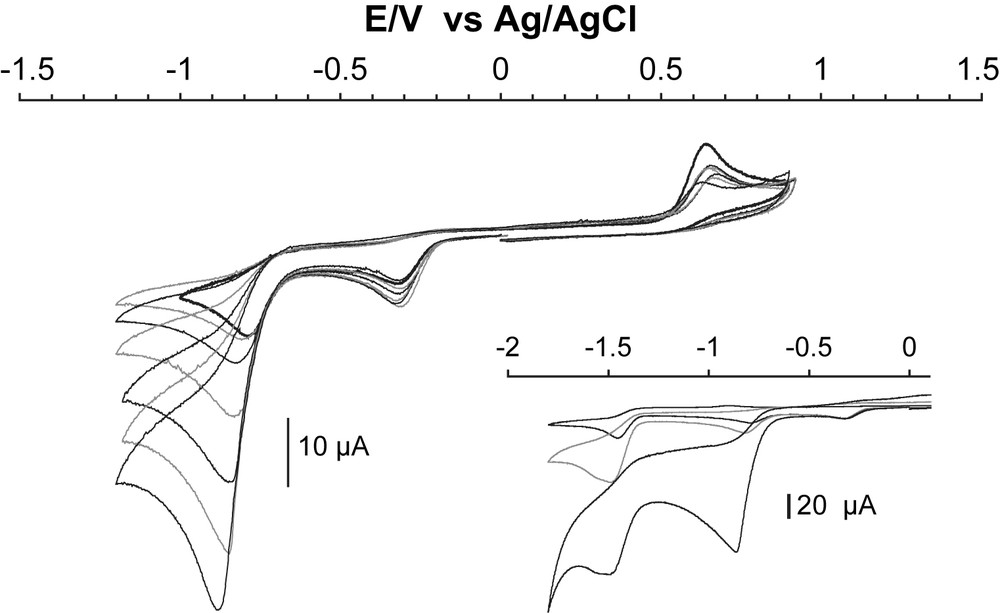
The cyclic voltammogram of [CpFe(CO)2(thf)](BF4) in DMF (Fig. 2) displays two irreversible one-electron cathodic waves at −0.33 V and −0.80 V vs Ag/AgCl that can be assigned to the stepwise reduction of iron(II) to iron(I) and iron(I) to iron(0), respectively. The first formed species is expected to be [CpFe(CO)2(thf)], from which THF is probably released by analogy with [CpFe(CO)2I]− which eliminates I− rapidly [15]. Such an EC mechanism likely explains the chemical irreversibility of this first process. The resulting 17-electron species [CpFe(CO)2] may rapidly dimerize to yield [CpFe(CO)2]2 (Eq. (1)) [18]. This reaction does, however, not occur rapidly enough at the vicinity of the glassy carbon electrode surface since, as indicated above, the cyclic voltammogram displays a second monoelectronic wave at −0.80 V vs Ag/AgCl, which is not observed in the case of an authentic sample of [CpFe(CO)2]2 under the same conditions.1
We assign the cathodic wave at −0.80 V vs Ag/AgCl (second process) to the reduction of [CpFe(CO)2] into [CpFe(CO)2]−. This species likely reacts with [CpFe(CO)2(thf)], diffusing from the bulk to yield the dimeric [CpFe(CO)2]2 (Eq. (2)) [19], as revealed by the observation of an irreversible bielectronic wave at −1.44 V vs Ag/AgCl on the same cyclic voltammogram. Indeed, the cyclic voltammogram of an authentic [CpFe(CO)2]2 sample (Fig. 2) displays a similar wave at −1.44 V vs Ag/AgCl under the same conditions, corresponding to the formation of [CpFe(CO)2]− from [CpFe(CO)2]2 through the cleavage of the metal–metal bond [20].
| 2[CpFe(CO)2] → [CpFe(CO)2]2 | (1) |
| [CpFe(CO)2(thf)]+ + [CpFe(CO)2]− → [CpFe(CO)2]2 + THF | (2) |
| [CpFe(CO)2]+ + [CpFe(CO)2]− → [CpFe(CO)2]2 | (2′) |
| [CpFe(CO)2]+ + [CpFe(CO)2H] → [CpFe(CO)2] + [CpFe(CO)2H]+ | (3) |
| [CpFe(CO)2H]+ + [CpFe(CO)2H] → [CpFe(CO)2] + [CpFe(CO)2]+ + H2 | (4) |
3.2 Electrocatalytic hydrogen evolution
Addition of increasing amounts of trichloroacetic acid (TCA, pKa = 3.5 in DMF) [21] to a solution of [CpFe(CO)2(thf)]+ in DMF results in two modifications: first, a catalytic wave assigned to hydrogen evolution develops on the monoelectronic wave initially located at −0.80 V vs Ag/AgCl and corresponding to the formation of [CpFe0(CO)2]− (Fig. 3). Secondly, the bielectronic wave at −1.44 V vs Ag/AgCl is replaced by a catalytic wave at the same potential (see the inset in Fig. 3), the intensity of which is not strongly dependent on the concentration of acid.2
A controlled-potential coulometry experiment was carried out to attest to the catalytic nature of the first process (Fig. 4). A carbon electrode was set to −1.0 V vs Ag/AgCl in a 0.1 mol L−1 solution of TCA in DMF. The background current passing through the cell in the absence of the catalyst is about 500 μA. Upon addition of [CpFe(CO)2(thf)](BF4) (1.0 mmol L−1), the current increases immediately, indicating hydrogen evolution catalysis. However, this current then levels off very rapidly after about 20 min while less than 4 C, corresponding to the achievement of about 2 turnovers, was passed through the cell. This denotes some deactivation of the catalyst.
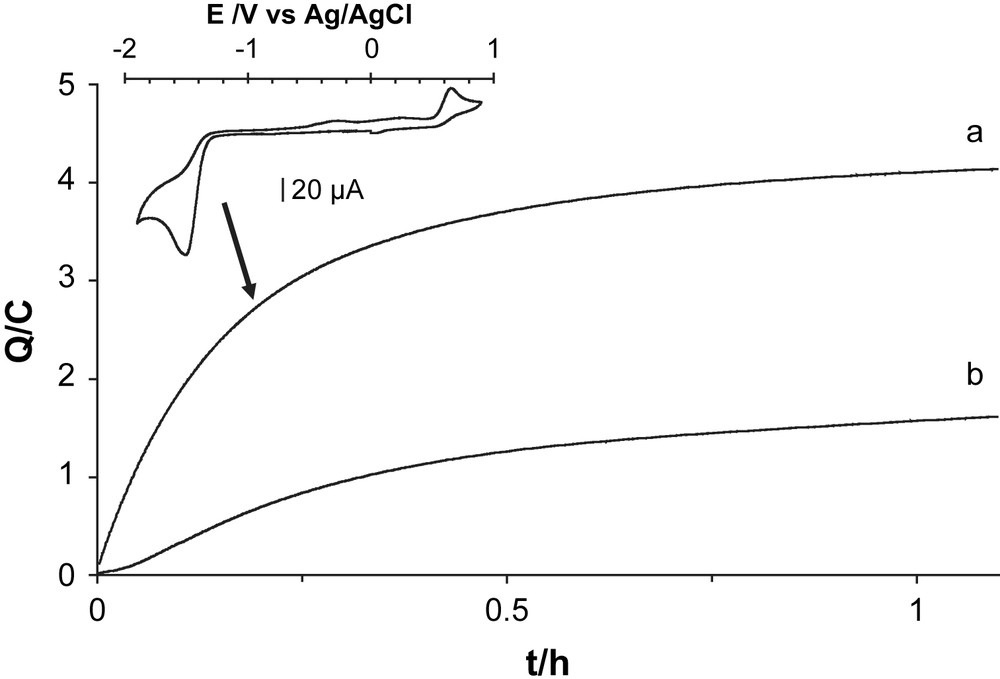
Coulometry for bulk electrolysis at −1.0 V vs Ag/AgCl of a DMF solution (10 mL) of TCA (0.1 mol L−1) and n-Bu4NBF4 (0.1 mol L−1) at a graphite electrode in the presence of [CpFe(CO)2(thf)](BF4) (1.0 mmol L−1, trace a), [CpFe(CO)2]2 (0.5 mmol L−1, trace b) after subtraction of the contribution due to direct reduction of TCA at the graphite electrode.
The cyclic voltammogram of the solution recorded after 20 min of electrolysis (inset in Fig. 4) did not display the cathodic waves at −0.33 V and −0.80 V vs Ag/AgCl anymore. By contrast, the catalytic process at −1.44 V vs Ag/AgCl is still present so that we can conclude that the electrolyzed solution exclusively contains the dimeric complex [CpFe(CO)2]2. This is confirmed by the color change of the solution from red to deep yellow.
In Fig. 4, we also provide the results for [CpFe(CO)2]2 in bulk electrolysis conditions: this compound has almost no catalytic properties for hydrogen electroproduction at −1.0 V vs Ag/AgCl in DMF in the presence of TCA (0.1 mol L−1).
4 Discussion
By analogy with the electrochemical response of various metal complexes under similar experimental conditions [7,8,22–24] the catalytic wave observed at −0.8 V vs Ag/AgCl is assigned to hydrogen evolution proceeding via the mechanism depicted in Fig. 5. The key intermediate is likely to be the known hydride derivative [CpFe(CO)2H] [17] resulting from the protonation of the Fe0 species. This is further supported by the fact that no catalytic wave was observed upon addition of a weaker acid such as Et3NH+ (chloride or tetrafluoroborate salt, pKa = 9.2 in DMF, data not shown). Actually Et3NH+ is not able to protonate [CpFe(CO)2]− as shown by the comparison of the pKas of Et3N (18.7) and [CpFe(CO)2H] (19.4) in CH3CN [17,25].
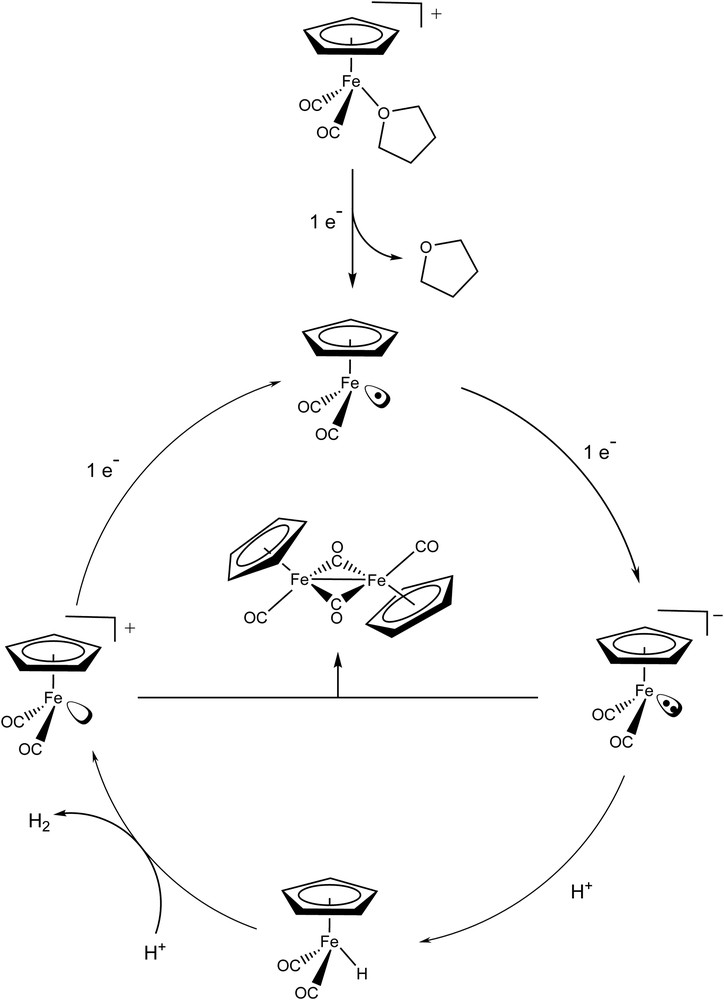
Proposed mechanism for H2-evolution catalyzed by [CpFe(CO)2(thf)]+.
Hydrogen evolution from a metal–hydride species can proceed from two main pathways: bimolecular reductive elimination or proton–hydride coupling. [CpFe(CO)2H] is known to be stable for long periods in neutral non-aqueous solutions and thus does not undergo fast bimolecular reductive elimination [17]. This allows us to propose that hydrogen evolution in our case proceeds via a heterolytic protonation step of this hydride regenerating an iron(II) species which can initiate a new cycle by reduction. Protonation of iron-hydride species generating hydrogen has already been reported for mononuclear [26] or binuclear systems [27–29].
However, the current enhancement (defined as the ratio ic/ip, with ic being the catalytic peak intensity and ip the peak intensity of a monoelectronic wave) is quite weak: up to 50 equiv of acid are needed to obtain a current enhancement of 3.7 which seems to be the upper limit. This limitation may be due either to a slow rate-determining step or to competitive side-reactions resulting in the inactivation of the catalyst. In the first hypothesis, the catalytic current observed in bulk electrolysis conditions is expected to be weak but constant during the first stages of the experiment. The rapid current decrease observed under such conditions allows us to decide for the second hypothesis. Furthermore we could electrochemically characterize the presence of dimeric [CpFeI(CO)2]2 in the solution after electrolysis and we could check that this compound is not active for catalytic hydrogen production under bulk electrolysis conditions at −1.0 V vs Ag/AgCl. We can thus conclude that dimerization and formation of [CpFeI(CO)2]2 is the major deactivation pathway. Finally, the catalytic behavior for hydrogen evolution observed at −1.44 V vs Ag/AgCl shows that [CpFe(CO)2]− keeps its entire activity when the deactivation pathway can be reverted. At this potential indeed, the dimer is electrochemically cleaved and [CpFe(CO)2]− is regenerated. This reaction is probably the rate-determining step in the catalytic mechanism, which explains why the catalytic current does not depend on the acid concentration.
Various recombination processes can account for dimerization. The most efficient process is certainly the reaction of the Fe0 species with FeII species, either introduced as starting compound or produced during the catalytic cycle (Eqs. (2) and (2′)) [19]. Two other processes may become significant at the time-scale of bulk electrolysis : (i) dimerization of the 17-electron intermediate [CpFe(CO)2] (Eq. (1)) [18] and (ii) a chain reaction, based on Eqs. (1), (3) and (4) initiated by adventitious amounts of iron(II) species and generating [CpFe(CO)2]2 together with H2 [17].
5 Conclusion
[CpFe(CO)2(thf)]+ catalyzes proton reduction from TCA at −0.8 V vs Ag/AgCl in DMF. From the standard potential of the TCA/H2 couple in DMF (−0.98 V vs Fc+/0) [21], a quite low overpotential of 350 mV may be deduced. These values are comparable with those of the most efficient diiron models of FeFe hydrogenases reported so far [30]. However, this compound suffers from a major drawback since it is rapidly inactivated. The identification of the inactivation pathway, namely dimerization to [CpFe(CO)2]2 is an important result regarding the optimization of the catalytic properties of such systems: these processes indeed may be avoided in future work through a careful design of bulky cyclopentadienyl ligands, by incorporation of the catalyst within protective cages [31] or by immobilization on a surface electrode.
1 We should, however, mention that the relative heights of these two waves are not always reproducible and depend on the surface state of the electrode.
2 As the process at −1.44 V vs Ag/AgCl corresponds to the formation of [CpFe0(CO)2]− from dimeric [CpFe(CO)2]2 generated near the electrode, we checked that an authentic sample of [CpFe(CO)2]2 displays the same catalytic behavior at this potential (data not shown).