

1 Introduction
During the last two decades, marine organisms such as sponges, tunicates, softcoral and starfish proved to be productive sources of structurally complex metabolites presenting unprecedented architectures [1]. These marine natural products often display unexpected biological properties that make them potential candidates for therapeutic applications. Among these fascinating molecules, the polyketide macrolides having a macrolactone as key structural feature represent an important class of biologically active compounds [2]. Many congeners of this family are potent cancer cell growth inhibitors and thus appear to represent promising leads for the development of new anti-cancer agents. Nevertheless, a limitation to their implementation in clinical evaluation is related to their low natural abundance, which is stimulating organic chemists to invent efficient synthetic routes to address this supply issue.
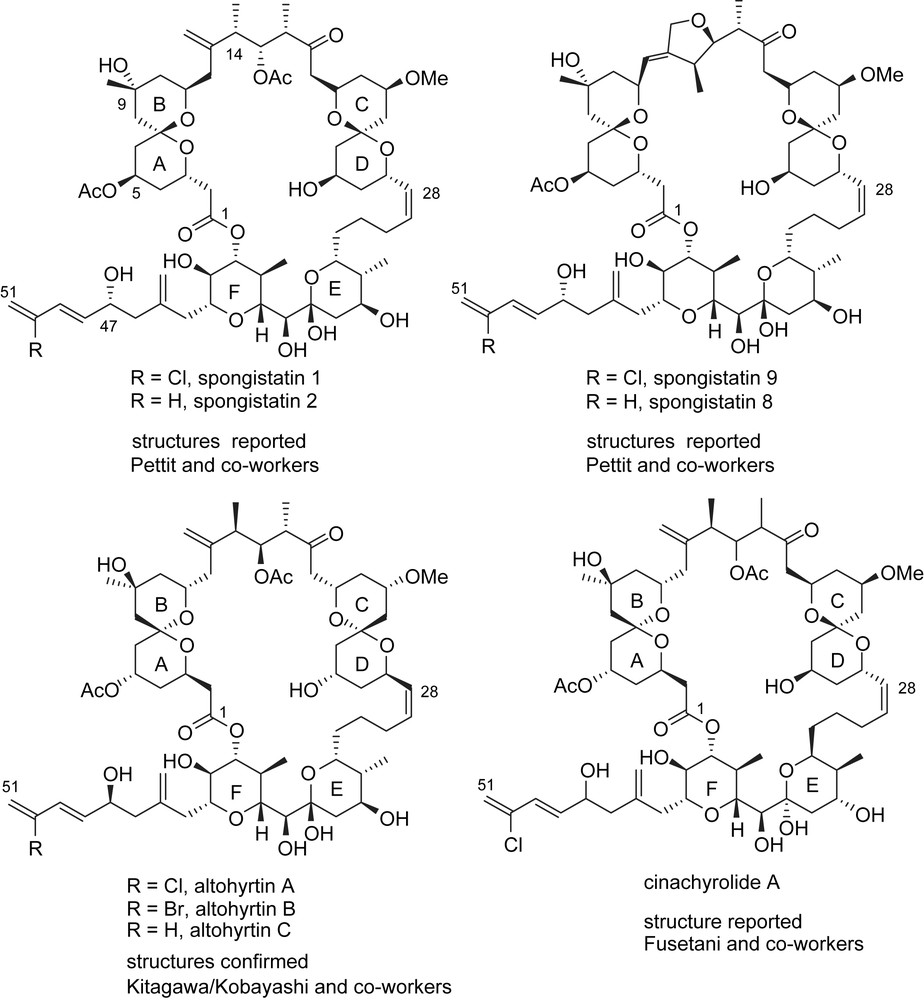
In 1993, the research groups of Pettit, Kitagawa/Kobayashi and Fusetani independently reported the isolation, from marine sponges, of a family of related highly oxygenated macrolactones containing highly substituted 6,6-spiroketals and tetrahydropyran rings [3]. These molecules were found to be among the most potent cancer cells' growth inhibitors tested to date by the U.S. National Cancer Institute on a panel of 60 human carcinoma cell lines, with GI50 values in the nanomolar and picomolar ranges. Despite these promising properties, further biological investigations were precluded by the extremely low quantities that can be obtained by extraction of marine organisms and the unacceptable ecological impact of larger scale isolation of the producing sponges. This scarce abundance, combined with remarkable structural complexity, prompted many research groups to address the challenge of the synthesis of these macrolides.
We give here an overview of the biological properties of spongistatins and related structures and of the different pathways that have been developed for their total syntheses and the synthesis of key subunits.
2 Isolation, structures and biological activities
In 1993, the group of Pettit reported the isolation of spongistatin 1 (Fig. 1) from an East Indian Ocean Perifera Spongia sp. (13.8 mg from 400 kg of sponge) [4]. The same year, Kitagawa and Kobayashi presented the isolation of altohyrtins A–C from Okinawan sponge Hyrtios altum (0.5 mg of altohyrtins A from 112 kg of sponge) [5], and the group of Fusetani isolated cinachyrolide A from the sponge Cinachyra sp. (1.1 mg from 6.6 kg of sponge), in Hachijo-jima Island [6]. Extensive NMR studies and circular dichroism allowed the full determination of the absolute and relative configurations of the altohyrtins [7]. For spongistatin 1 and cinachyrolide A, the stereostructure was only partially determined. At that time, the differences mainly concerned the relative configurations of AB, CD spiroketals and E-ring, as well as the stereochemistry at C(14), C(15) and C(47). Further considerable efforts led to the isolation of spongistatins 2–9 [8].
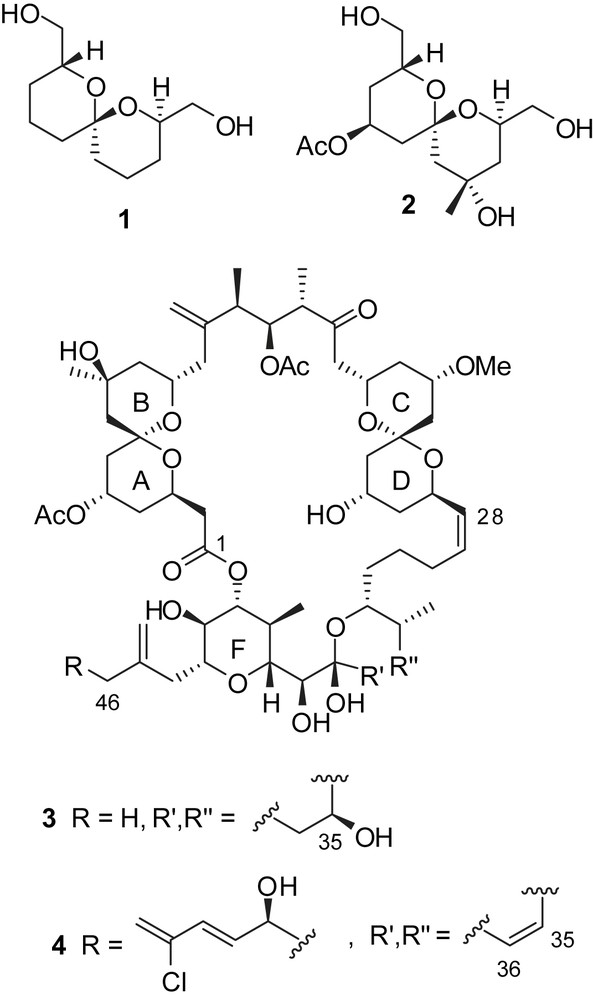
The most potent congener of this family is spongistatin 1 which exhibited typical GI50 values of 0.15 nM for in vitro assays on tumor cell lines. In addition, an extraordinary potency was observed on a panel of highly chemoresistant cell lines with GI50 values down to 0.03 nM [4,8a]. Further investigations revealed promising activity at low concentration against in vivo human melanoma and ovarian carcinoma xenografts [9]. The mode of action most probably results from the inhibition of mitosis and microtubule assembly to tubulin. The binding of spongistatins to a distinct region of the vinca domain is responsible for the inhibition of tubulin polymerization [10]. Investigations on the electron crystallographic structure of tubulin [11] revealed the presence of a putative binding pocket on the tubulin surface which could accommodate spongistatin 1 and provide a suitable environment for hydrophobic interactions. Advanced modeling studies of the interactions of spongistatin 1 with the candidate binding site indicated that the spiroketal subunits might be the likely tubulin binding elements. Based on this hypothesis, Uckun and co-workers proposed the synthesis of a simple spiroketal pyran (1, SPIKET-P) designed by docking studies on the putative spongistatin binding pocket (Fig. 2) [12]. According to their studies, spiroketal-diol 1 induced apoptosis in human breast cancer cells at the 10 nM level, caused tubulin depolymerisation at the 500 nM level and affected GTP-induced assembly of bovine brain microtubule protein [13]. Prompted by these remarkable biological results on such a structurally simple surrogate of spongistatins, Smith proposed the synthesis of an analogue of spongistatins AB spiroketal (2) that was also submitted to biological evaluation [14]. Neither derivative 2 nor the parent compound 1 displayed significant activity against several human cancer cell lines. Moreover, using different assays, it was demonstrated that these two compounds did not affect tubulin polymerization and polymer assembly. Simple mimics such as 1 and 2 are thus unlikely to be responsible for the potent activity of spongistatins. Structural modifications of spongistatin 1 have also been reported. For instance, the side chain truncation at C(46) (3) resulted in a drastic loss of growth inhibitory activity on several human tumor cell lines, while E-ring dehydration (4) led to increased cytotoxicity (GI50 values < 0.08 nM) [15].
The first total syntheses of altohyrtin C and altohyrtin A by the groups of Evans [16] and Kishi [17], respectively, confirmed the stereochemical assignments proposed by Kitagawa and Kobayashi and demonstrated the identity of spongistatin 2 to altohyrtin C and spongistatin 1 to altohyrtin A.
The structure of these appealing molecules is based on a 42-membered macrolactone embedded with two spiroketal subunits (AB and CD) and two highly substituted tetrahydropyrans (E and F), and functionalized by a sensitive trienic side chain, for a total of 24 stereocenters. Spongistatins 5, 7–9 contain an additional 5-membered ring within the skeleton. Interestingly, while the AB spiroketal benefits from a double conformational anomeric effect (axial–axial conformation), the CD spiroketal stands in an axial–equatorial conformation (single anomeric effect), but probably gains some stabilization from intramolecular hydrogen bonding.
All strategies toward the total synthesis of spongistatins share some common disconnections. A late macrolactonization using Yamagushi's conditions [18] is envisaged to close the macrocyclic core at C(1)–C(41)–OH and the (Z)-alkene at C(28)–C(29) offers the possibility of Wittig-type fragment coupling (Fig. 3).
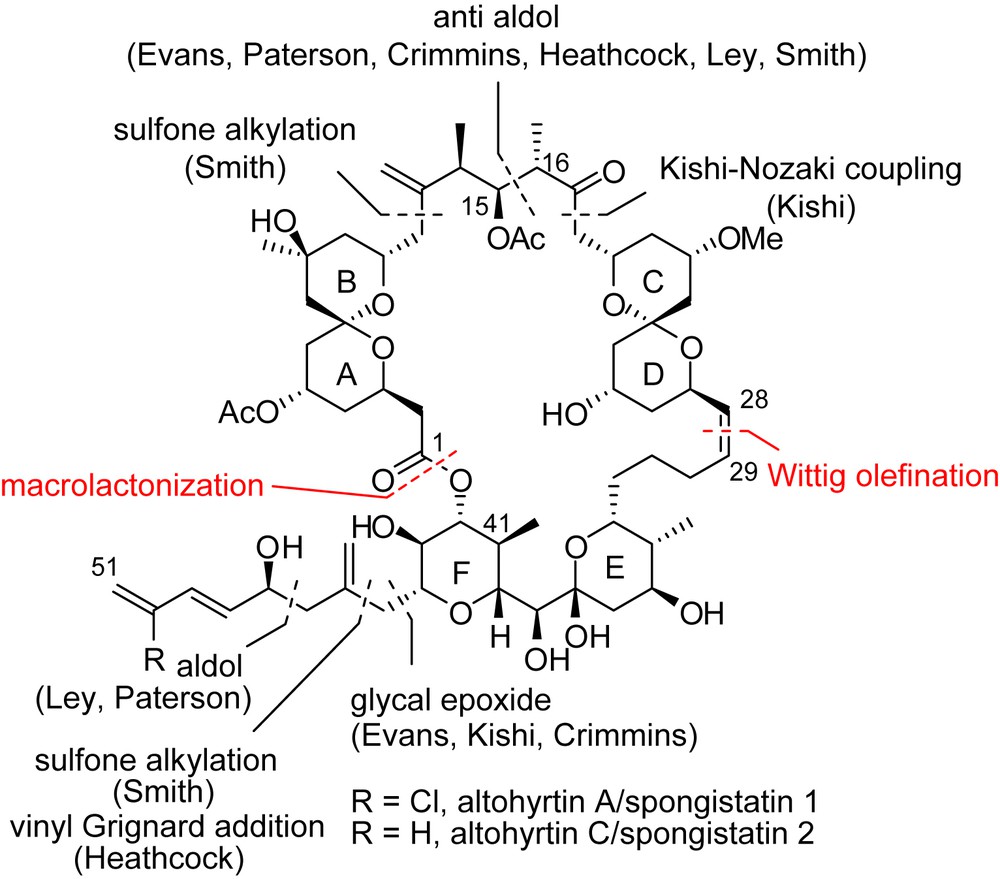
Then different approaches have been proposed for the completion of the ABCD bis-spiroketal subunit, the construction of the highly oxygenated EF rings and the attachment of the trienic side chain. Total syntheses of spongistatins/altohyrtins have been reported by seven groups (Evans [16], Kishi [17], Smith [19,20], Paterson [21], Crimmins [22], Heathcock [23] and Ley [24]) each presenting different practical solutions for the assembly of the ABCD and EF key fragments. The bis-spiroketal subunit can be constructed through an anti-aldol reaction to form the C(15)–C(16) bond or via a Kishi–Nozaki coupling to produce the C(17)–C(18) linkage. Sulfone alkylation at C(14) also gives rise to the same fragment. Appending of the sensitive trienic side chain was proposed through various methodologies: sulfone alkylation or vinyl Grignard addition at C(44), opening of glycal epoxide at C(43) or aldolisation to form the C(46)–C(47) bond.
3 Total syntheses of altohyrtins/spongistatins
3.1 Total synthesis of altohyrtin C/spongistatin 2 by Evans [16]
Evans and co-workers proposed a versatile strategy as far as stereochemistry is concerned because of the discrepancies existing at that time between altohyrtins and spongistatins. Assembly of AB and CD spiroketals was envisaged by acid mediated cyclization of linear keto polyol precursors. Synthesis of the C(1)–C(15) fragment started, on the one hand, by protection of alcohol 5, reduction of the ester and Wittig homologation to afford Weinreb amide 6 (Fig. 4). 1,4-Addition to its benzaldehyde hemiacetal led to the corresponding 1,3-syn protected diol (>95:5 dr) which was further derived to aldehyde 7. On the other hand, diastereoselective alkylation of oxazolidinone 8 and removal of the chiral auxiliary allowed conversion into allylsilane 9. SnCl4 promoted addition to aldehyde 15 afforded the antipodal C(11) configuration which was inverted by Mitsunobu reaction.
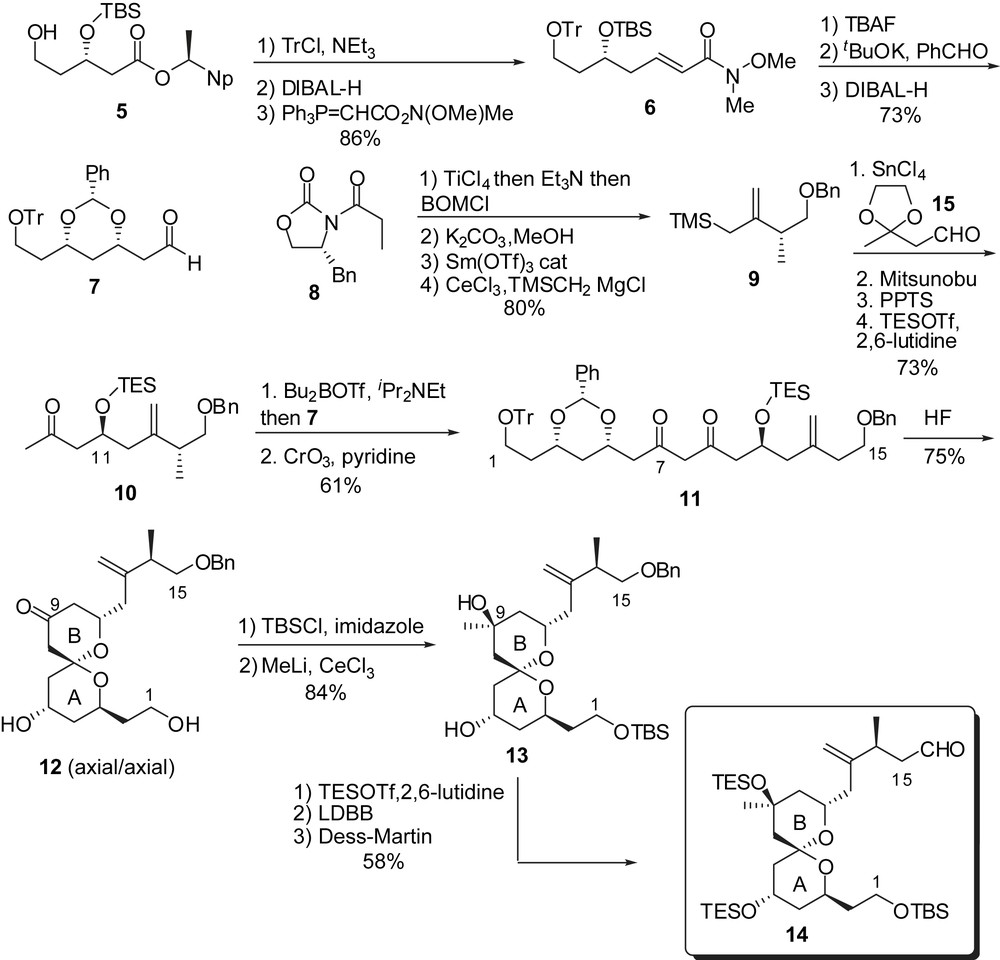
Coupling of boron enolate derived from 10 to aldehyde 7 afforded a mixture of diastereoisomeric aldols which were oxidized to diketone 11. Treatment with HF induced deprotection and spiroketalization to the thermodynamically favoured anomer 12. The tertiary alcohol at C(9) was installed though stereoselective addition of MeLi and further manipulation led to the AB-spiroketal subunit 14.
Construction of the CD spiroketal involved addition of the boron enolate derived from methyl ketone 16 (obtained in a similar route to that of 7) to aldehyde 17, followed by methylation of the intermediate aldol to afford ketone 18 with a high diastereoselectivity (96:4 dr) (Fig. 5). CSA promoted spiroketalization led to an initial mixture of axial/axial and axial/equatorial spiroketals 19a and 19b (8:1). Equilibration in the presence of Mg(OCOCF3)2 (chelation with C(25) alcohol and C(27) anomeric oxygen) resulted in a 1:2.2 mixture of 19a and 19b, from which the desired isomer was isolated. The C(17) ester was then converted into the corresponding ethyl ketone which was engaged in a boron aldol condensation to aldehyde 14 to form the C(15)–C(16) bond with 90:10 Felkin–Anh stereoselectivity. Selective desilylation of C(1) and C(5) alcohols allowed conversion of the primary alcohol into a methoxyacetate group and installation of acetates at C(5) and C(15) positions. Removal of the trityl group without isomerization of the CD spiroketal could only be achieved in the presence of Me2AlCl. The resulting alcohol was then oxidized to aldehyde 23.
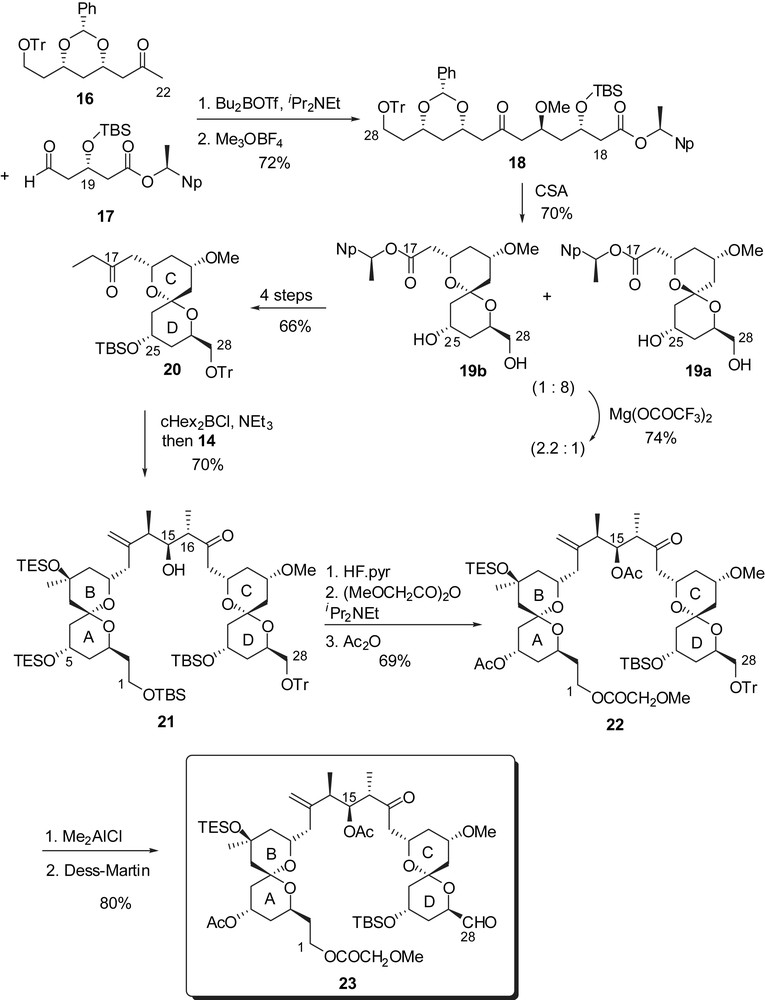
Assembly of the EF-ring subunit was envisaged through acylation of an E-ring sulfone with an activated F-ring anomeric carboxylic acid. Synthesis of the E-ring fragment was performed through Fukuyama reduction of thioester 24 and acid-catalyzed silyl ether cleavage/acetalization to afford, after protection, methyl ketal 25 (Fig. 6). Protection of the C(35) alcohol, followed by transformation of the terminal alkene into the corresponding benzyl ether and anomeric sulfone formation led to the precursor of the acylation step (26). The F-ring subunit resulted from silver promoted lactonization of thioester 27 followed by dehydration at C(42),C(43) and transformation of the benzyl ether into an activated benzotriazolyl amide 29. The coupling step proceeded through addition of the E-ring sulfone anion derived from 26, to amide 29. Methanolysis of the resulting adduct afforded the EF bis(pyran) 30. Diastereoselective reduction with KBHEt3 installed the C(38) alcohol with excellent control (>99:1 dr), which was subsequently silylated. The benzyl ether was transformed into the corresponding phosphonium salt 31 as precursor of a Wittig olefination.
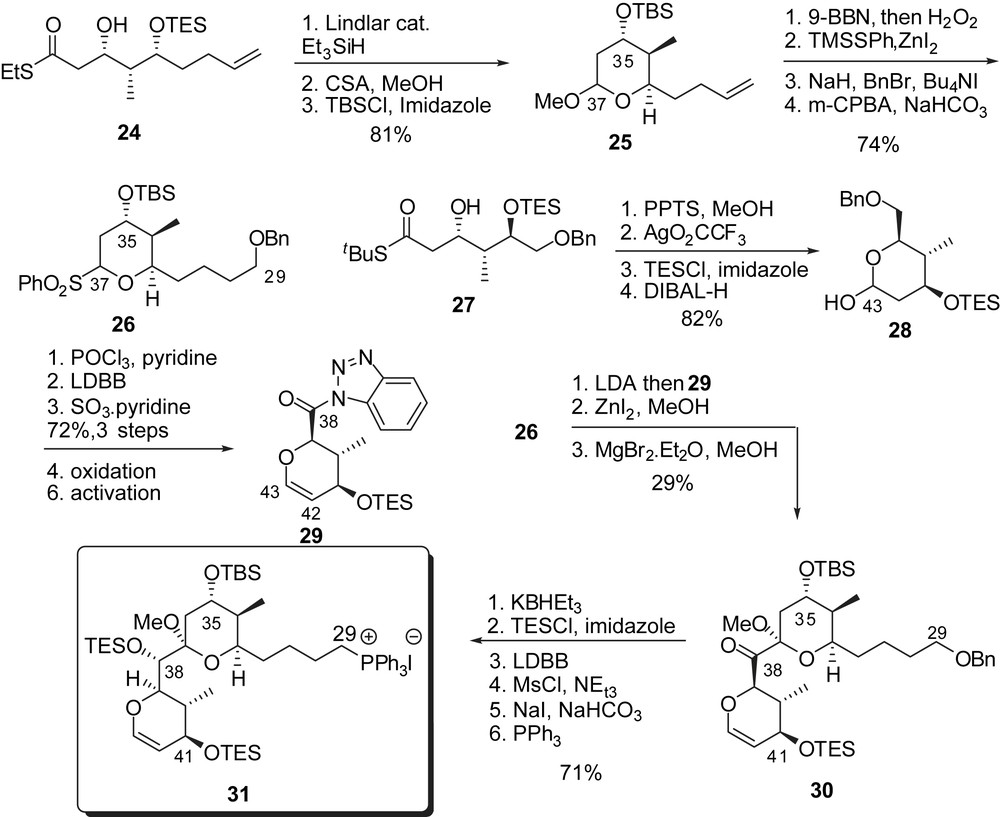
Assembly of spongistatin 2 started by Wittig reaction of 31 with aldehyde 23 providing the corresponding (Z)-olefin (>95:5 dr) which was further elaborated to intermediate 32 (Fig. 7). Stereoselective epoxidation with dimethyldioxirane allowed introduction of the trienic side chain through regioselective addition of excess 33 to afford the protected seco-acid 34. Protecting group manipulations followed by regioselective macrolactonization onto the C(41) alcohol and final removal of silyl ethers completed the first total synthesis of spongistatin 2/altohyrtin C in 0.8% overall yield for the longest linear sequence starting from ethyl glyoxalate (32 steps).
3.2 Total synthesis of altohyrtin A/spongistatin 1 by Kishi [17]
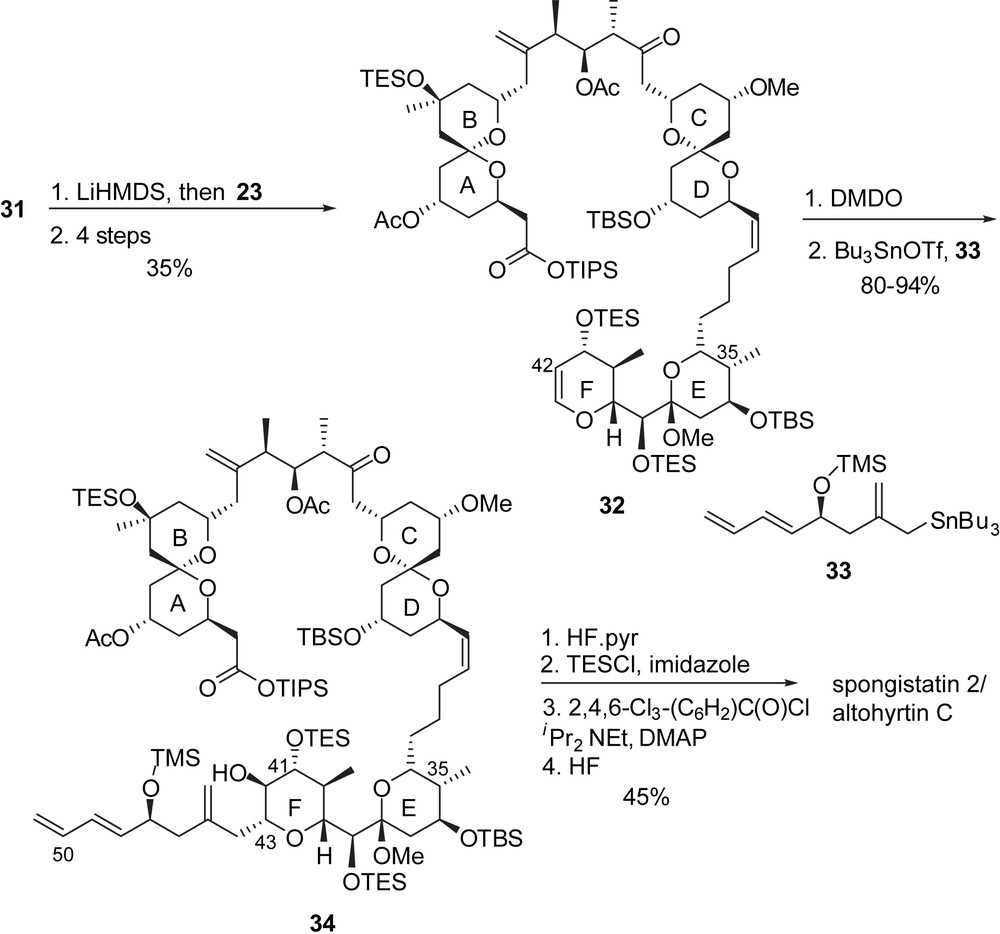
In their approach, Kishi and co-workers proposed the assembly of the bis(spiroketal) subunit through a Takai–Oshima–Kishi–Nozaki coupling to build the C(17)–C(18) bond between a AB derived aldehyde and a D fragment vinyl iodide. The preparation of AB and D-ring precursors was based on the regioselective opening of epoxides with carbon nucleophiles (dithiane anions and organocuprates). Synthesis of the C(1)–C(17) fragment involved addition of 1,3-dithiane anion on epoxide 35 followed by silylation of the resulting alcohol (Fig. 8). Deprotonation of the dithiane moiety allowed addition to epoxide 37 to install the tertiary alcohol at C(9). Cleavage of silyl ether and subsequent basic treatment afforded a terminal epoxide at C(11),C(12). Vinyl iodide 40, derived from the alkyne precursor 39, was transformed into the corresponding cuprate and added in situ to epoxide 38. Further cleavage of silyl ethers provided the precursor of the spiroketalization (41). NIS-promoted dithiane hydrolysis followed by acidic treatment induced spiroketalization to the axial/axial AB subunit 42. Further elaboration and protecting groups' manipulation allowed oxidation of the alcohol at C(17) to provide the functionalized AB-spiroketal aldehyde 44.
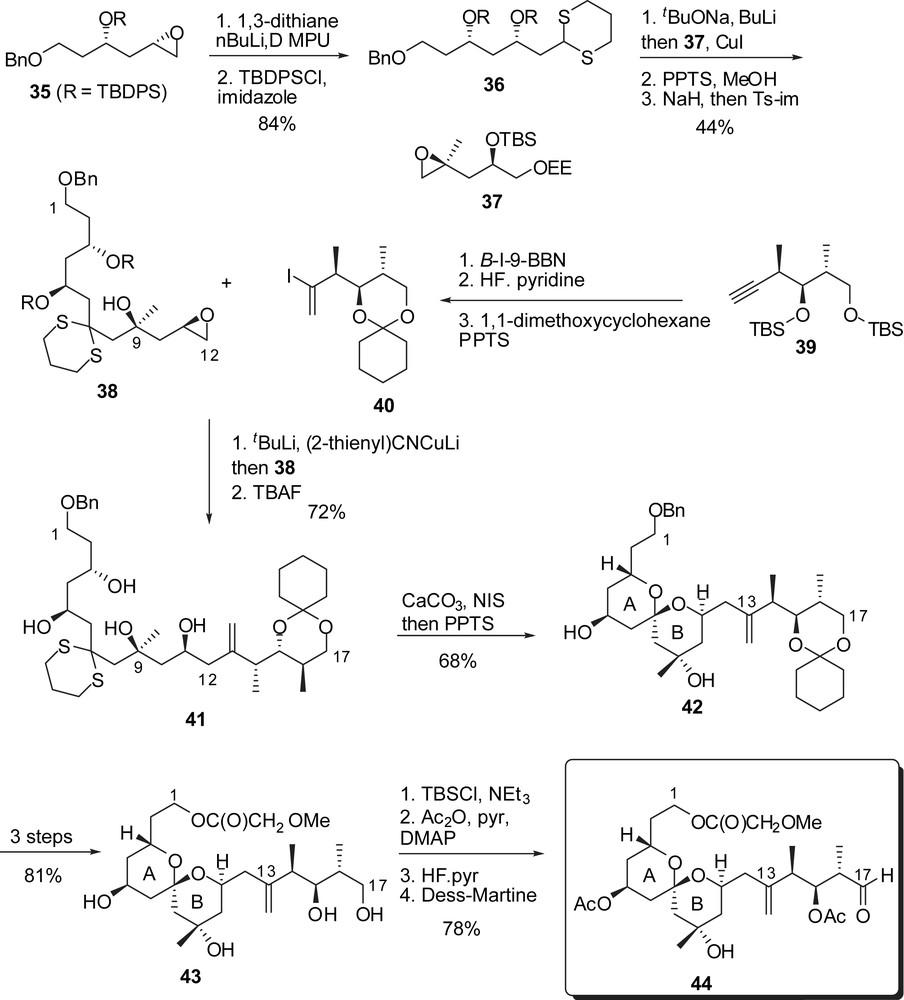
The D-ring fragment was also elaborated from the addition of the dithiane anion derived from 45 to a terminal epoxide providing the C(18)–C(28) skeleton (Fig. 9). NIS-assisted cleavage of the dithiane moiety induced formation of a methyl ketal which was further elaborated into the protected alkyne 48. The vinyl iodide moiety was then introduced through hydrostannylation, and protecting group exchanges led to the D-ring intermediate 49. The key Cr(II)/Ni(II)-promoted coupling with aldehyde 44 proceeded in high yield and the resulting allylic alcohol was oxidized to enone 50. Intramolecular Michael addition led to the desired configuration at C(19) but to the unwanted axial/axial configuration of the CD spiroketal. Acidic removal of the silyl ether at C(25) induced equilibration to a 1:1 mixture of the desired isomers 53 and 52 that could be further recycled into 53. Several steps were then required to install the suitable protecting groups for the endgame sequence and to produce a silyl protected carboxylic acid at C(1). Finally the C(28) alcohol was oxidized to the corresponding aldehyde, providing the ABCD subunit 54, ready to undergo Wittig coupling.
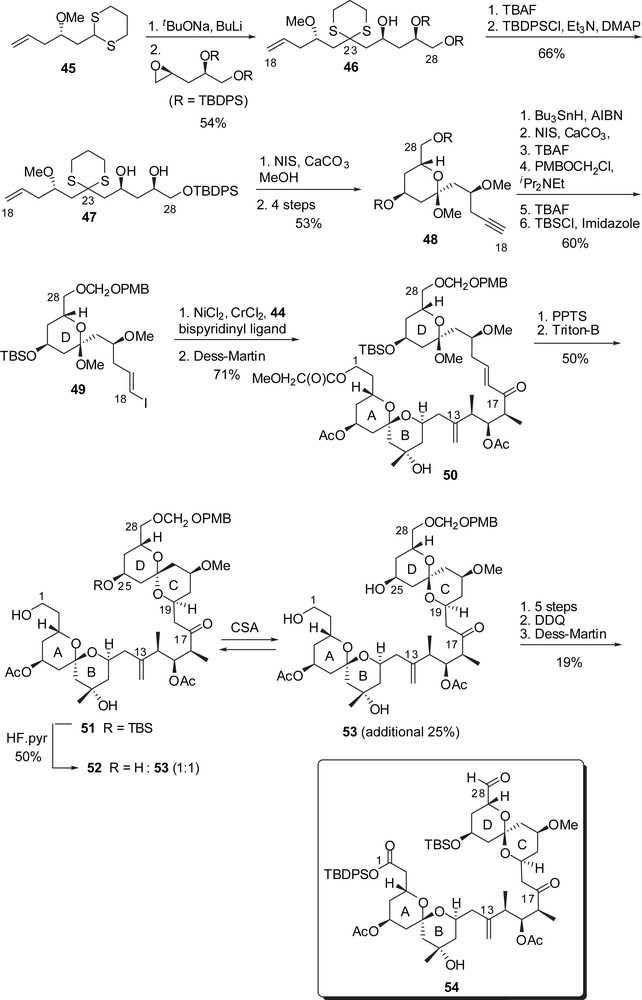
The synthesis of the EF fragment was envisaged through an early appending of the trienic side chain on an F-ring glycal epoxide followed by coupling to an E-ring iodopyran. A sequence of asymmetric crotyl- and allylborations under Brown's conditions [25] provided the homoallylic alcohol 55 as the C(29)–C(37) skeleton in high yields and selectivities (Fig. 10). Introduction of a silyl ether at C(35) position and Birch reduction of the benzyl ether allowed installation of an aldehyde at C(37). Dehydrative cyclization of 56, followed by iodination at C(37) position delivered the F-ring iodoglycal 58. Similarly, semi-protected triol 59 was elaborated into the F-ring glycal 61. Addition of an excess of allyl cuprate derived from 62 on the F-ring epoxide at C(42),C(43) allowed regioselective attachment of the C(44)–C(48) fragment. Hydrolysis of the acetonide followed by oxidation to an aldehyde moiety at C(48) provided the precursor for the introduction of the chloro diene moiety. Addition of allylindium species derived from 66 and subsequent dehydration in the presence of Martin's sulfurane afforded, after further elaboration, the functionalized F-ring fragment 67.
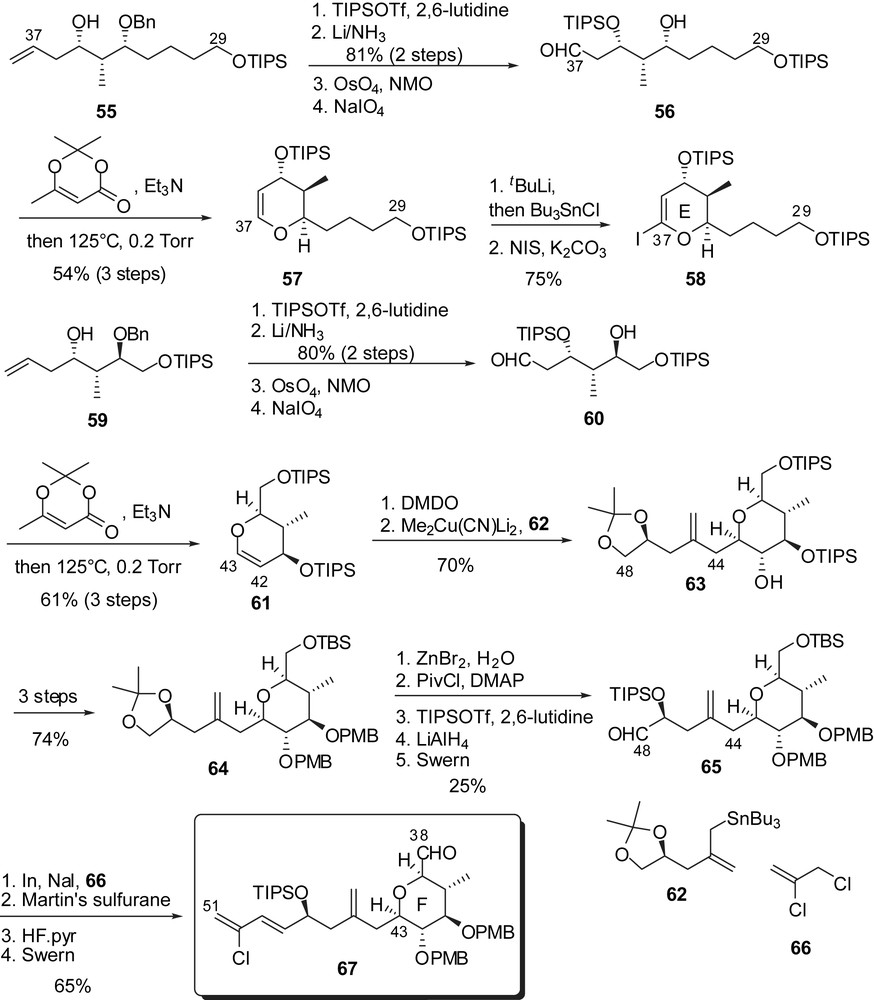
Generation of the Grignard reagent from iodoglycal 58, followed by addition to aldehyde 67 generated the EF-ring subunit and installed the C(38) alcohol with high stereoselectivity (Fig. 11). Iodomethanolysis of the E-glycal and reductive dehalogenation resulted in the formation of a methyl ketal at C(37) (68). Protecting group manipulations and formation of a phosphonium salt at C(29) delivered the precursor for the Wittig coupling to the ABCD fragment. This reaction led to the corresponding (Z)-olefin 70 which was transformed into the corresponding seco-acid by selective deprotection (partial methyl ketal hydrolysis at C(37) occurred simultaneously). Selective macrolactonization with alcohol at C(41) under Yamagushi's conditions and subsequent silyl ethers cleavage provided altohyrtin A/spongistatin 1 in 39 linear steps and 0.005% overall yield, starting from 2-(triisopropylsilyloxy)ethanal.
3.3 Total synthesis of altohyrtin A/spongistatin 1 by Paterson [21]
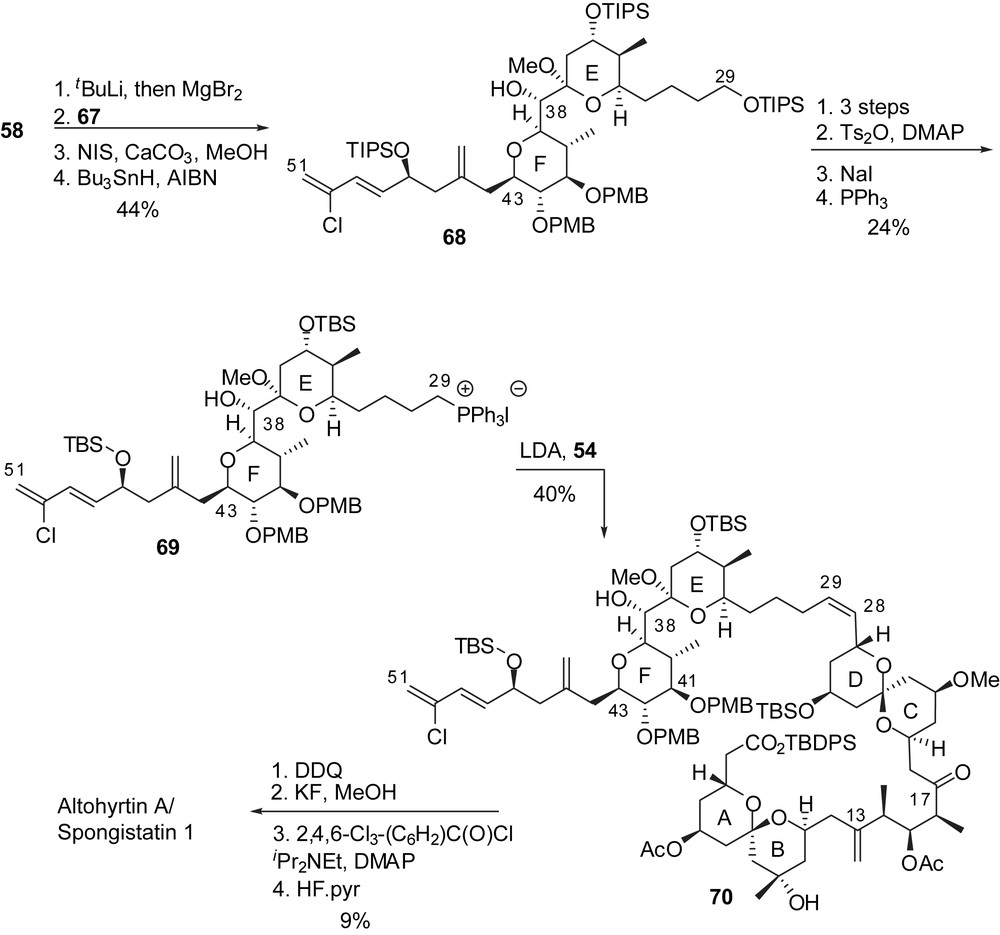
The strategy of Paterson and co-workers to prepare the key subunits of spongistatin 1 was based on the application of stereoselective boron aldol couplings using both reagent and substrate controls to deliver 1,3- and 1,5-dioxygenated moieties. The C(1)–C(15) skeleton arose from addition of the boron enolate from methyl ketone 71 to aldehyde 72, thus installing the 5,9-anti relationship of the alcohols (Fig. 12). Selective removal of the triethylsilyl groups induced spirocyclization to the unique thermodynamically favoured spiroketal. Oxidation at C(9) followed by equatorial addition of Grignard reagent introduced the carbinol moiety on the B ring (74). Further elaboration installed a protected carboxylic acid at C(1) and an aldehyde moiety at C(15) to deliver the fully elaborated AB spiroketal 76. A similar pathway was adopted for the synthesis of the CD subunit, starting by the aldol condensation of boron enolate derived from methyl ketone 77 to aldehyde 78, thus ensuring the anti relationship of hydroxyls at C(21) and C(25). Acidic cleavage of silyl ethers led to spirocyclization to provide a 1:5 mixture of the desired axial/equatorial spiroketal 81 and the undesired thermodynamically favoured axial/axial isomer 80. Several cycles of equilibration/separation could provide larger quantities of the desired spiroketal, up to 69% yield. Further transformations installed an ethyl ketone at C(17). Enolisation of 82 and in situ condensation of the resulting boron enolate to the AB fragment 76 afforded a 9:1 stereoselectivity in favour of the anti-aldol. This optimized key step could deliver more than 400 mg of aldol 83 and was then used by several research groups to achieve the assembly of the ABCD northern hemisphere of spongistatins/altohyrtins.
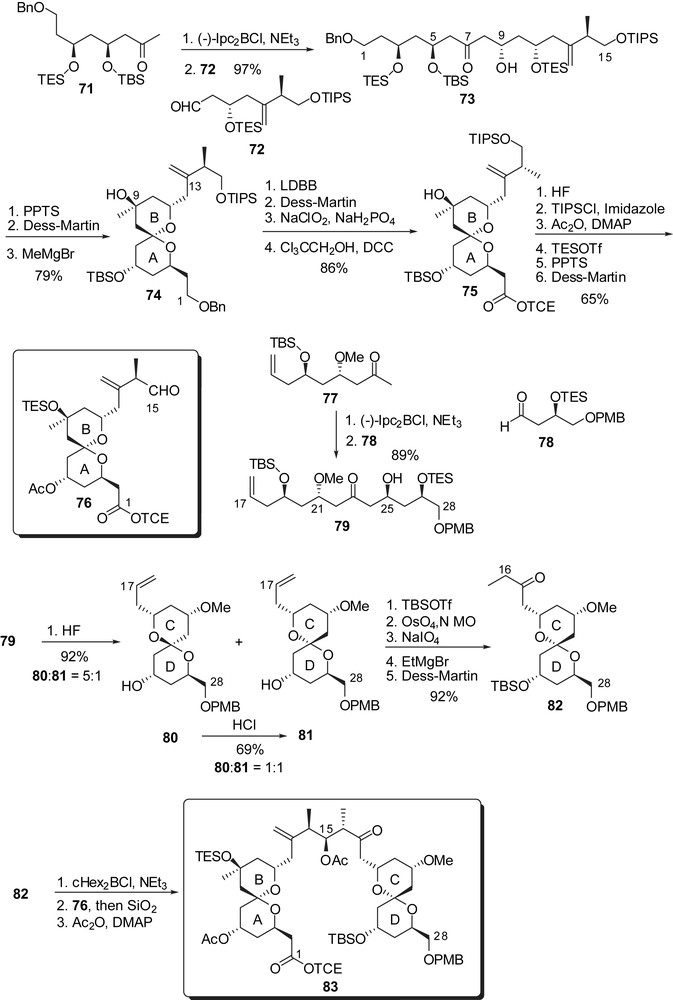
In their strategy toward the EF rings' subunit, Paterson and co-workers also envisaged a boron aldol condensation to assemble a C(37) methyl ketone F-ring to an E-ring precursor aldehyde (Fig. 13). The C(36)–C(46) skeleton was prepared from aldehyde 84 through a sequence of HWE olefination/stereoselective dihydroxylation of the intermediate α,β-unsaturated ester. A second HWE olefination delivered enone 86. Acidic removal of the acetonide moiety was accompanied by an intramolecular hetero Michael addition to produce a C(43) epimeric mixture of tetrahydropyrans. Subsequent equilibration in basic medium followed by Petasis methylenation and oxidation at C(37) afforded the F-ring subunit 87. Boron aldol condensation to aldehyde 88 appended the C(29)–C(35) skeleton with a good 9:1 stereoselectivity at C(35). Methyl ketal formation was performed under mild acidic conditions and the resulting EF rings' intermediate was further transformed into methyl ketone 90. Another boron aldol coupling resulted in the introduction of the chloro diene moiety. The C(29) chloride was finally converted into a phosphonium salt, ready to undergo endgame reactions.
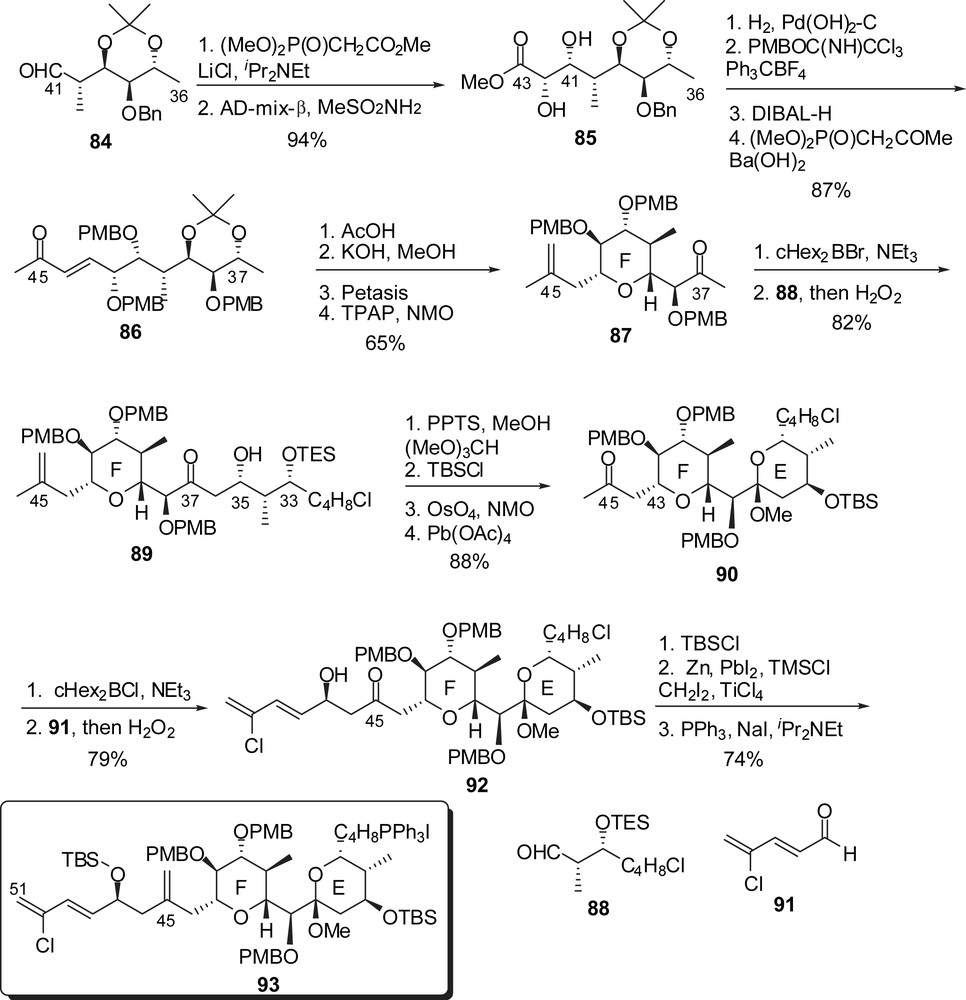
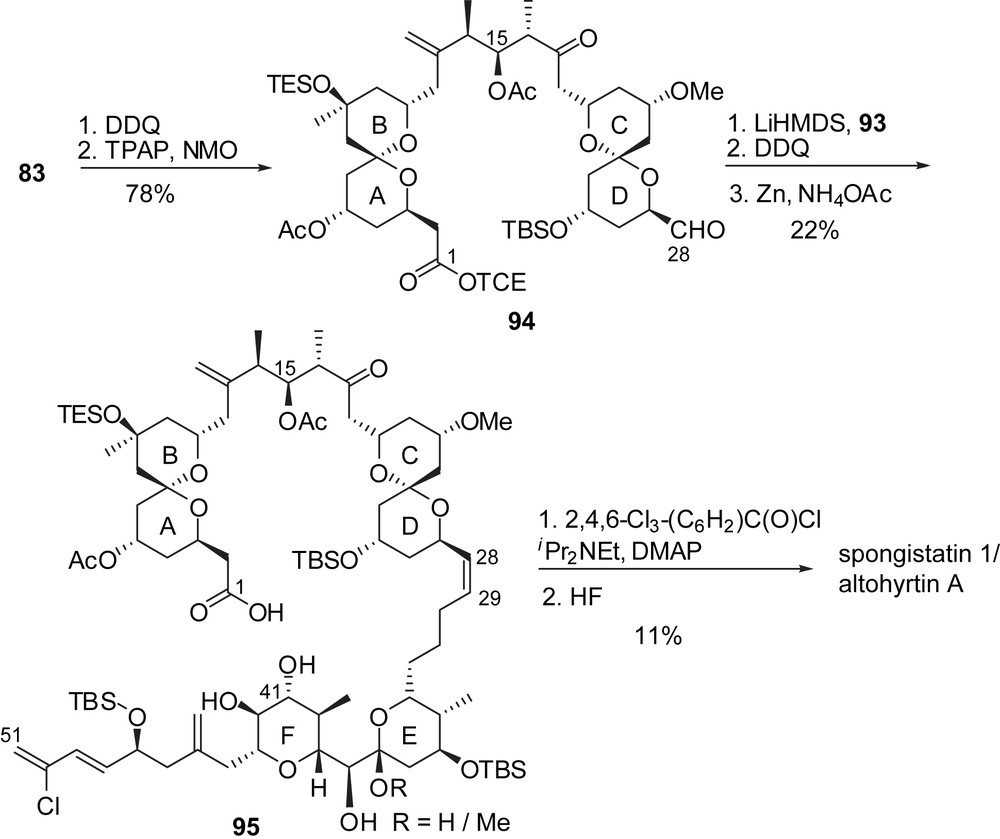
Cleavage of the C(28) p-methoxybenzyl ether of the ABCD hemisphere 83 allowed transformation into the corresponding aldehyde 94 which was submitted to Wittig coupling with iodophosphonium 93 to give the (Z)-C(28)–C(29) double bond with a 97:3 selectivity (Fig. 14). Selective macrolactonization on the C(41) alcohol and final deprotection delivered spongistatin 1/altohyrtin A in 1.6% overall yield and 33 linear steps for the longest sequence, starting from (3S)-3-methyl-4-(triisopropyloxy)butan-2-one. The E-ring dehydrated analogue 4 was isolated as a minor product after the deprotection step.
3.4 Total synthesis of altohyrtins A and C/spongistatins 1 and 2 by Crimmins [22]
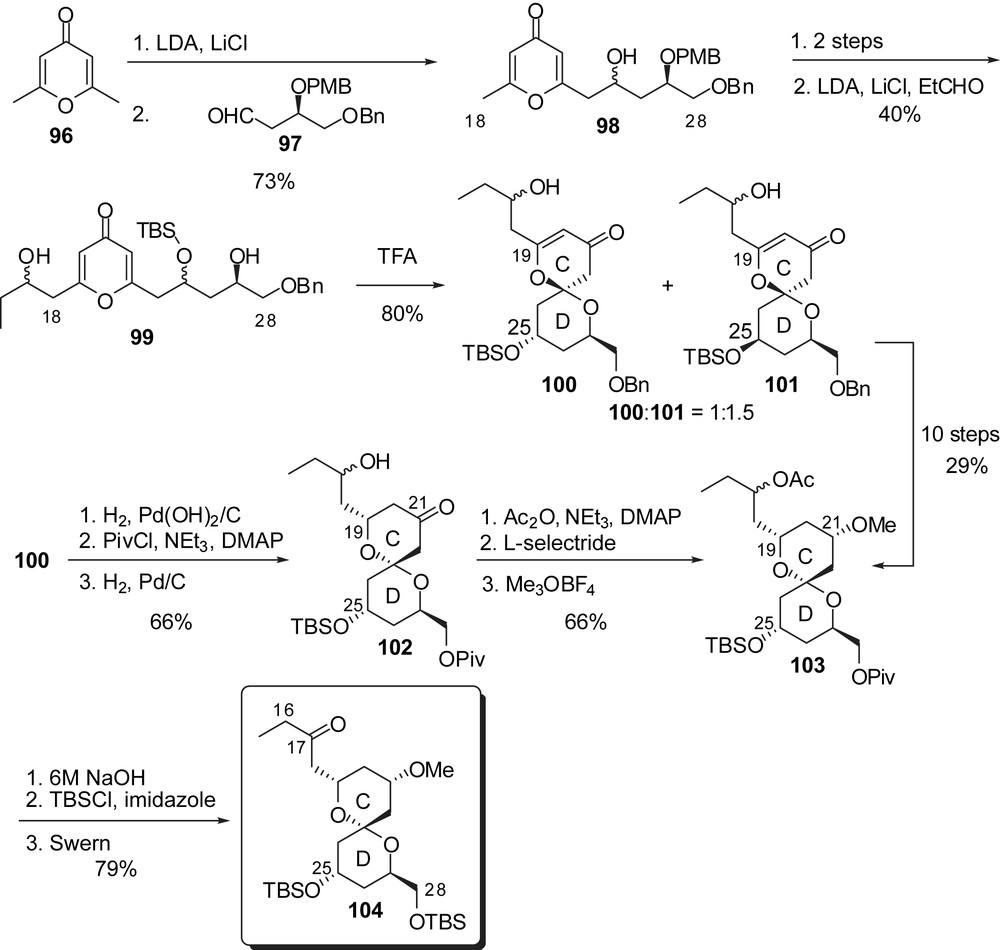
The strategy of Crimmins and co-workers for the synthesis of spongistatins 1 and 2 was based on a first coupling of the CD spiroketal with the EF-ring subunit followed by an anti-aldol coupling to introduce the AB fragment. Both AB and CD spiroketals were elaborated from pyrone precursors (Figs. 15 and 16). Preparation of the C(16)–C(28) skeleton started by addition of lithiated 2,6-dimethyl-γ-pyrone to aldehyde 97 to produce a mixture of stereoisomeric adducts 98. Chain elongation at C(18) provided intermediate 99. Acidic treatment led to spiroacetalization to yield a mixture of the desired C(25) equatorial isomer 100 and its axial epimer 101 in a 1:1.5 ratio. Further hydrogenation of the double bond induced a ring inversion to produce the C(23) axial/equatorial arrangement of the spiroketal and the expected C(19) equatorial substituent (102). Diastereoselective reduction of the carbonyl moiety at C(21) and subsequent protection of the resulting equatorial hydroxyl group afforded intermediate 103. The undesired isomer of the spirocyclization could also be converted in 10 steps into 103. Final elaboration of the CD subunit included protecting group manipulations and oxidation at C(17) to give ethyl ketone 104.
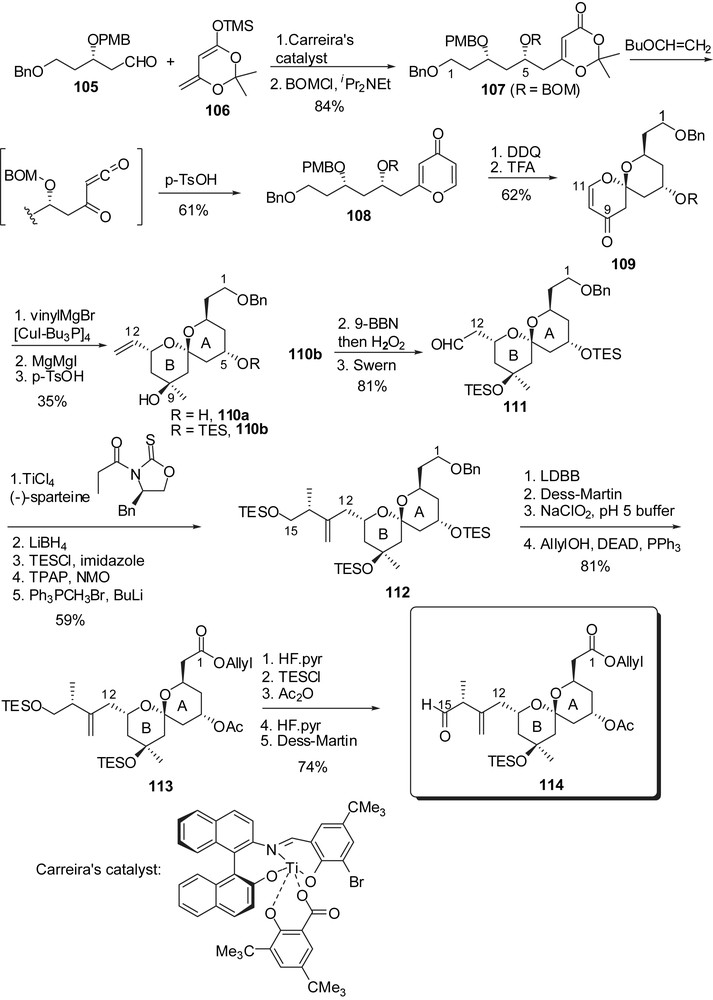
The optimized preparation of the C(1)–C(15) fragment (Fig. 16) started with Mukaiyama aldol condensation of silyl dienolate 106 to aldehyde 105 in the presence of Carreira's catalyst to provide a 10:1 stereoselectivity in favour of the syn isomer which was protected as 107. Synthesis of the key pyrone intermediate made use of a hetero Diels–Alder reaction of an acyl ketene generated from dioxinone 107 with butyl vinyl ether to provide pyrone 108, after acidic treatment of the intermediate butyl acetal. Removal of the p-methoxybenzyl group allowed acid promoted spirocyclization to spiroenone 109, presenting the axial/axial arrangement at C(7). Cuprate addition at C(11) resulted in a 5:1 stereoselectivity and further transformation into a methyl carbinol at C(9) afforded 110a. Its silylated derivative 110b was submitted to hydroboration of the vinyl moiety followed by oxidation to aldehyde 111. Installation of the C(12)–C(15) side chain proceeded through an asymmetric aldol procedure using a titanium enolate derived from a oxazolidinethione derivative. Further elaboration installed the C(1) carboxyl moiety and the C(15) aldehyde to deliver 114 as the precursor for the key anti-aldol coupling to generate the C(15)–C(16) linkage.
Elaboration of the EF-ring fragment also exploited two successive diastereoselective aldolisations using titanium and boron enolates derived from oxazolidinethione/oxazolidinone to give the tetraol derivative 115 (Fig. 17). Oxidative cleavage of the olefin followed by dehydration of the resulting hemiketal delivered enol ether 116. Further elaboration installed a methyl ketone moiety at C(37) that was engaged in a stereoselective aldol addition to aldehyde 118. Subsequent acidic treatment of the intermediate aldol induced hemiketal formation to provide derivative 119. Protecting group manipulations and transformation of the C(29) hydroxyl group into a phosphonium salt 121, via tosylate 120, led to the precursor necessary for the Wittig coupling. Interestingly, Crimmins and co-workers introduced the AB and CD spiroketals in a two step sequence, first olefination with aldehyde 122 (derived from intermediate 104) to form the C(28)–C(29) double bond and then an anti-boron aldol addition to aldehyde 114 for the generation of the C(15)–C(16) linkage. The endgame transformations included introduction of the C(44)–C(51) trienic side chains according to the procedure of Evans, macrolactonization on the alcohol at C(41) and final deprotection to provide spongistatin 1/altohyrtin A and spongistatin 2/altohyrtin C. These syntheses proceeded in about 38 steps from 1,3-propanediol.
3.5 Total synthesis of altohyrtin C/spongistatin 1 by Smith (second-generation approach) [19,20]
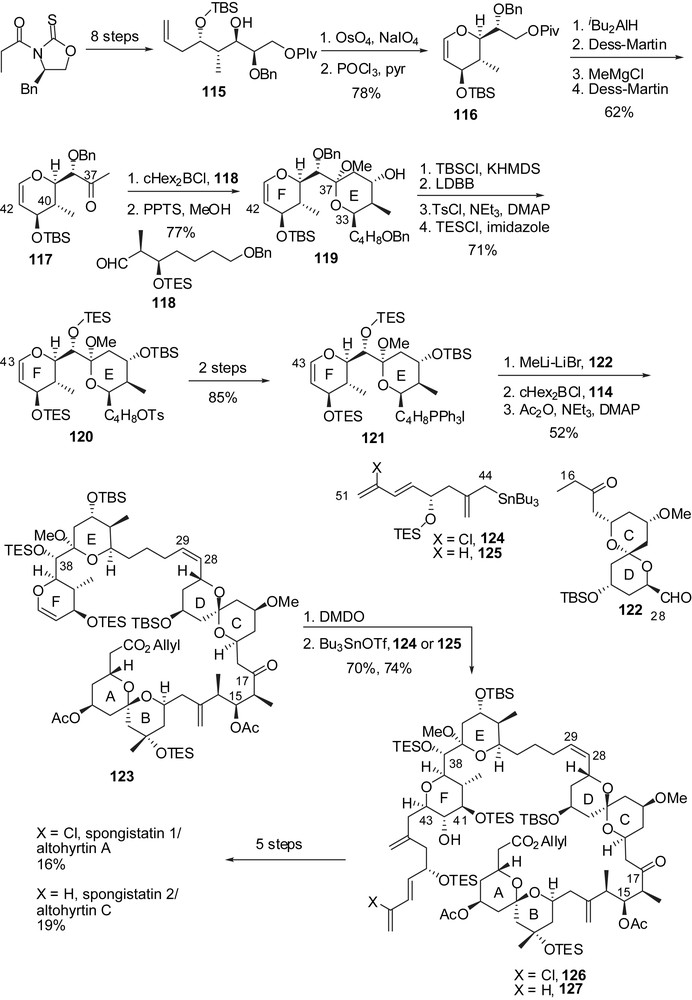
After the development of a first approach to spongistatin 2/altohyrtin C [19], Smith and co-workers proposed a more efficient second-generation synthesis of spongistatin 1 that will be discussed below [20]. Construction of both AB and CD spiroketals was envisaged through multicomponent linchpin union of silyl dithianes with epoxides [26]. For the preparation of the C(1)–C(15) fragment, methyl ketone 128 was converted into the corresponding trisyl hydrazone and then coupled to epoxide 129, after deprotonation (Fig. 18). The terminal silyl ether was cleaved allowing formation of an epoxide at C(8),C(9). The linchpin union of 2-triethylsilyl-1,3-dithiane with successively epoxide 133 and the lithium alcoholate derived from epoxide 131 delivered the C(1)–C(15) skeleton of spongistatin 1. Simultaneous removal of dithiane and silyl ethers induced spiroketalization to afford the targeted axial/axial spiroketal 135 as the sole isolated isomer. Protecting group manipulations followed by oxidation of the alcohol at C(1) and formation of an aldehyde at C(15) completed the synthesis of the AB-spiroketal subunit (137).
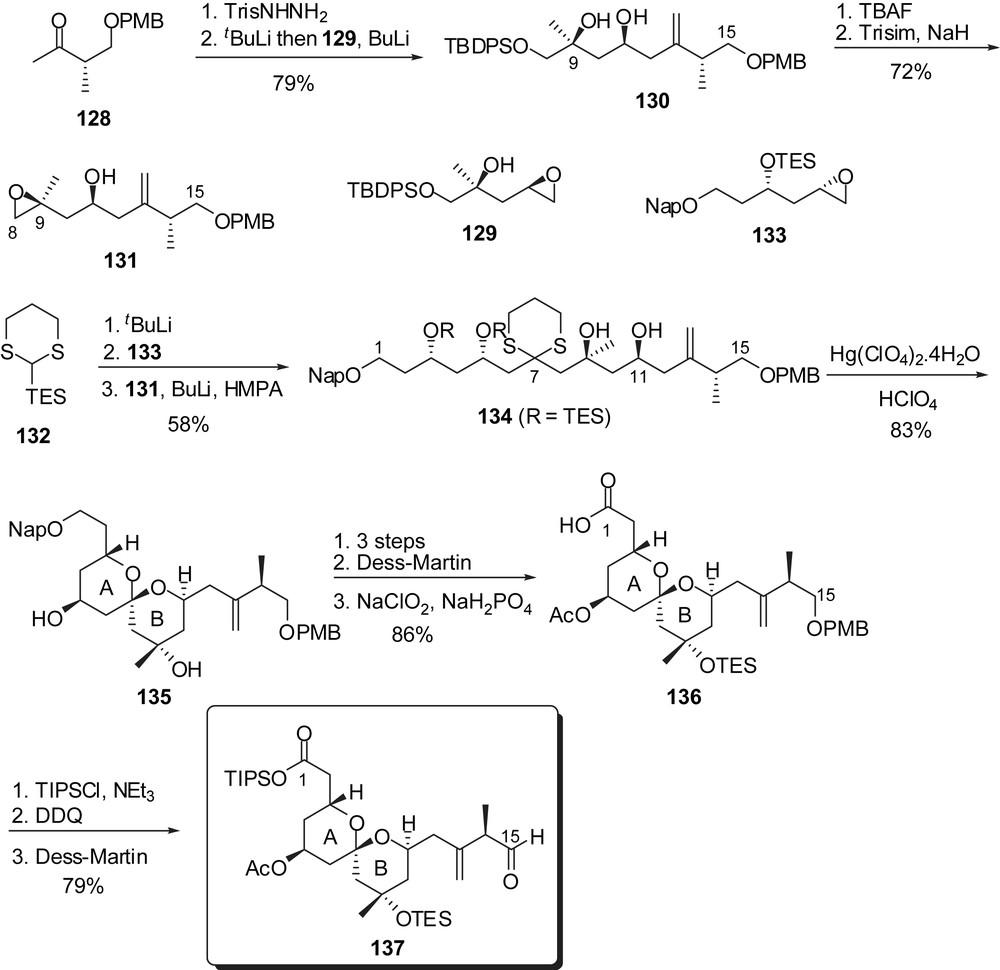
Similarly, for the assembly of the CD spiroketal, the efficient linchpin union involved the successive addition of epoxides 138 and 139 on dithiane 132 to afford the C(16)–C(28) skeleton 140 (Fig. 19). Methylation of the alcohol at C(21) followed by removal of both dithiane and silyl ethers induced spiroketalization to a mixture of the undesired axial/axial isomer 141 and the targeted axial/equatorial spiroketal 142 (dr = 4:1). Dihydroxylation of the olefin allowed equilibration in the presence of calcium perchlorate/perchloric acid and subsequent cleavage of the diol moiety resulted in the formation of the desired spiroketal 143, separable from the minor isomer (dr = 1:4). As experimented by Evans, the assembly of the ABCD bis-spiroketal fragment was accomplished through an anti-boron aldol reaction followed by further elaboration leading to the C(1)–C(28) subunit 145.
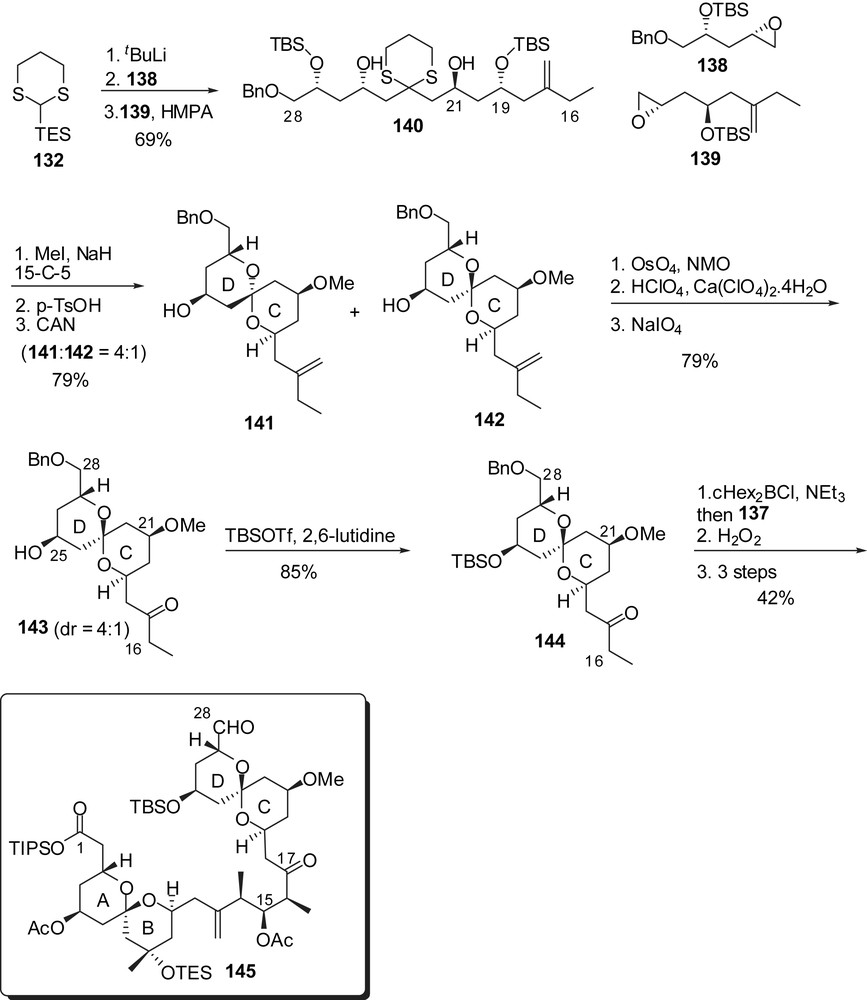
Smith and co-workers proposed the preparation of the EF-ring fragment through the addition of a dithiane anion prefiguring the E-ring to an F-ring aldehyde. The latter was elaborated from homoallylic alcohol 146 which was first converted into fully protected triol 147 (Fig. 20). Sharpless asymmetric dihydroxylation installed the C(39) configuration and subsequent basic treatment induced cyclization to an F-tetrahydropyran (dr = 6:1), that was further submitted to oxidation at C(38) to deliver aldehyde 148. The C(29)–C(37) skeleton was derived from homoallylic alcohol 149 which underwent a sequence of iodocarboxylation/methanolysis to provide epoxide 150 with high stereoselectivity for the installation of the C(35) stereocenter (>12:1 dr). 1,3-Dithiane addition and protecting group manipulations afforded the precursor of the E-ring (151). Coupling with aldehyde 148 proceeded with excellent selectivity (>20:1 dr) to install the C(38) alcohol. Cleavage of both acetonide and dithiane moieties resulted into an E-ring hemiketal which was further protected to produce the EF-ring segment 153. Enol ether formation was then achieved through triflation of the C(42) alcohol and subsequent elimination of triflic acid.
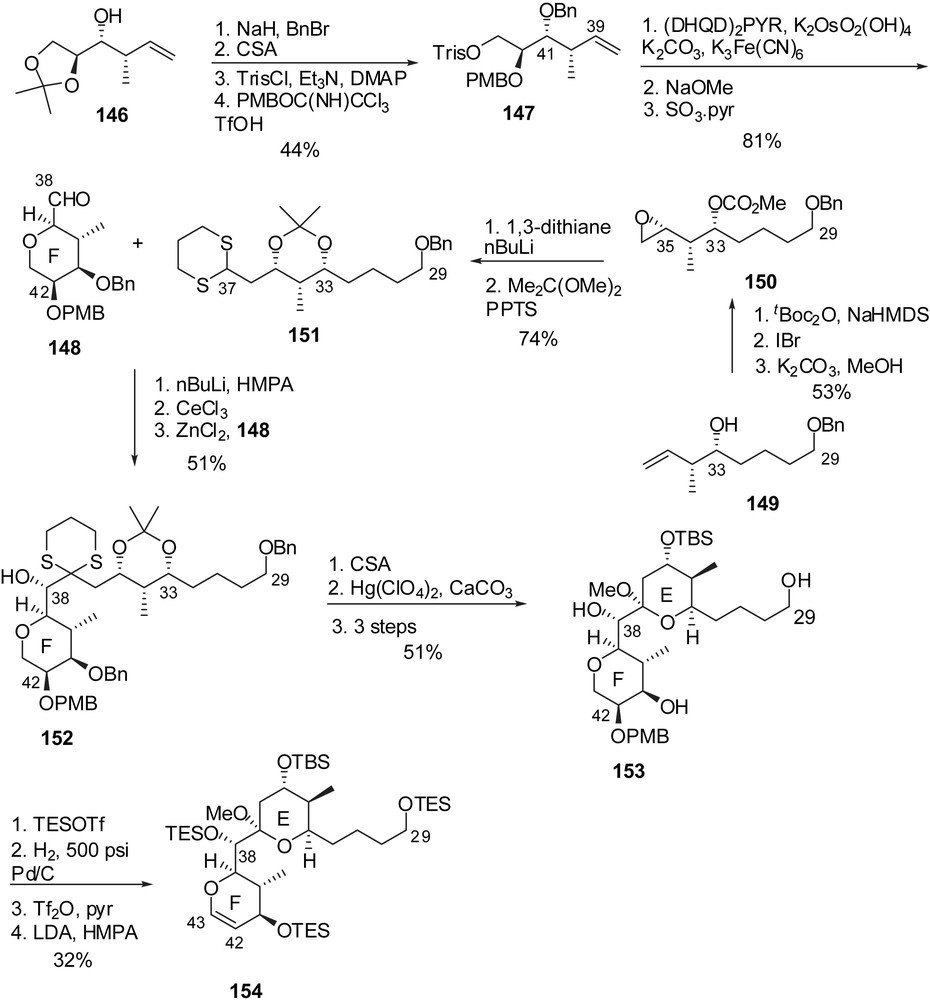
The attachment of the fully elaborated trienic side chain was performed according to Evans precedent [16] through regio- and stereoselective addition of allylstannane 155 on the epoxide derived from glycal 154 (Fig. 21). The resulting adduct was further elaborated into phosphonium salt 157. The endgame Wittig olefination with aldehyde 145 and careful adjustment of final protecting groups afforded seco-acid 158. Macrolactonization under Yamagushi's conditions and subsequent cleavage of silyl ethers delivered spongistatin 1/altohyrtin A in 0.5% overall yield and 29 steps for the longest linear sequence, based on the EF subunit.
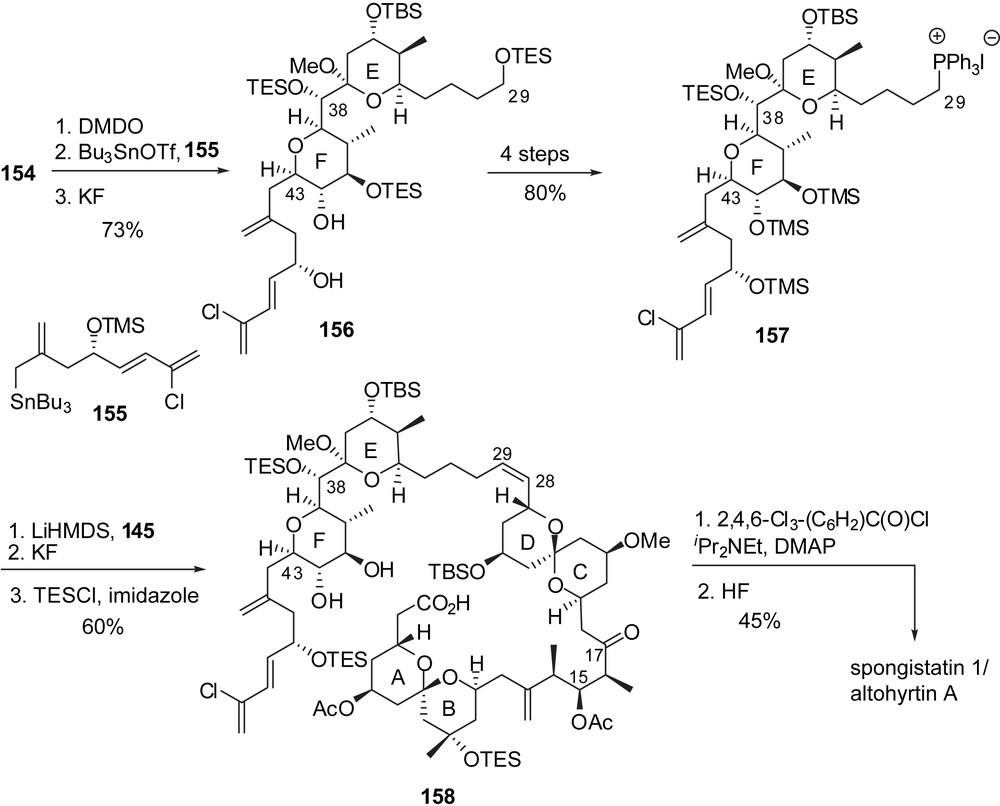
3.6 Total synthesis of altohyrtin C/spongistatin 2 by Heathcock [23]
The highly convergent strategy developed by Heathcock and co-workers was the first pathway that allowed the production of 250 mg of altohyrtin C/spongistatin 2, which represented a considerable improvement over the previously reported pathways. Such a scale had never been reached before, either from extraction of sponges or by synthetic routes. The synthesis of AB and CD fragments was envisaged from antipodal chiral pool building blocks and relied on the spirocyclization of β-diketone precursors. Aldol condensation of methyl ketone 159 to aldehyde 160 and subsequent oxidation delivered the C(1)–C(12) skeleton (Fig. 22). Acidic treatment induced spiroacetalization to a single stereoisomer 162, with an axial/axial arrangement. Installation of the methyl carbinol at C(9) and adjustment of protecting groups allowed further introduction of the C(12)–C(15) side chain through addition of lithium vinyl reagent derived from 164. Formylation of the resulting alcohol and reduction of the so-formed allylic formate led to the AB-spiroketal subunit 166.
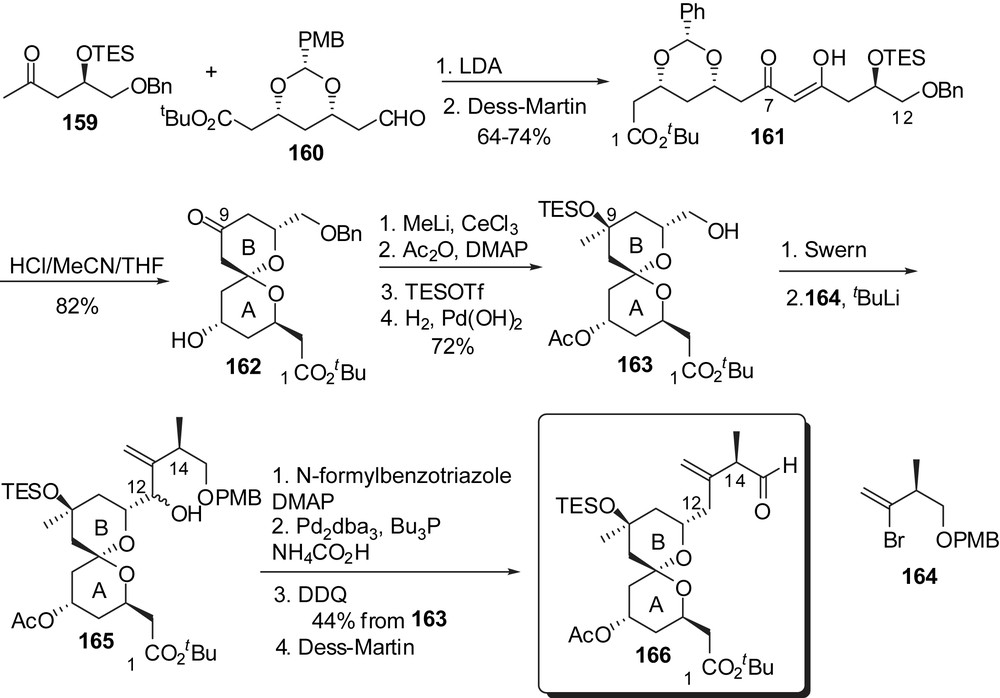
For the preparation of the CD fragment a similar approach was developed from methyl ketone 167 (antipodal building block to 159) (Fig. 23). Spirocyclization from β-diketone 167, in the presence of ZnBr2, proceeded with a complete stereoselectivity in favour of the axial/equatorial kinetic spiroketal 170, which represents a significant improvement over the strategies presented above. Reduction of the ketone at C(21) was performed in a similar manner as in the synthesis of Crimmins and the sequence was completed by installation of the ethyl ketone functionality at C(17).
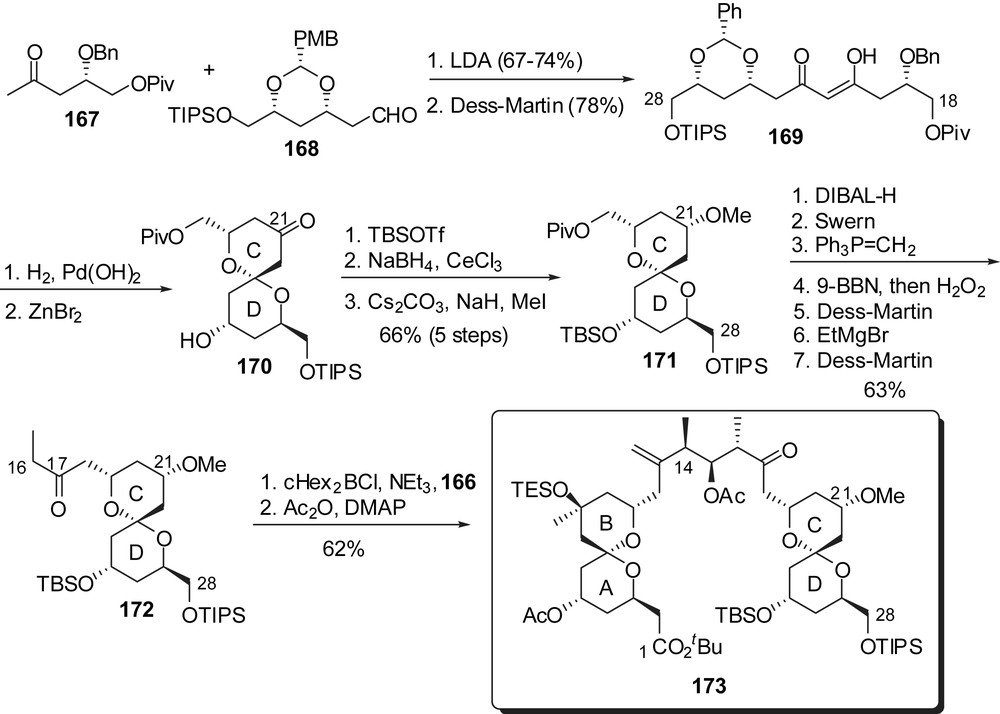
Both AB and CD units were then assembled by an anti-boron aldol reaction to afford, after acetylation, the C(1)–C(28) fragment 173.
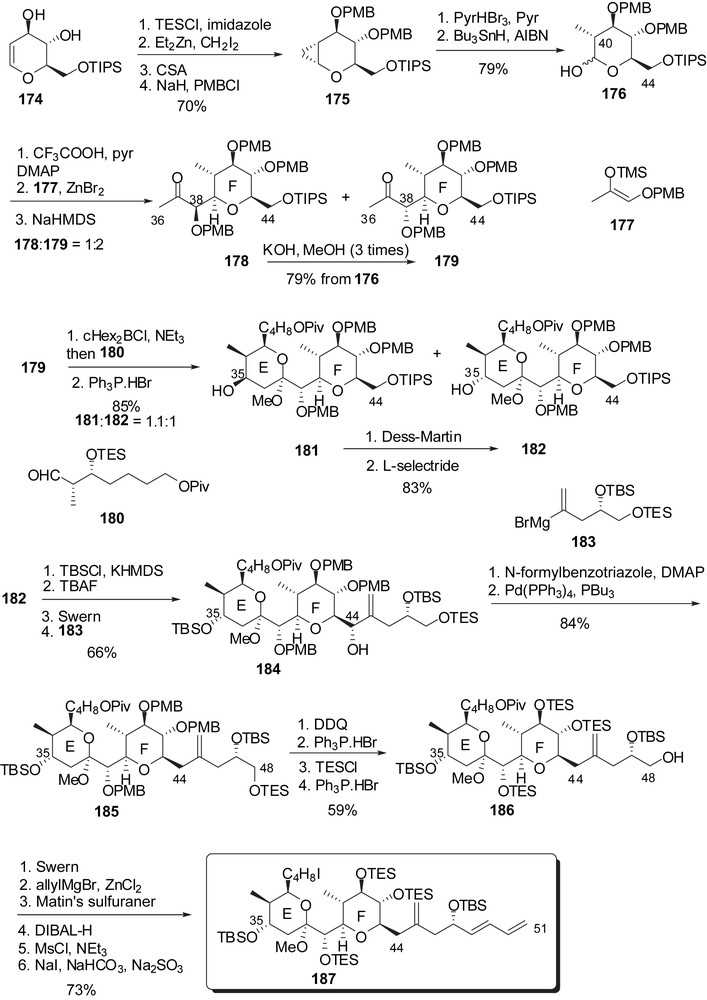
Construction of the EF subunit relied on a C-glycosylation of a glycal F-ring precursor to generate the C(36)–C(44) skeleton followed by aldol condensation of the methyl ketone at C(37) to an E-ring precursor aldehyde (Fig. 24). Glucal 174 [27] was selectively protected and submitted to cyclopropanation. The C(40) methyl group was then generated through a sequence of hydrobromination of the cyclopropyl group/reduction of the intermediate bromide. C-Glycosylation with silyl enol ether 177, followed by anomeric equilibration under basic conditions provided a 1:2 mixture of the desired isomer 179 and its C(38) epimer 178. The later could be converted into 179 after successive equilibrations in the presence of KOH. Boron aldol condensation to aldehyde 180 followed by acid promoted cyclization afforded methyl ketal 182 and its C(35) epimer 181 in a 1:1.1 ratio. A two step recycling of 181 provided more of the desired EF rings. Coupling with Grignard reagent 183 followed by deoxygenation at C(44) installed the C(44)–C(48) side chain. The terminal diene was then introduced through allyl Grignard addition on the C(48) aldehyde followed by dehydration of the resulting allylic alcohol to deliver the elaborated C(29)–C(51) fragment 187.
The endgame connections involved Wittig coupling of the phosphonium salt derived from 187 to the ABCD subunit 173, followed by removal of silyl ethers at C(41) and C(42) to allow a final macrolactonization/deprotection sequence (Fig. 25). These last steps provided 0.25 g of spongistatin 2/altohyrtin C with a longest linear sequence of 37 steps from (S)-malic acid. The remarkable optimization of the synthetic route to the EF subunit was illustrated by a 6.8% overall yield for the production of 187 from commercially available tri-O-acetyl-d-glucal. This achievement constituted an important step on the way to deliver multigram quantities of these fascinating macrolides for in vivo biological evaluations.
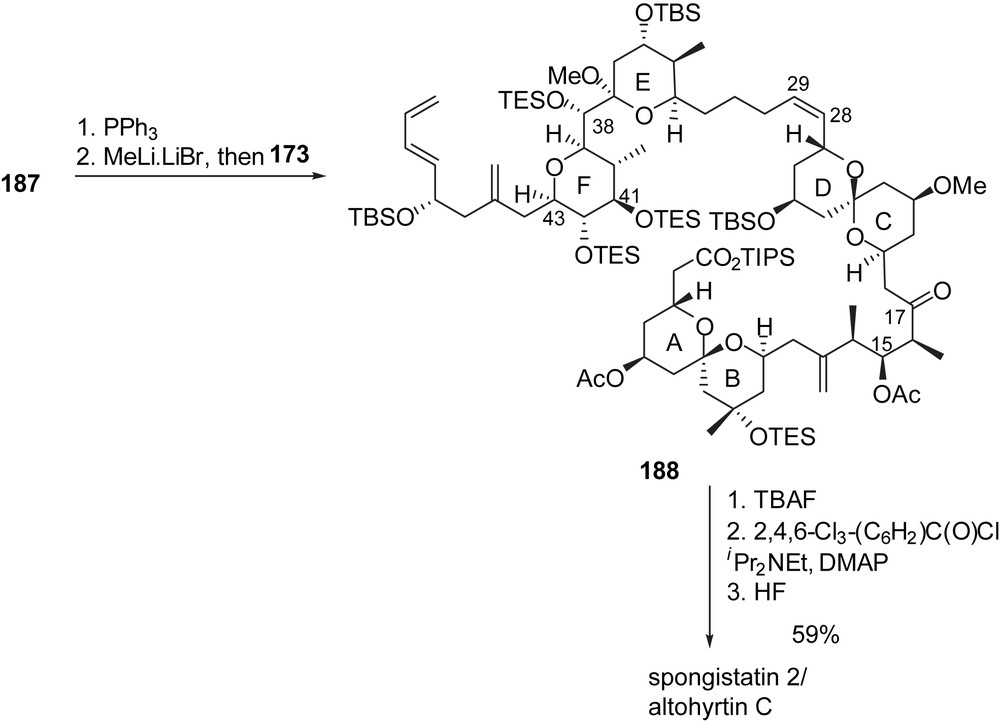
3.7 Total synthesis of altohyrtin A/spongistatin 1 by Ley [24]
Guided by the latent C2 pseudo-symmetry of spongistatin 1 about the C(15) atom, Ley and co-workers designed a synthetic pathway where both AB and CD spiroketals derive from a common linear precursor. For that purpose, addition of the acetylide anion of a 1:1 C(5) epimeric mixture 190 to aldehyde 189, followed by oxidation of the intermediate alcohol led to ynone 191 (Fig. 26). Double conjugate addition of 1,3-propanedithiol and subsequent acidic treatment provided a 1:0.7:0.26 mixture of three spiroketals presenting the AB axial/axial, CD axial/axial and CD axial/equatorial arrangements, respectively. The undesired spiroketal 194 was efficiently converted into the axial/axial CD isomer through equilibration, thus giving access to 193 and 195 as precursors of AB and CD spiroketals of spongistatin 1. Removal of the dithiane moiety on 193 and stereoselective addition of MeLi installed the C(9) carbinol group. Further transformation included carbon homologation to introduce the C(1) carboxyl moiety and oxidation at C(15) to deliver the elaborated AB subunit 198, ready to undergo aldol condensation with a CD fragment. In parallel, dithiane cleavage on 195 and alcohol silylation allowed Luche reduction at C(21) to afford the corresponding equatorial alcohol. Subsequent methylation of the C(21) hydroxyl group, followed by selective deprotection of the primary alcohol allowed oxidation to the corresponding carboxylic acid. The olefin was then submitted to ozonolysis to introduce the C(17) ketone moiety and further smooth decarboxylation delivered the CD spiroketal subunit 201.
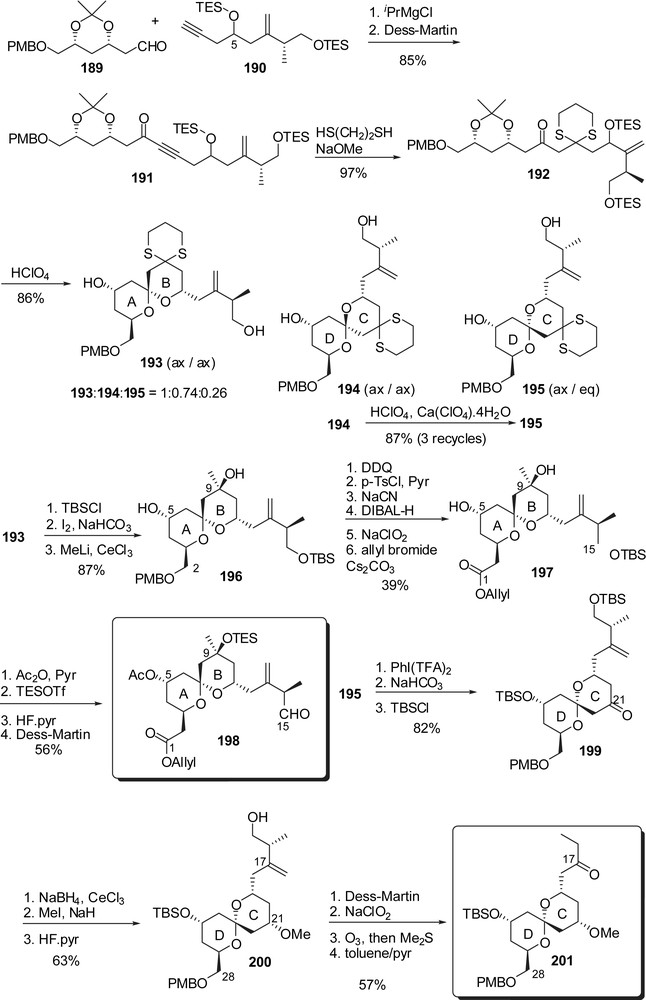
For the synthesis of the southern hemisphere, Ley and co-workers envisaged ring F as a central core on which both the C(29)–C(35) and C(47)–C(51) fragments are sequentially appended (Fig. 27). Preparation of the F-ring started from protected pyranoside 202 which underwent a Mukaiyama-type C-glycosylation with 2-trimethylsilyloxy-propene followed by basic promoted anomeric epimerization and protection of the resulting oxo moiety at C(37) to provide the F-ring precursor 203. Further elaboration and carbon homologation at C(44) via iodide displacement by cyanide anion and further transformation into an intermediate carboxylic acid, allowed Weinreb amide introduction (205). The C(38) hydroxyl was then installed by oxidation of the silyl enol ether derived from methyl ketone at C(37) to provide 206 as a single isomer. Stereoselective aldol condensation of boron enolate derived from methyl ketone 206 to aldehyde 207 gave the corresponding syn-adduct 208. Acid promoted ketalization followed by addition of MeLi to the Weinreb amide moiety delivered the EF rings' methyl ketone 209. A second boron aldol coupling to chlorodienyl aldehyde 210 gave access to the fully elaborated southern hemisphere 211.
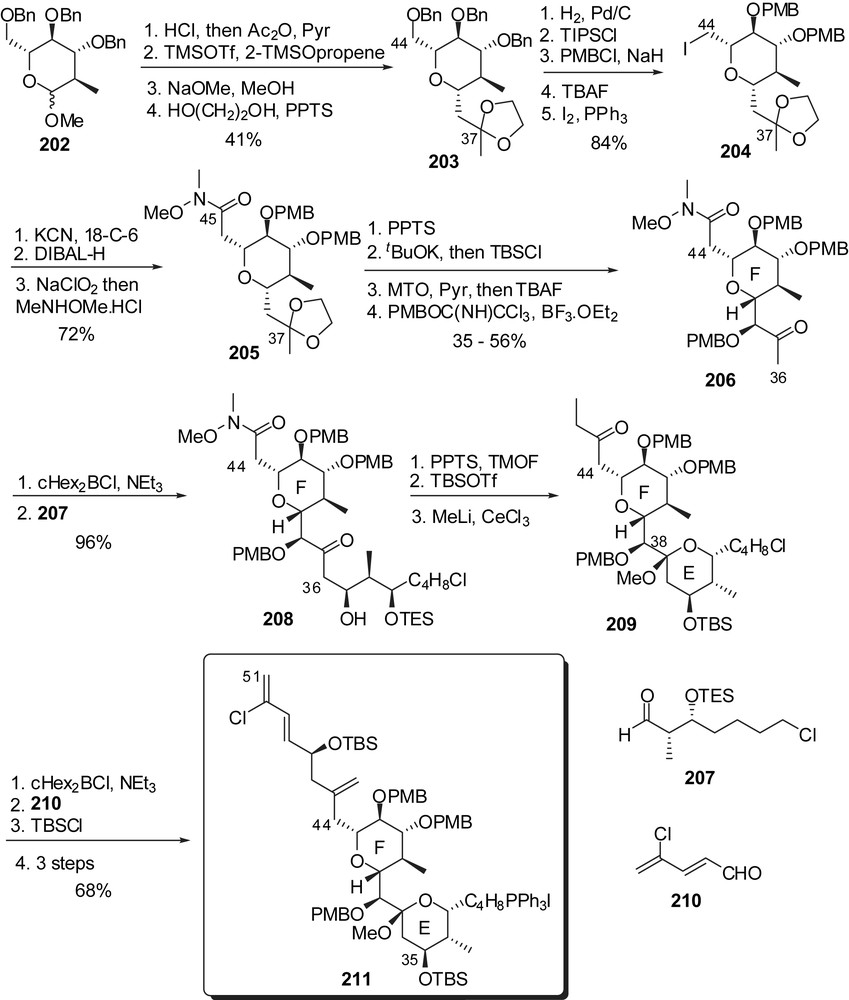
The endgame transformations started with assembly of the ABCD bis-spiroketal subunit by the well established anti-aldolisation to construct the C(15)–C(16) linkage (Fig. 28). Wittig coupling to the EF rings' fragment 211, macrolactonization and HF mediated final deprotection completed the total synthesis of spongistatin 1/altohyrtin A. This approach allows reduction in the number of steps toward the ABCD hemisphere as both spiroketals arise from a common precursor. Starting from (S)-glycidol and epichlorohydrin, the ABCD segment 212 was produced in 46 steps.
At present seven research groups succeeded in the total syntheses of spongistatins 1 and 2, but further efforts are still undergoing to provide multigram quantities of these fascinating macrolides, required for further biological investigations.
4 Synthetic approaches toward key subunits of spongistatins/altohyrtins
In parallel to the remarkable efforts devoted to the total synthesis of these macrolides several research groups developed strategies toward key subunits, mainly both AB and CD spiroketals and EF rings' fragments.
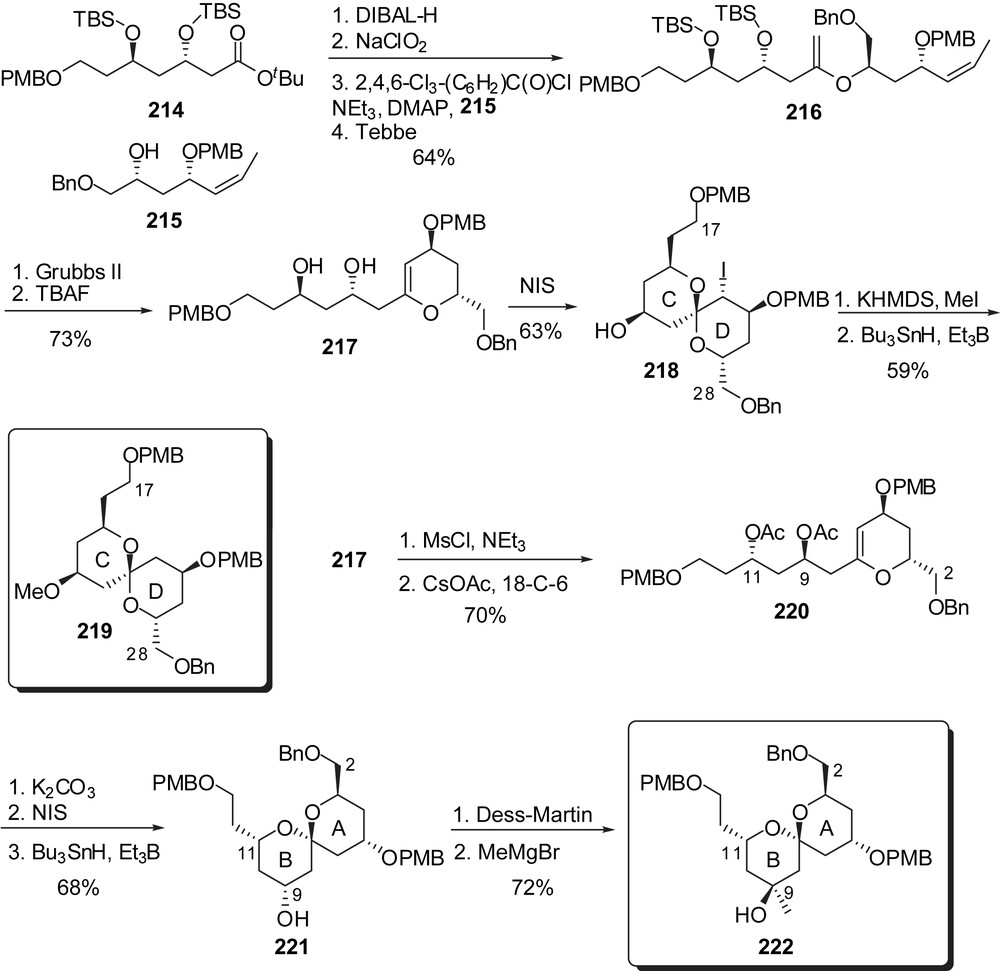
A new approach toward AB and CD subunits was explored by Roush, exploiting a common glycal intermediate and making use of an iodo-spiroketalization process to generate the spiroketals. The C(17)–C(28) skeleton was assembled through the coupling of the carboxylic acid derived from 214 to the secondary alcohol of 215, using Yamagushi's conditions followed by Tebbe olefination to generate enol ether 216 (Fig. 29) [28]. Ring closing metathesis and subsequent silyl ethers' cleavage afforded dihydroxy glycal 217 as the common precursor for both AB and CD fragments. NIS-promoted iodocyclization provided the desired axial/equatorial CD spiroketal 218 as the major isomer (8:1 dr), which was further transformed into the fully elaborated C(17)–C(28) fragment of spongistatin 1. Intermediate 217 was in turn submitted to a double inversion process at C(9) and C(11) positions followed by the intramolecular iodo-spiroketalization reaction which delivered, this time, the AB spiroketal possessing two stabilizing anomeric effects 221 [29]. Installation of the methyl carbinol at C(9) completed the preparation of a C(2)–C(13) fragment of spongistatin 1 (222).
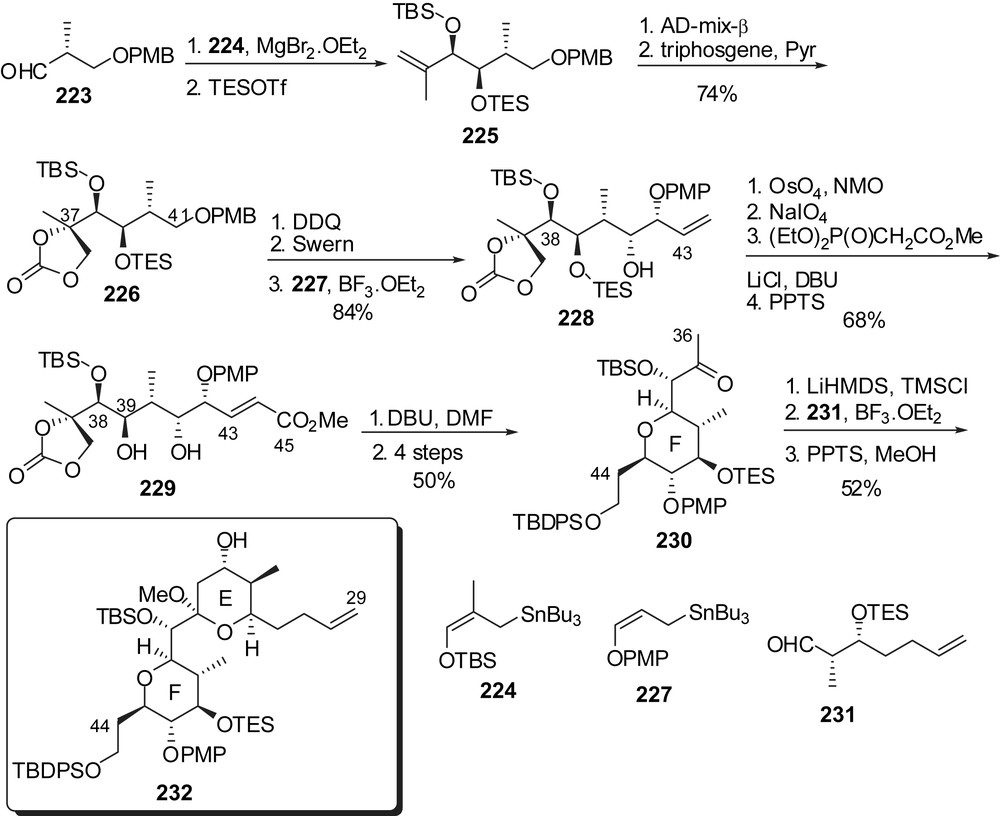
Roush also reported an alternative route to the central F-ring through successive diastereoselective γ-alkoxyallyl stannylations (Fig. 30) [30]. Chelation controlled addition of allylstannane 224 to aldehyde 223 proceeded with an excellent stereoselectivity (>20:1 dr) to provide homoallylic alcohol 225. Stereoselective dihydroxylation of the olefin followed by installation of a carbonate moiety at C(37) and oxidation at C(41) delivered the aldehyde precursor for a second addition of allylstannane 227 which also proceeded with high stereocontrol (228, >20:1 dr). Further elaboration to α,β-unsaturated ester 229 was required for an intramolecular 1,4-addition from the alcohol at C(39) to produce F-ring 230. The E-ring precursor was then appended through Mukaiyama aldol coupling of silyl enol ether derived from 230 to aldehyde 231, with a 2.5:1 stereoselectivity. Methyl ketal formation in the presence of PPTS afforded the elaborated C(29)–C(45) fragment of spongistatin 1/altohyrtin A (232).
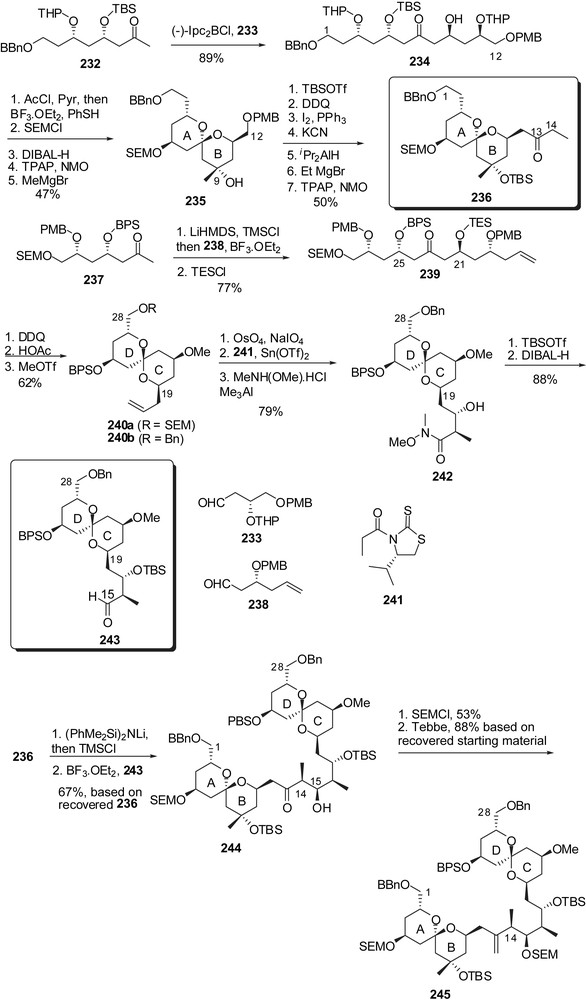
Paquette and co-workers explored the assembly of the ABCD northern hemisphere through a Mukaiyama aldolisation to generate the C(14)–C(15) linkage, which contrasts with the anti-aldol condensation used by many research groups for the creation of the C(15)–C(16) bond. The C(1)–C(12) skeleton was generated through addition of boron enolate from methyl ketone 232 to aldehyde 233 (Fig. 31) [31]. Lewis acid promoted removal of THP ethers induced spirocyclization to a single axial/axial isomer. Protecting groups' manipulation followed by methyl carbinol formation at C(9) gave spiroketal 235. The C(12) alcohol was iodinated to allow nucleophilic displacement with cyanide anion. A sequence of reduction/ethyl Grignard addition/oxidation of the intermediate C(13) alcohol installed the C(13) ethyl ketone moiety to provide the fully elaborated AB spiroketal 236. The CD spiroketal precursor was assembled through a Mukaiyama aldol condensation of silyl enol ether derived from 237 to aldehyde 238 to install the C(21)–C(25) anti configuration. Removal of the p-methoxybenzyl ethers led to the formation of the desired axial/equatorial spiroketal 240a. On the benzylated analogue 240b, homologation at C(21) through stereoselective aldolisation using chiral auxiliary 241 and subsequent Weinreb amide introduction led to the intermediate spiroketal 242. Silylation of the alcohol at C(17) and reduction using DIBAL-H gave the CD spiroketal subunit 243. Mukaiyama aldolisation on fragments 236 and 243 proceeded with high stereocontrol to afford the C(14),C(15) syn arrangement. Olefination with Tebbe reagent finally gave rise to the ABCD portion of spongistatin 1/altohyrtin A (245).
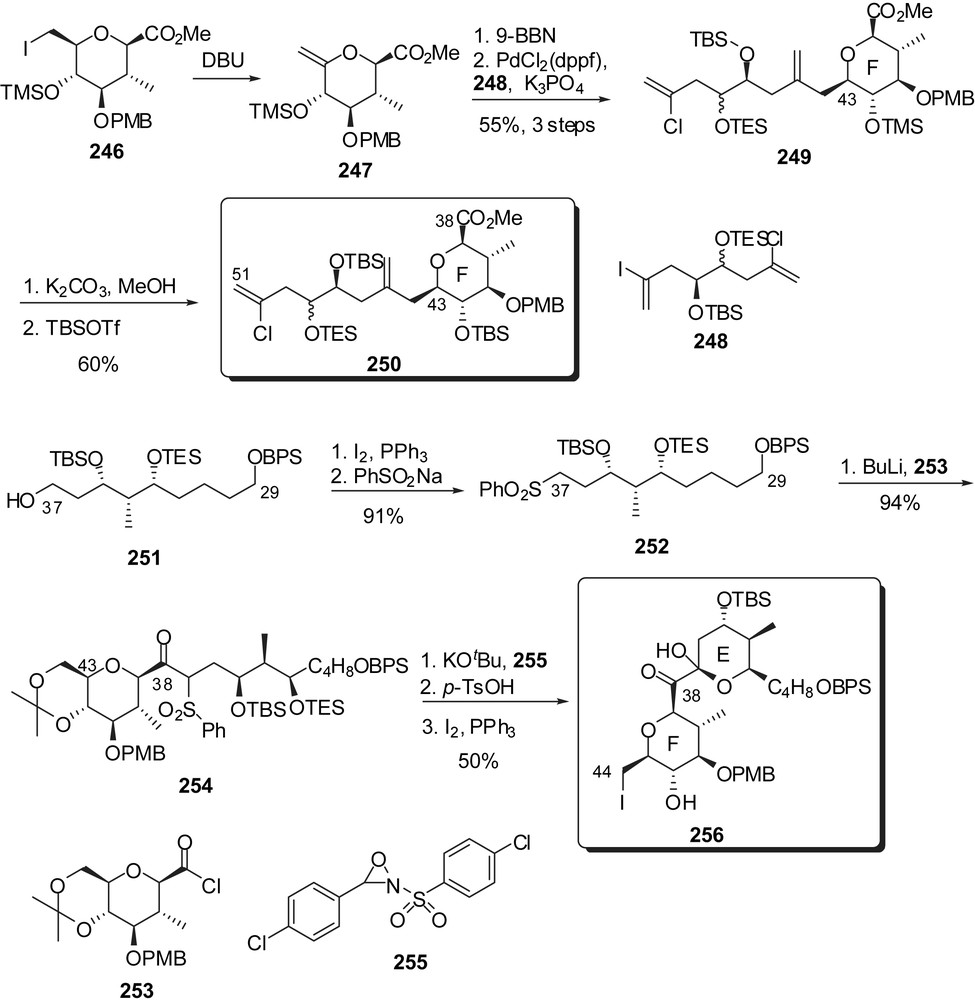
Synthetic efforts, by the same group, toward the EF rings' subunit revealed the possibility to append the trienic side chain through a palladium promoted cross-coupling at C(44) (Fig. 32) [32]. For that purpose, the F-ring exo-methylene derivative 247 was converted into an intermediate borane reagent that was coupled to vinyl iodide 248 to afford 249 as a single equatorial isomer at C(43). Silyl protecting group interconversion delivered the C(38)–C(51) skeleton of spongistatin 1 (250). For the assembly of the EF rings' fragment, addition of a sulfone anion derived from 252 to acyl chloride 253 provided the C(29)–C(44) skeleton. Oxidation with oxaziridine 255 to generate an intermediate α-diketone followed by acidic treatment induced cycle E formation. Iodination at C(44) completed the sequence to deliver 256 as the precursor of the side chain introduction as explored above.
Rather than using a Mukaiyama aldolisation to generate the C(14)–C(15) linkage of spongistatin 1, Nakata and co-workers explored the coupling of a crotyl stannane C(1)–C(14) AB fragment to CD spiroketal aldehyde at C(15) [33d]. Preparation of these fragments was based on dithiane anions' addition to epoxides (Fig. 33) [33a–c]. This strategy, applied to dithiane 257 and epoxide 258, delivered the C(1)–C(13) skeleton 259 in good yield. Protecting group adjustment and dithiane cleavage induced spiroketalization toward the axial/axial isomer 260. Reductive cleavage of the pivaloyl group followed by oxidation of the resulting alcohol afforded aldehyde 261. Vinyl Grignard addition followed by oxidation of the intermediate allylic alcohol and methylenation installed the diene moiety of 262. Further elaboration generated the crotyl stannane moiety at C(14). The CD spiroketal partner arose from the coupling of the lithium anion derived from 264 to epoxide 265 and subsequent spiroketalization on the unmasked ketone at C(23). A mixture of the undesired isomer 267 and of the axial/equatorial spiroketal 268 was obtained and further transformed into the corresponding p-methoxybenzylidene acetals 269 and 270. At that stage, the undesired isomer could be isomerized up to a 2:3 ratio in favour of 270. Spiroketal 270 was finally transformed into the C(15) aldehyde 271. The crucial coupling to crotyl stannane 263 afforded a good 83% yield of the ABCD fragment 272, but presenting wrong configurations at C(14) and C(15). Several steps were then required to restore the natural configuration of these stereocenters and to deliver Smith ABCD hemisphere 145. Even if this strategy allowed assembly of the two spiroketals' subunits, further elaboration into the natural C(14)–C(17) fragment could only be achieved in low yields.
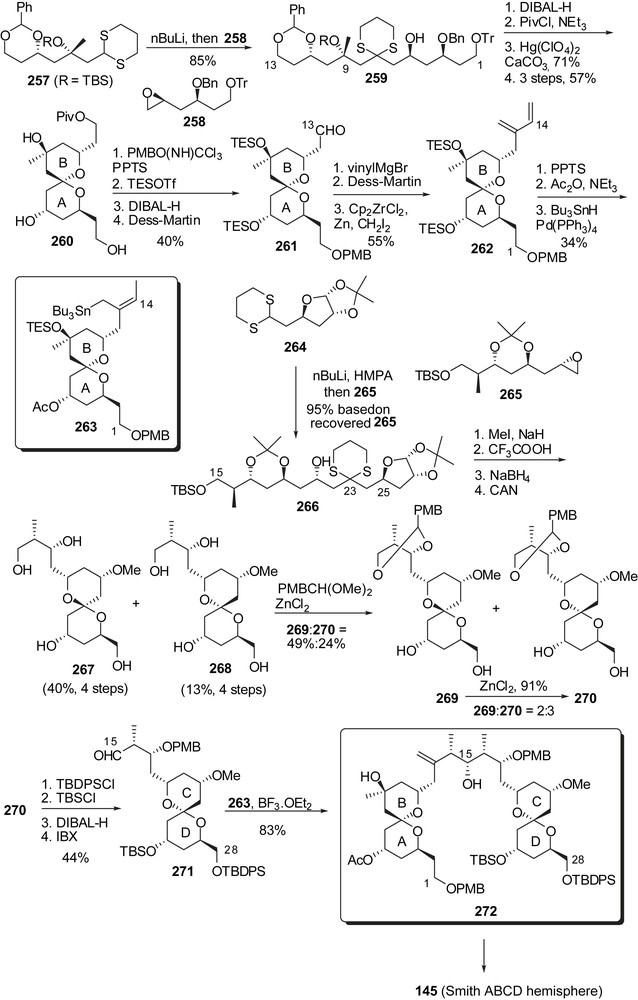
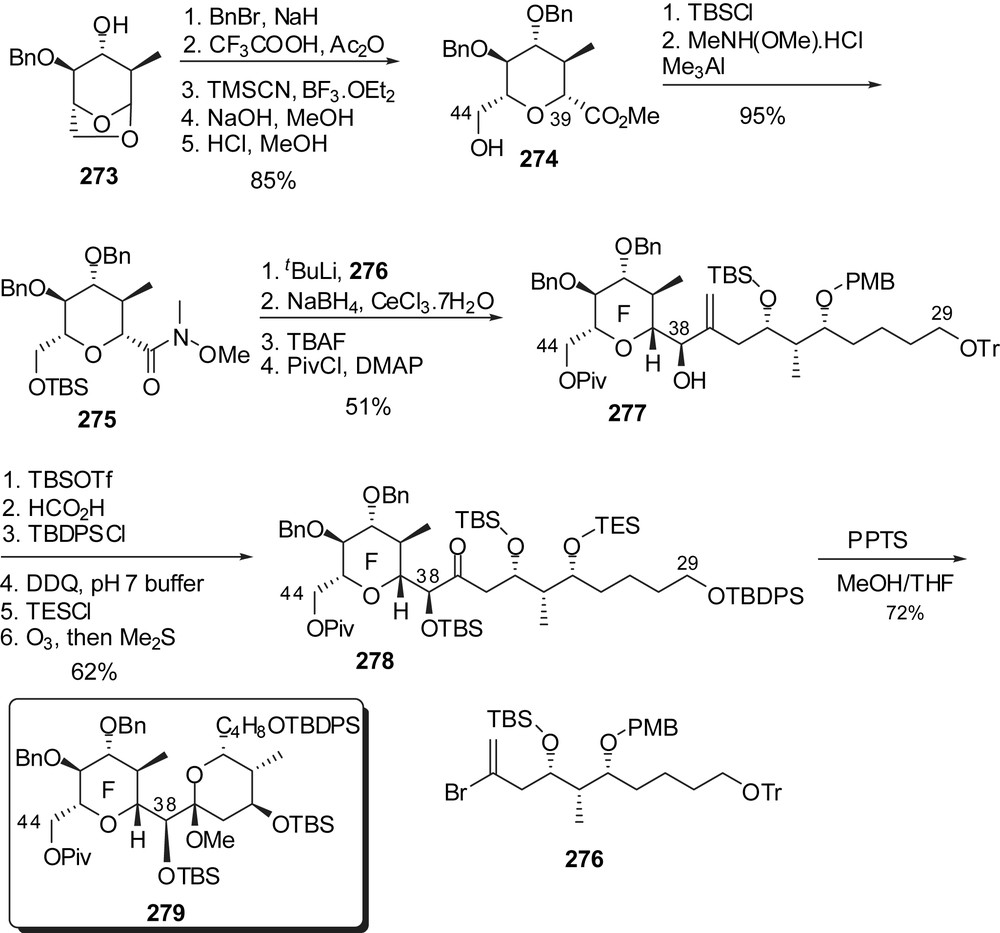
Nakata and co-workers also developed a synthetic route toward the EF rings' subunit [34] making use of a coupling between a C(29)–C(37) vinyl bromide to an F-ring C(38) Weinreb amide (Fig. 34). For that purpose anhydrosugar derivative 273 was opened to an anomeric mixture of acetates which were reacted with TMSCN. The resulting epimeric mixture of cyanides was hydrolyzed under basic conditions and at the same time equilibrated to the C(39) equatorial isomer 274 as the major product. Silylation of the C(44) hydroxyl group and Weinreb amide formation allowed coupling to the vinyl lithium reagent derived from 276. Further diastereoselective reduction at C(38) and protecting group manipulations afforded the C(29)–C(44) skeleton 278. Acid mediated methyl ketal formation generated the EF rings' derivative 279.
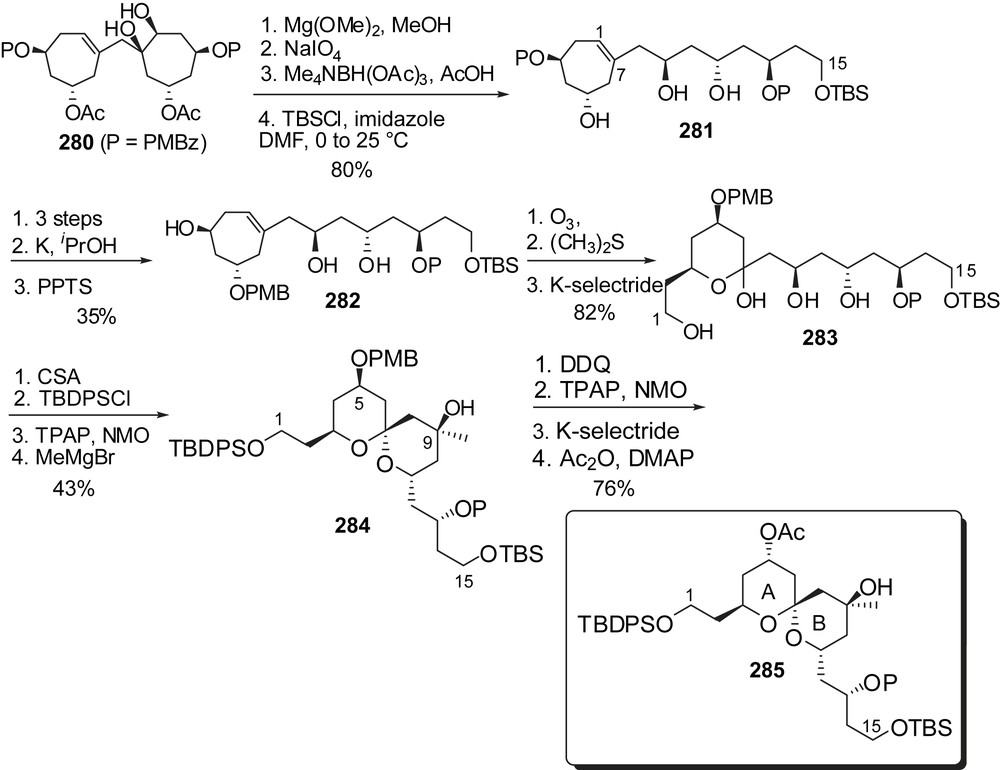
Studies toward the synthesis of the AB spiroketal of spongistatins have also been reported by Vogel and co-workers and are based on the stereoselective functionalization of a meso-methylenebis(cyclohept-3-ene-1,6-diyl diester) [35]. In this approach all 15 carbon atoms of the skeleton are generated since the early stages of the sequence (Fig. 35). Oxidative cleavage of diol 280 [36], followed by diastereoselective reduction of the intermediate keto aldehyde installed the C(7)–C(15) fragment. Ozonolysis of the remaining olefin and in situ selective reduction of the resulting aldehyde delivered hemiketal 283. Acidic treatment induced spirocyclization to a single axial/axial isomer 284. Further elaboration included methyl carbinol installation at C(9) and inversion of the C(5) configuration to deliver an AB spiroketal fragment 285.
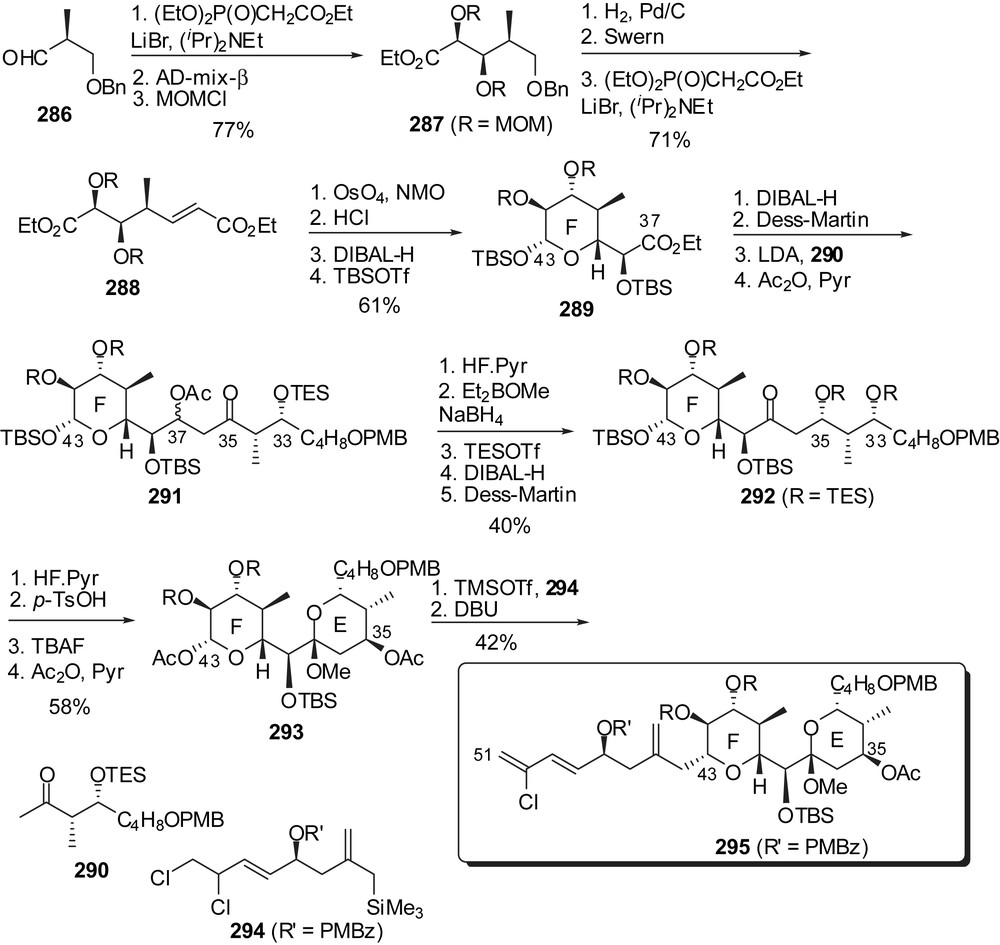
The same group disclosed a synthetic route to a fully elaborated EF-ring subunit making use of successive sequences of HWE olefination/stereoselective dihydroxylation of the resulting α,β-unsaturated ester to build the F core on which are then appended the E-ring precursor and the trienic side chain (Fig. 36) [37]. HWE olefination of aldehyde 286 followed by stereoselective dihydroxylation and protection of the intermediate diol provided the protected triol 287. Cleavage of the benzyl ether and subsequent oxidation allowed a second similar sequence. Acidic treatment of the intermediate semi-protected tetrol induced lactonization to close the F-ring. Reduction to the corresponding lactol and silylation afforded tetrahydropyran 289 presenting all substituents in equatorial position. This pathway to the F core proceeded with a high 29% overall yield from aldehyde 286 and could be scale to gram quantities of 289. Introduction of the E-ring precursor was performed through aldol condensation of the lithium enolate derived from methyl ketone 290 to the aldehyde at C(37). The 95:5 C(37) epimeric mixture of aldols 291 was further elaborated into the C(33) ketone 292, thus allowing methyl ketal formation to deliver the EF-ring fragment 293. Introduction of the trienic side chain involved β-C-glycosylation (4:1 dr) at C(43) with allylsilane 294 followed by in situ elimination of HCl to afford the fully elaborated C(29)–C(51) fragment of spongistatin 1 295.
Further synthetic strategies were developed toward isolated fragments of spongistatins. Roberts and co-workers disclosed the synthesis of the C(24) to C(37) perimeter of spongistatin 1 (Fig. 37) [38]. Brown crotylation of aldehyde 296 followed by protection of the intermediate homoallylic alcohol delivered 298 in moderate yield and 86% diastereoselectivity. Iodocyclization, followed by methanolysis of the resulting iodocarbonate afforded epoxide 299 in high stereoselectivity (15.6:1 dr). Silylation of the secondary alcohol followed by nucleophilic opening of the oxirane with lithio dithiane 300 provided the C(29)–C(37) skeleton of spongistatin 1. Preparation of the C(24)–C(36) perimeter started from (S)-malic acid dimethyl ester which was transformed into ynone 303. Stereoselective reduction in the presence of oxaborilidine 304 and catechol borane proceeded with syn stereoselectivity (95:5 dr). Further elaboration afforded alkyne 305. Silyl ether cleavage followed by oxidation allowed crotylation with borane 297 to deliver the C(24)–C(36) skeleton of spongistatin 1 306. The same group reported pyranose based approaches to the F-ring of spongistatin 1 [39]. Benzylation of anhydrosugar 273 was followed by Lewis acid promoted hydrolysis of the 1,6-anhydro bridge. α-C-Glycosylation with allyltrimethylsilane and subsequent ozonolysis under basic conditions afforded methyl ester 307 with a 7:4 diastereoselectivity in favour of the α-anomer. Protecting groups' interconversion followed by reduction of the ester moiety and protection of the resulting aldehyde gave dithiane 308 as a C(38)–C(45) F-ring fragment.
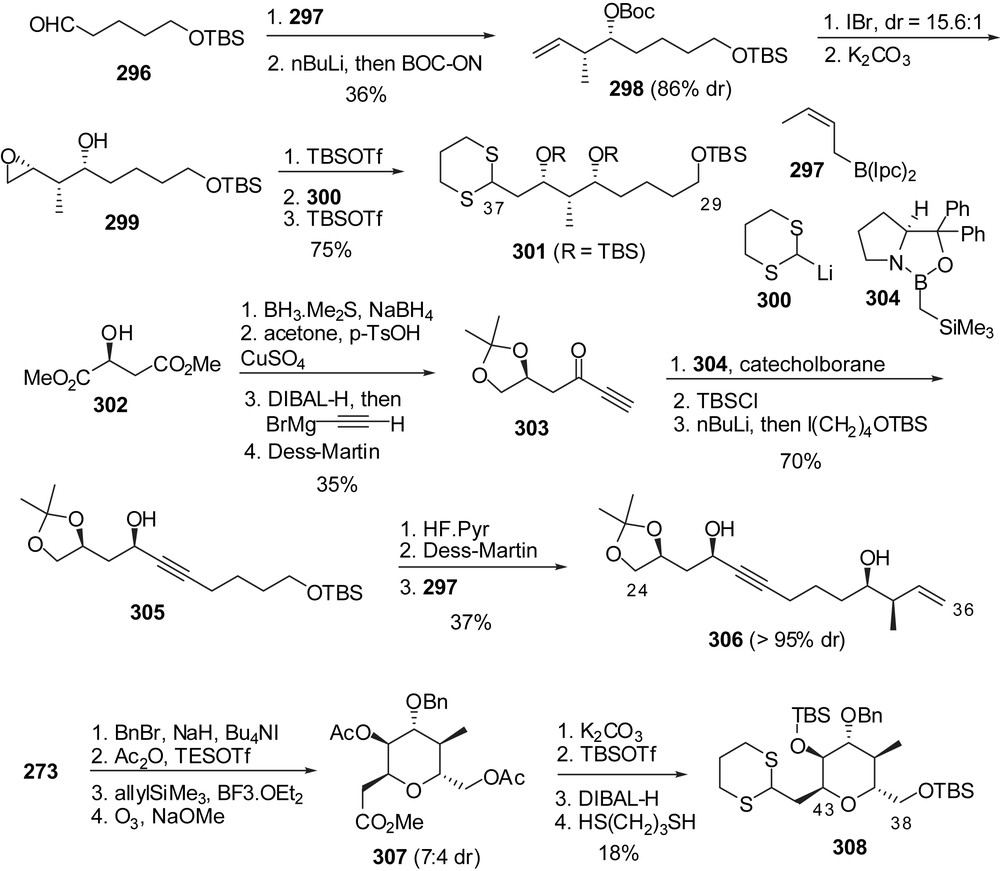
The group of Hoffmann has proposed the elaboration of 8-oxabicyclo[3.2.1]oct-6-en-3-one derivatives into functionalized tetrahydropyrans, in particular in several fragments related to spongistatins [40]. Bicyclic ketone 309 was reduced with complete selectivity to the corresponding endo-alcohol (Fig. 38). Protection followed by asymmetric hydroboration allowed desymmetrization of the system to afford diol 310 in excellent enantiomeric purity. A sequence of oxidation/Baeyer–Villiger ring expansion/acidic treatment delivered methoxyacetal 311. This intermediate was then converted into lactone 312 which is a relevant unit to C ring of spongistatin 1. Interestingly, changing the nature of the reducing agent of the bicyclic ketone and the stereochemistry of the hydroboration reagent led to other isomers such as lactone 313 which is relevant to B and D rings of spongistatin 1. Based on the same methodology, anomeric sulfide 315 was converted into separable isomeric sulfones 316 and 317 as C ring segments of spongistatins.
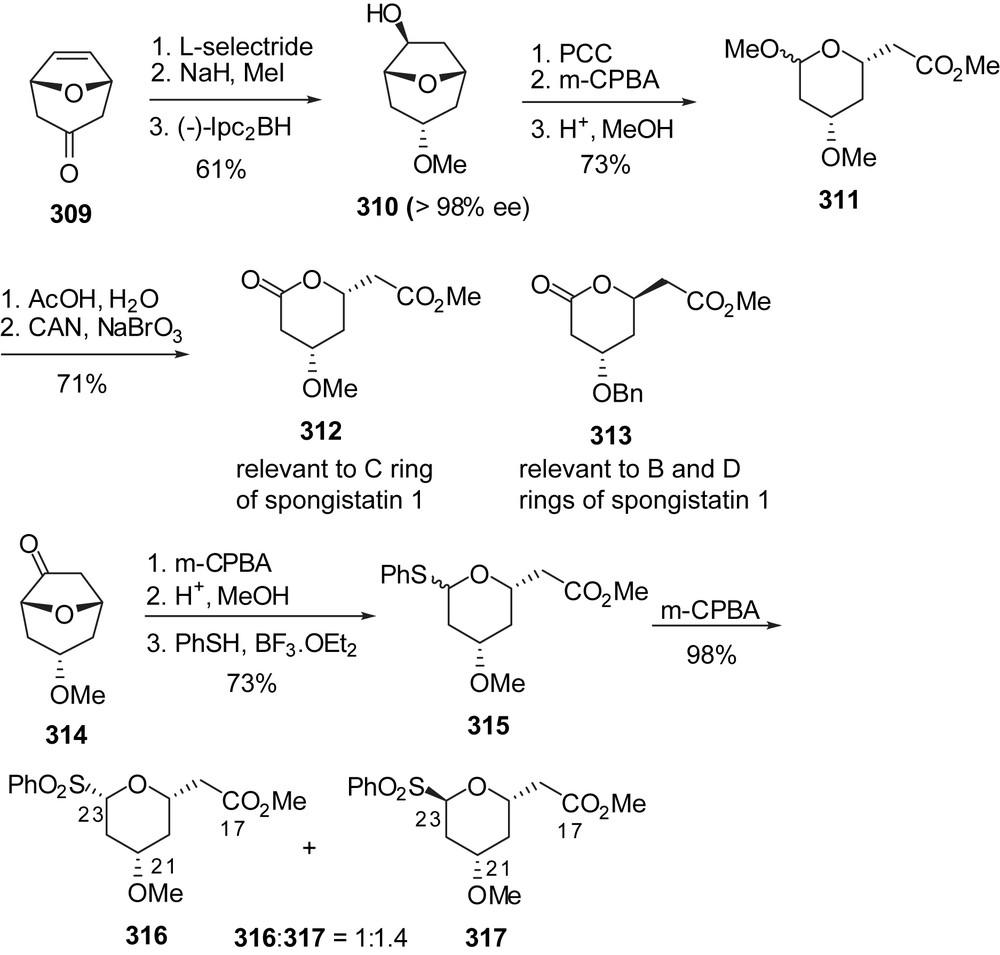
Approach toward the E-ring fragment of spongistatin 1 was developed from racemic methyl substituted bicyclic ketone 318 (Fig. 39). Stereoselective reduction to the exo-alcohol using SmI2 and subsequent benzyl ether formation allowed asymmetric hydroboration of the olefin moiety for the separation of diastereoisomeric intermediates 319 and 320, after oxidation. Both ketones were isolated in high enantiomeric purity. As described above, Baeyer–Villiger oxidation and lactone ring opening delivered methyl ester 321 as the C(31)–C(37) core of spongistatin 1. Further elongation of the side chain through allyl Grignard addition and final transformation into a C(37) triphenylphosphonium tetrafluoroborate delivered the C(29)–C(37) E-ring 324. Alternatively, a phenyl sulfone moiety could also be introduced at the C(37) position as illustrated by the preparation of derivatives 325 and 326.
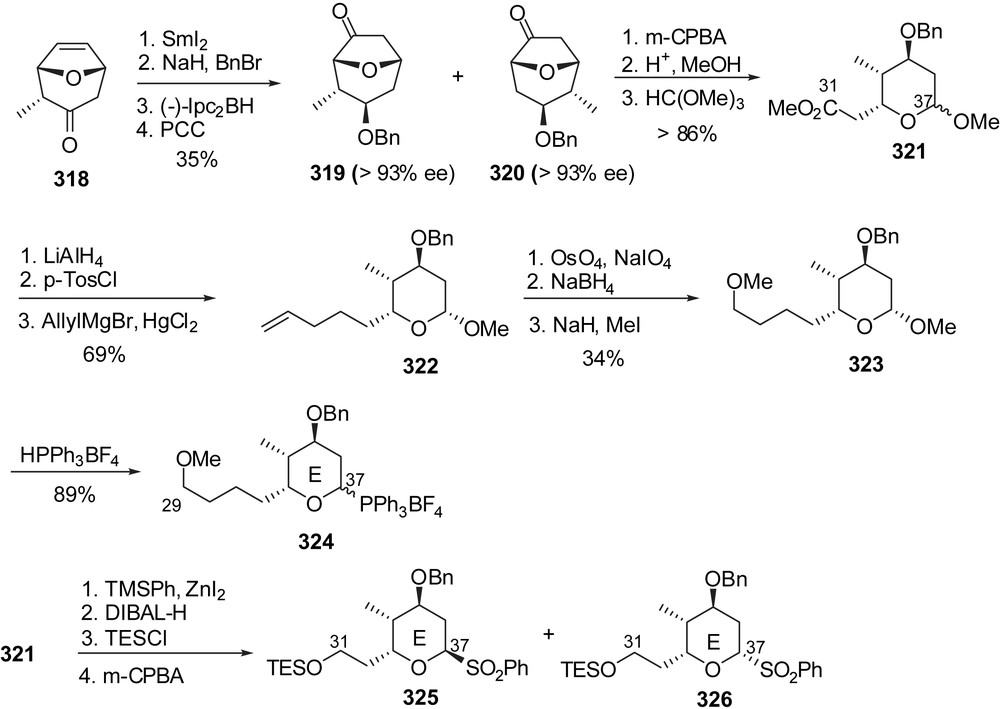
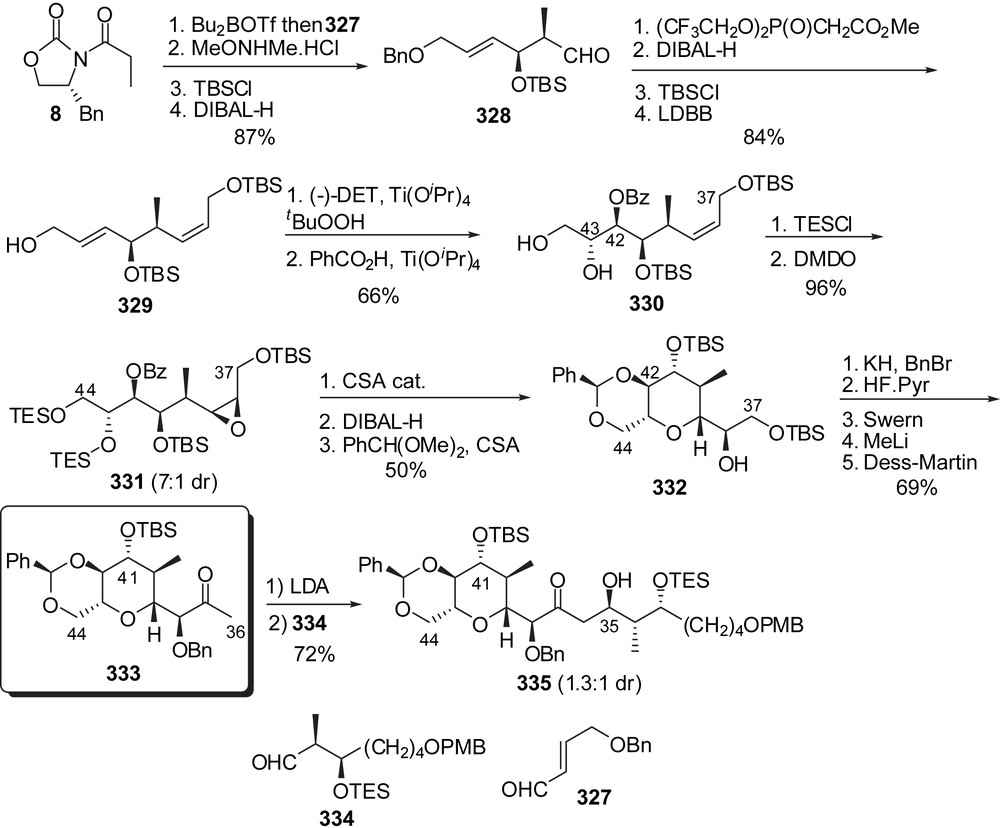
The preparation of a fully elaborated F-ring fragment of spongistatins was envisaged by Anderson and co-workers through cyclization of an epoxide precursor (Fig. 40) [41]. Evans aldolisation to aldehyde 327 followed by Weinreb amide formation and controlled reduction gave rise to aldehyde 328 with excellent stereoselectivity. Wadsworth–Horner–Emmons olefination and further elaboration provided diene 329. Sharpless asymmetric epoxidation of the allylic alcohol followed by regioselective ring opening with benzoic acid resulted in the formation of the C(37)–C(44) skeleton of spongistatin 1 (330) with the required anti relationship between alcohols at C(42) and C(43). After silylation of the diol moiety, a second epoxidation proceeded with a 7:1 diastereoselectivity in favour of the syn isomer 331. Selective removal of the triethylsilyl groups induced intramolecular epoxide opening to form the F-ring subunit. Reductive cleavage of the benzoyl protecting moiety and subsequent benzylidene acetal formation on the diol at C(42),C(44) delivered intermediate 332. Final elaboration included introduction of a C(37) methyl ketone group as precursor of the appending of the E-ring skeleton. Unfortunately aldol condensation to aldehyde 334 gave a mixture of C(35) epimeric alcohols with a 1.3:1 ratio in favour of the undesired configuration. This stereoselectivity could not be improved.
An innovative approach to the AB-spiroketal subunit of spongistatin 1 was proposed by Barrett through double allylation of aldehydes and further transformation into C2-symmetric spiroketals (Fig. 41) [42]. Treatment of aldehyde 336 with bis(diisopinocampheylborane) derivative 337 provided olefin 338 in 48% yield. In a single step, it was possible to assemble a C11 hexaol. The olefin was readily transformed into ketone 339 that underwent spirocyclization under acidic condition to deliver the C2-symmetric spiroketal 340 as the major isomer (58%). This intermediate represents an AB fragment of spongistatin 1.
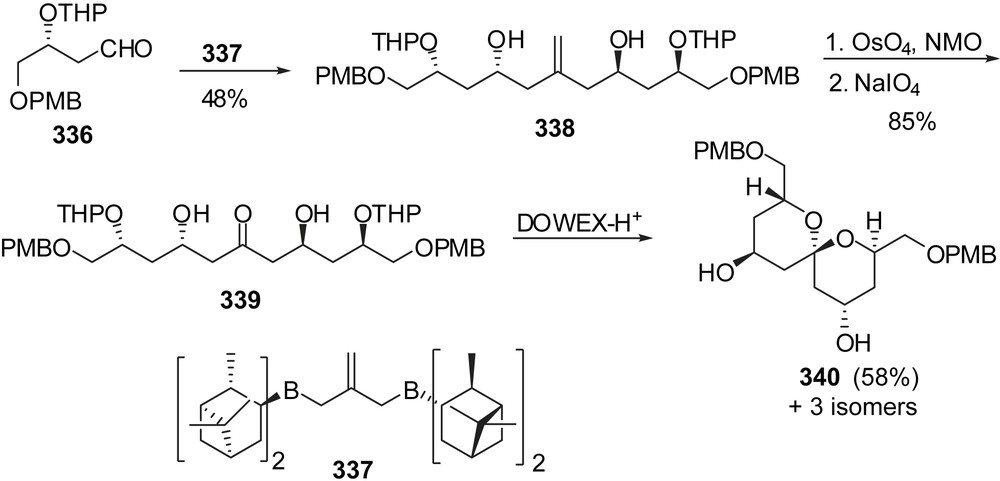
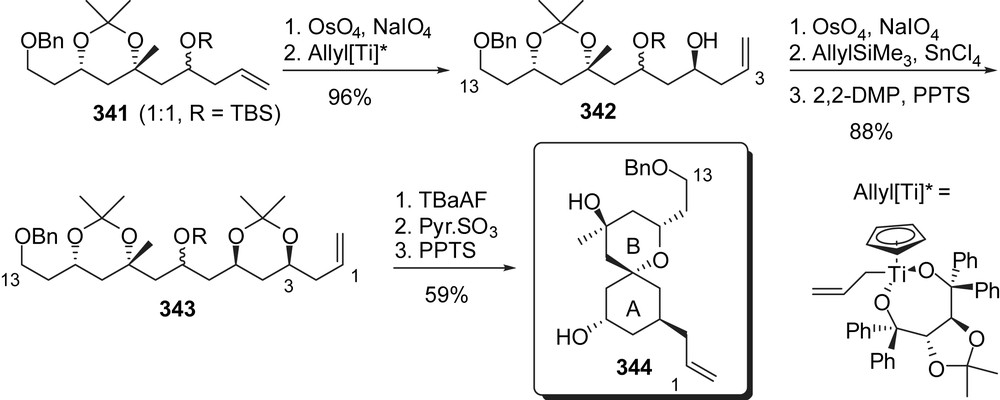
Stereoselective allylmetalations have also been proposed by the group of Cossy [43] for the preparation of a C(1)–C(13) AB spiroketal fragment of spongistatins (Fig. 42). The (1:1) C(7) epimeric mixture of homoallylic alcohols 341 was submitted to a sequence of oxidative cleavage of the double bond/enantioselective allyl titanation to install the C(5) alcohol with a high 98:2 selectivity. Oxidation of the olefin followed by a diastereoselective allylmetalation produced the C(3),C(5)-syn diol 343 in high yield. Oxidation at C(7) followed by acidic treatment induced removal of both acetonide moieties and concomitant spiroketalization to the thermodynamic AB spiroketal 344.
5 Conclusions
This variety in strategic approaches to key fragments of spongistatins illustrates the remarkable synthetic challenge associated to these appealing macrolide structures. Despite the successes already reached by efficient total syntheses of spongistatins 1 and 2, the access to multigram quantities of these molecules still remains an issue that has to be met in order to perform in vivo studies. The preparation of libraries of analogues [44,45] as well as optimization and/or exploration of novel routes that may save steps are undergoing in several research groups.