

1 Introduction
Cancer is one of the most severe worldwide health problems. In 2005, 7.6 million of the 58 million deaths in the world were attributed to cancer [1]. Since metastasis is often responsible for fatal outcomes among cancer patients, substantial progress in the ability to treat this disease will critically depend on the discovery of new anti-metastatic drugs and the development of more effective clinical strategies [2].
Recently, the discovery of migrastatin, a new anti-migratory agent, has stimulated the development of a diverse array of analogues with activity against tumor cell migration. This review will present the isolation and structure elucidation of migrastatin, as well as the syntheses and applications of migrastatin and its analogues.
2 Isolation and structure elucidation
In 2000, Imoto et al. reported the first isolation of migrastatin 1 from a cultured broth of the strain Streptomyces sp. MK929-43F1 which was collected in Japan [3]. The gross structure of migrastatin was proposed to be a 14-membered macrolactone incorporating three double bonds and featuring a pendant alkyl glutarimide side-chain. Later, in 2002, the absolute configuration of migrastatin was assigned by X-ray crystallographic analysis of the N-p-bromophenacyl derivative 2, and the absolute configuration was also confirmed by the modified Mosher's method [4]. The macrocyclic core of migrastatin features an interesting syn–syn stereotriad at C8–C10 with the (8S, 9S, 10R) configuration and incorporates a (Z)-trisubstituted double bond (Fig. 1).

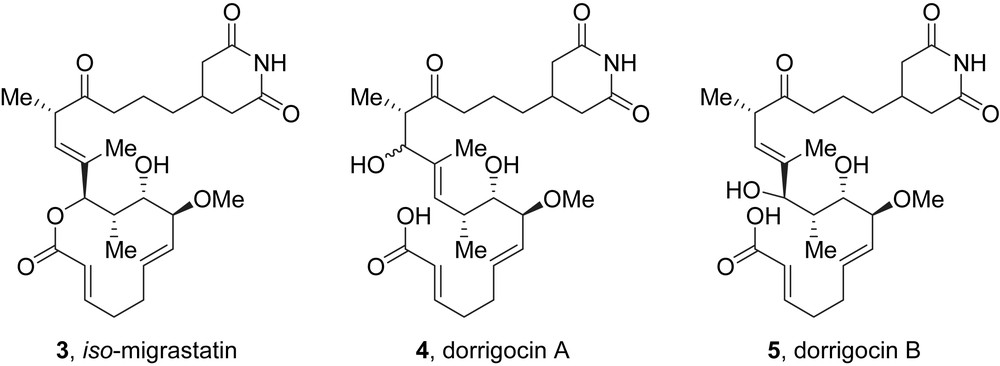
The same year, in 2002, Licari et al. isolated migrastatin as well as iso-migrastatin 3 from a culture of Streptomyces platensis NRRL 18993 [5]. This strain was already known for producing dorrigocins A 4 and B 5, which are linear isomers of migrastatin (Fig. 2). More recently, Shen et al. used gene-inactivation of the S. platensis polyketide synthase to demonstrate that migrastatin and dorrigocins were actually shunt metabolites of iso-migrastatin [6].
3 Biological activity of migrastatin and analogues
In earlier studies, Imoto et al. reported that migrastatin inhibited the migration of esophageal cancer EC17 cells and mouse melanoma B16 cells with IC50s of 1.0 μg/mL and 3.0 μg/mL, respectively, in wound healing assays [3]. Although other glutarimide containing natural products possess antibiotic activities [7], migrastatin has no effect on the biosyntheses of proteins, RNA, or DNA. However, Imoto et al. found that in their original isolation of migrastatin, teleocidin-related impurities were present. As teleocidin-related compounds are well known to inhibit tumor cell migration, the study of the inhibition of tumor cell migration by pure migrastatin was carried out, and it was shown that migrastatin actually inhibited the migration of EC17 cells with an IC50 of 10 μg/mL [8]. In this study, the authors also found that migrastatin displayed low cytotoxicity, and it was a weak anti-proliferative agent on a variety of tumor cells with IC50s around 100 μg/mL or higher. Furthermore, Danishefsky et al. showed that migrastatin had a modest IC50 of 29 μM on the migration of 4T1 mouse breast tumor cells in a chamber cell migration assay [9]. The metabolic stability of migrastatin in mouse plasma was verified, and no lactone opening was observed [10]. The migration of human umbilical vein endothelial cells was also moderately inhibited with an IC50 of 65 μM [10].
More recently, Imoto et al. reported that migrastatin was capable of suppressing multidrug resistance [11]. Indeed, migrastatin inhibits P-glycoprotein and consequently sensitizes drug resistant P-glycoprotein-overexpressing tumor cells to anticancer drugs like taxol, vinblastine, and vincristine. It was proposed that migrastatin interacts with P-glycoprotein at the azidopine- and vinblastine-binding site. In a recent study, Nakae et al. revealed that migrastatin acts as a muscarinic acetylcholine receptor antagonist [12]. However, the biological target and mode of action of migrastatin remain unknown.
Inspired by the anti-metastatic potential of migrastatin, a variety of simplified analogues, the syntheses of which will be presented later (vide infra), were evaluated for structure–activity relationship studies (Fig. 3). Danishefsky et al. have screened the biological activity of analogues 6–21 and they have reported that macrolactones 8 and 9 were 1000 times more active than migrastatin itself on the migration of 4T1 cells with IC50s of 22 nM and 24 nM, respectively (Table 1) [9,10]. Whereas 1 was reported to be stable in mouse plasma, lactones 8 and 9 showed rapid lactone opening. Hydroxy-acid 14, which is the hydrolyzed form of lactone 9, still possesses an IC50 of 378 nM [10]. When macroketone 11, which was stable in mouse plasma, was tested on 4T1 cell migration, an IC50 of 100 nM was obtained [10].
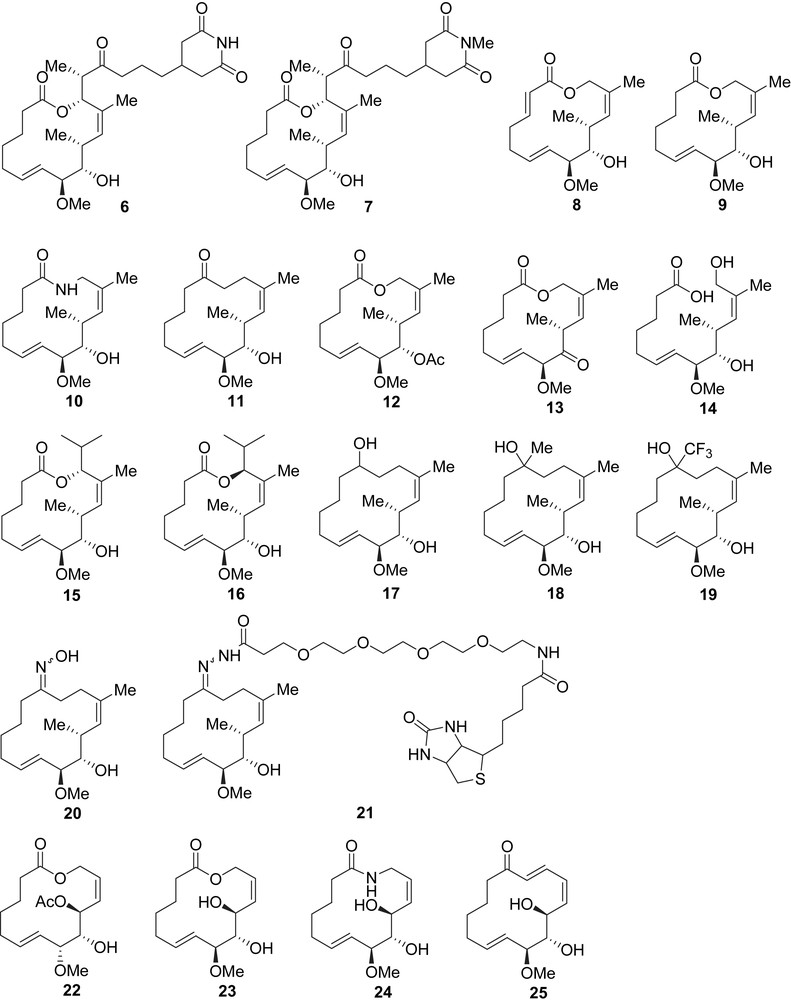
Chamber cell migration assay with 4T1 cells [10]
| Compound | IC50 |
| 1 | 29 μM |
| 6 | 10 μM |
| 7 | 7.0 μM |
| 8 | 22 nM |
| 9 | 24 nM |
| 10 | 255 nM |
| 11 | 100 nM |
| 12 | 192 nM |
| 13 | 223 nM |
| 14 | 378 nM |
| 15 | 227 μM |
| 16 | 146 μM |
| 17 | 8.9 μM |
| 18 | 3.1 μM |
| 19 | 101 nM |
| 20 | 2.3 μM |
| 21 | 331 nM |
When analogues 8, 9, and 11 were evaluated on human endothelial cell migration (HUVECs), lower inhibitory activities were observed, but analogues 8 and 9 still display higher activities than migrastatin 1 (Table 2) [10]. It was also shown that macrolactone 8 had good activity against the migration of human cancer cells such as HT 29, Ovcar3, and RL without interfering with cell proliferation [10].
Chamber cell migration assay with HUVECs [10]
| Compound | IC50 |
| 1 | 65 μM |
| 8 | 150 nM |
| 9 | 125 nM |
| 11 | 12 μM |
In another study, Danishefsky et al. demonstrated that macrolactam 10 and macroketone 11 were potent metastasis inhibitors in in vivo mouse models. These simplified migrastatin analogues appear to inhibit Rac activation, thus blocking the formation of lamellipodia and inactivating the metastatic process [13].
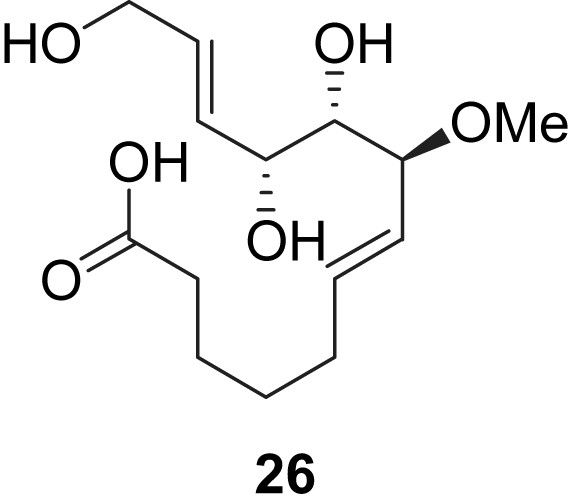
More recently, Murphy et al. reported their results in the evaluation of new migrastatin analogues 22–25 as potent anti-metastatic agents, but these macrocyclic compounds displayed lower activities than Danishefsky's analogues [14]. However, during their studies, Murphy et al. disclosed that the simplified dorrigocin analogue 26 was a potent inhibitor of the migration of gastric adenocarcinoma (AGS) cells at nanomolar concentrations (Fig. 4).
The inhibition of tumor cell migration is an important process that could contribute to the eradication of cancer. Further studies concerning the mode of action of migrastatin and its analogues will probably help the chemists to design improved migrastatin analogues for applications in cancer therapies.
4 Synthesis of migrastatin and analogues
4.1 Total synthesis of migrastatin
To date, two syntheses of migrastatin have been reported in the literature [10,15,16] as well as one semisynthesis achieved from iso-migrastatin [17].
4.1.1 Synthesis of Danishefsky et al. [10,15]
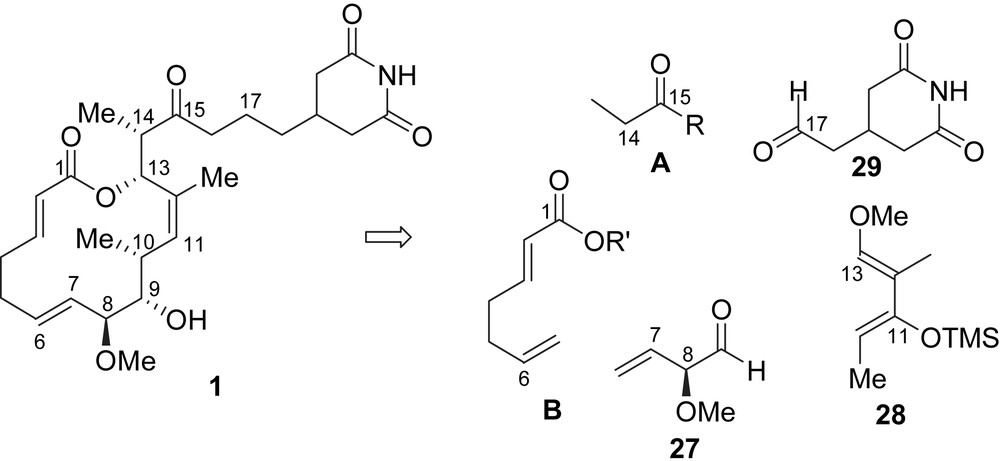
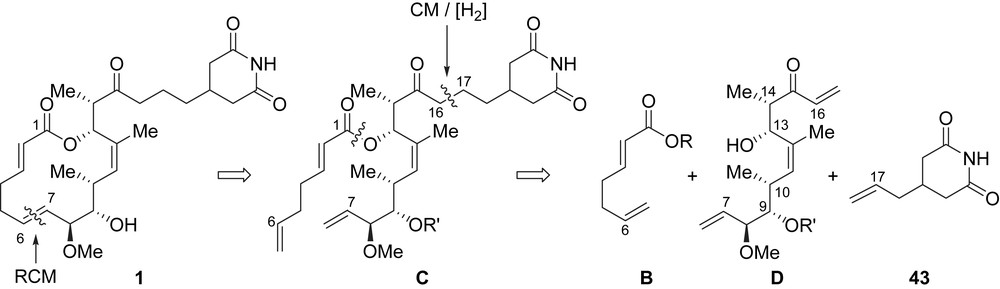
The strategy of Danishefsky et al. for the elaboration of migrastatin relied on the preparation and assembly of five subunits A, B, 27–29. The three stereogenic centers present at C8–C10 would be installed through a stereoselective Lewis Acid-Catalyzed Diene Aldehyde Condensation (LACDAC) between chiral aldehyde 27 and substituted butadiene 28. The obtained C7–C13 fragment would next be elongated by using a condensation with a chiral propionamide of type B, which would establish the C13 and C14 stereocenters. The glutarimide side-chain would be attached to the C7–C16 fragment by using a Horner–Wadsworth–Emmons (HWE) olefination with aldehyde 29 followed by the reduction of the generated enone. Finally, a fragment of type B would be linked at C13 to introduce the non-conjugated diene moiety, and a RCM (ring-closing metathesis) would establish the macrolactone core (Scheme 1).
The synthesis of migrastatin started with the preparation of allylic alcohol 34 which possesses three of the five stereogenic centers present in migrastatin as well as the (Z)-trisubstituted double bond (Scheme 2). In order to achieve the chelation-controlled LACDAC, the key aldehyde 27 was prepared from commercially available 2,3-O-isopropylidene-l-tartrate (30) in four steps. Indeed, diester 30 was reduced with DIBAL-H to the corresponding dialdehyde and subsequently treated with the in situ generated divinylzinc to afford a diol which was obtained with a good diastereoselectivity (>90%). After methylation of the obtained diol (NaH, MeI) and removal of the acetonide protecting group (HCl, MeOH), tetraol 31 was obtained in 60% yield from tartrate 30. The oxidative cleavage of diol 31 was achieved using Pb(OAc)4, leading to aldehyde 27 which was directly engaged in an α-chelation controlled LACDAC with diene 28. The formation of dihydropyrone 32 (87% yield from diol 31), as a single diastereoisomer incorporating the syn–syn stereotriad required for the synthesis of migrastatin, can be rationalized by an exo mode of approach of the diene as a result of the chelation with TiCl4 of both the carbonyl group and the α-methoxy group of the aldehyde [18]. Dihydropyrone 32 was then transformed to diol 33 in three steps. The carbonyl group was first reduced with LiBH4, the resulting alcohol was subjected to a Ferrier-rearrangement upon treatment with CSA, producing a lactol which was finally reduced with LiBH4 to yield diol 33.
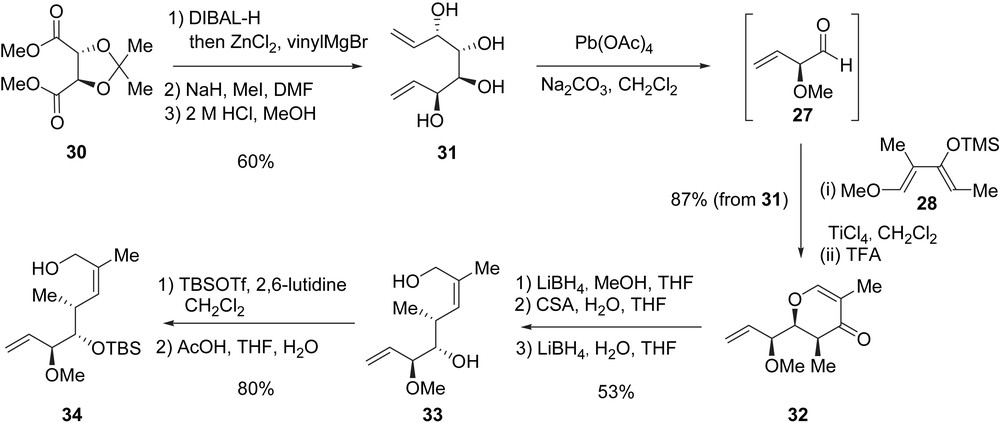
In their first approach to this fragment, Danishefsky et al. differentiated between the primary versus the secondary hydroxyl groups in diol 33 by using a three-step sequence involving an acetylation of the primary hydroxyl group, a silylation of the free secondary hydroxyl group, and finally, the removal of the acetate at the primary position. Unfortunately, this sequence suffered from low yielding upon scaling-up the synthesis. In contrast, a better procedure involved the protection of both hydroxyl groups with TBSOTf and selective cleavage of the primary silyl ether (AcOH, THF, H2O) yielding alcohol 34 (80% from 33).
The synthesis of fragment C7–C21, compound 40, is delineated in Scheme 3. After oxidation of 34 to the corresponding aldehyde, an Evans anti-aldol reaction (MgCl2, Et3N, TMSCl) [19] with oxazolidinone 35 allowed the isolation of imide 36 in 67% yield. The protection of alcohol 36 as a triethylsilyl (TES) ether followed by the removal of the chiral auxiliary led to the formation of primary alcohol 37 (83%) which was transformed to phosphonoacetate 38 after three additional steps. The HWE olefination between 38 and aldehyde 29 under Masamune–Roush conditions [20] installed the glutarimide side-chain of migrastatin, allowing the formation of enone 39 as a single olefin isomer in 57% yield over the last four steps. Fragment C7–C21 40 was finally obtained in 82% yield after the conjugate reduction of enone 39 with the Stryker reagent {[(Ph3P)CuH]6} and the selective cleavage of the TES silyl ether at C13.
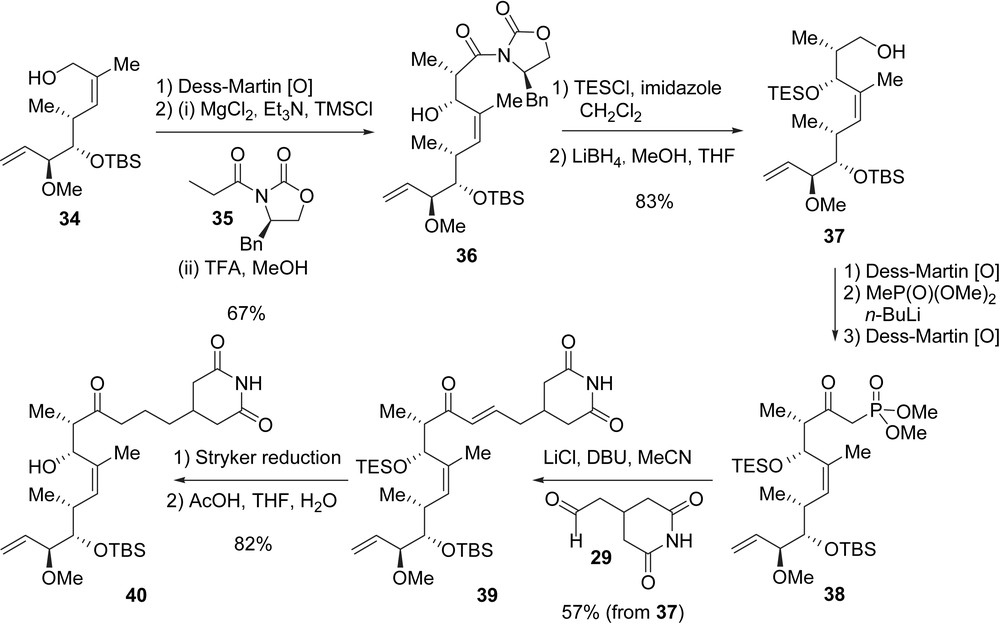
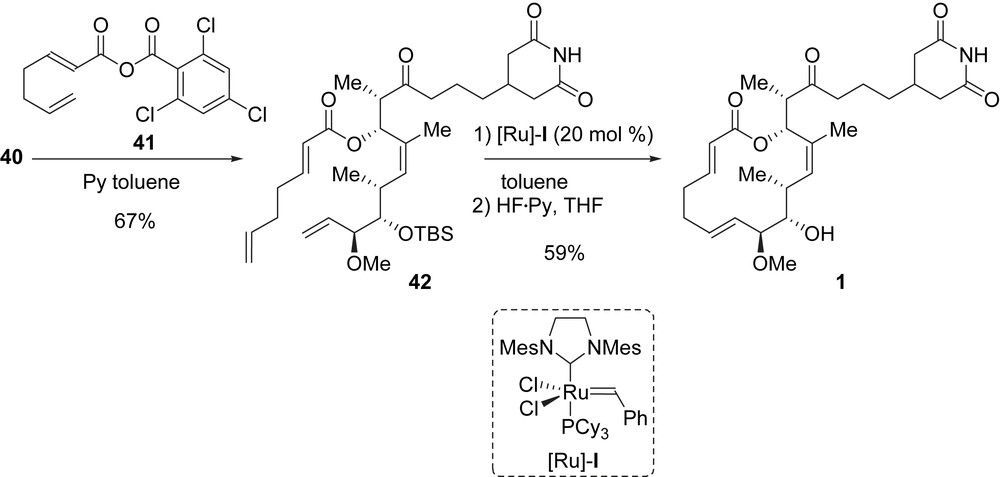
To complete the synthesis of migrastatin, secondary alcohol 40 was reacted with the previously prepared mixed anhydride 41 under Yamaguchi acylation conditions to give dienic ester 42 [21]. Danishefsky et al. envisioned other acylation conditions, but only the Yamaguchi protocol gave satisfactory results. Finally, the macrolactone core of migrastatin was established by applying a RCM to diene 42, and the final cleavage of the TBS ether at C9 furnished 1 (Scheme 4). Migrastatin was obtained after 23 steps in the longest linear sequence from tartrate 30 with an overall yield of 2.3%.
4.1.2 Synthesis of Cossy and Reymond [16]
In this approach to migrastatin, the macrolactonization was also envisioned by applying a RCM to a diene of type C, which would be in turn prepared by the Yamaguchi acylation of B and D (Scheme 5). The key step in this synthesis was based on the use of a cross-metathesis (CM) to install the alkyl glutarimide side-chain thus creating the C16–C17 bond. Such CM would eventually be useful for installing other side-chains thereby affording straightforward access to migrastatin analogues. The stereogenic centers at C9, C10 and C13, C14, present in fragment D, would be controlled by using two enantio- and diastereoselective crotylmetalations. The use of these reactions implies the synthesis of a fragment of type D and allylglutarimide 43 (Scheme 5).
The synthesis of fragment D started from the commercially available methyl ester 44, which was transformed to alcohol 45 in four steps (58%). After oxidation of 45 under Swern conditions, the obtained crude aldehyde was treated with but-2-enyl-[(tri-n-butyl)]stannane 46 in the presence of MgBr2·OEt2 to give the syn–syn stereotriad 47 with a good stereocontrol (dr = 9/1) and in 87% yield for the last two steps [22]. After protection of the secondary alcohol as a silyl ether and oxidative cleavage of the double bond, 47 was transformed to aldehyde 48 which was directly treated with Still–Gennari phosphonate 49 to produce unsaturated ester 50 in 74% yield with good (Z)-stereocontrol of the C11–C12 double bond (Z/E = 97/3) [23]. In order to introduce the last two stereogenic centers at C13 and C14, ester 50 was transformed into α,β-unsaturated aldehyde 51 in two steps (DIBAL-H then MnO2), and then directly treated with the highly face-selective crotyltitanium complex Ti-(S,S)-I leading to 52 in 75% yield (from 50) with a good diastereoselectivity (dr = 9/1) [24]. In order to install the glutarimide side-chain by CM, alcohol 52 was transformed in five steps to the vinyl ketone 53. At this stage, a chemoselective oxidative-cleavage of the terminal C15–C16 double bond was required while leaving untouched the internal C11–C12 double bond. To protect the internal double bond from osmylation, the hydroxyl group present at C13 in compound 52 was first protected with TESCl, and then the C15–C16 double bond was selectively transformed into an aldehyde (OsO4, NMO then NaIO4) which was easily transformed to vinyl ketone 53 after two additional steps (vinylMgBr then Dess–Martin oxidation). Enone 53 was obtained in 53% yield from alcohol 52 (Scheme 6).
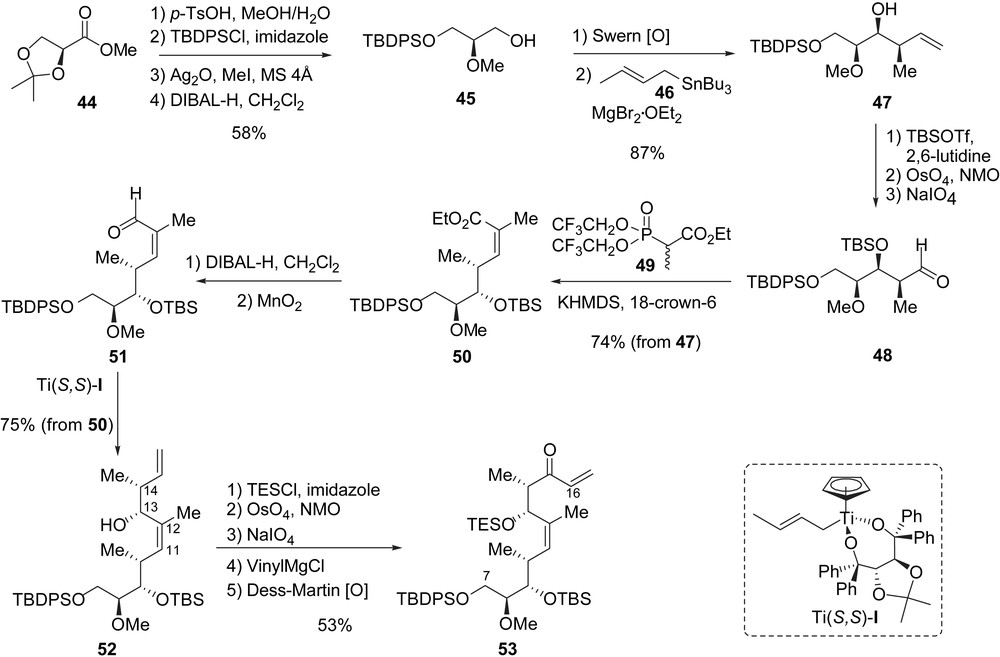
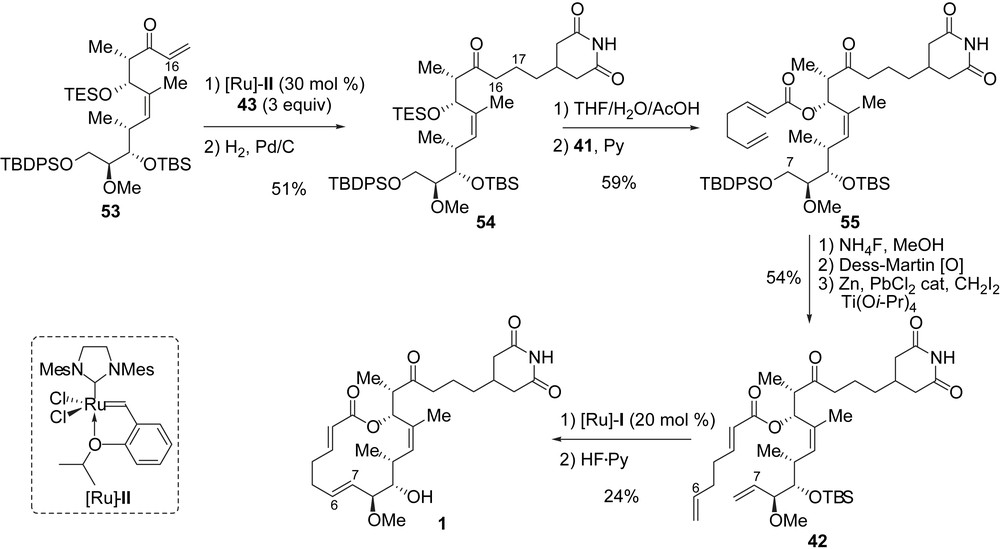
Worthy of note is the great importance of the triethylsilyl protecting group at C13 which shielded the internal C11–C12 double bond from osmylation (52 to 53) [25]. As well, this protecting group masked the C11–C12 double bond from the following CM and hydrogenation steps that had to be used to complete the synthesis of migrastatin. Enone 53 was first coupled with allylglutarimide 43 by using a chemoselective CM reaction catalyzed by the Hoveyda–Grubbs catalyst [Ru]-II (53% corrected yield, 60% conversion), and the resulting cross-product was chemoselectively hydrogenated (96%) at C16–C17 to produce 54. With 54 in hand, the endgame was clear as a diene of type C had to be prepared. First, the TES ether was selectively cleaved to afford a secondary alcohol, which was acylated using the mixed anhydride 41 under the conditions described previously by Danishefsky et al. to yield ester 55 in 59% yield for the last two steps. The latter was transformed to diene 42 in 44% yield after three steps that included a selective cleavage of the primary silyl ether at C7 with NH4F in methanol, a Dess–Martin oxidation of the obtained alcohol to an aldehyde, and a Takai methylenation [26]. As described previously, the total synthesis of migrastatin was completed with the RCM of diene 42 and removal of the TBS protective group (24% yield) (Scheme 7). Migrastatin 1 was obtained in 26 steps (longest linear sequence) from the commercially available ester 44 with an overall yield of 0.5%.
4.1.3 Semisynthesis of Shen et al. [17]
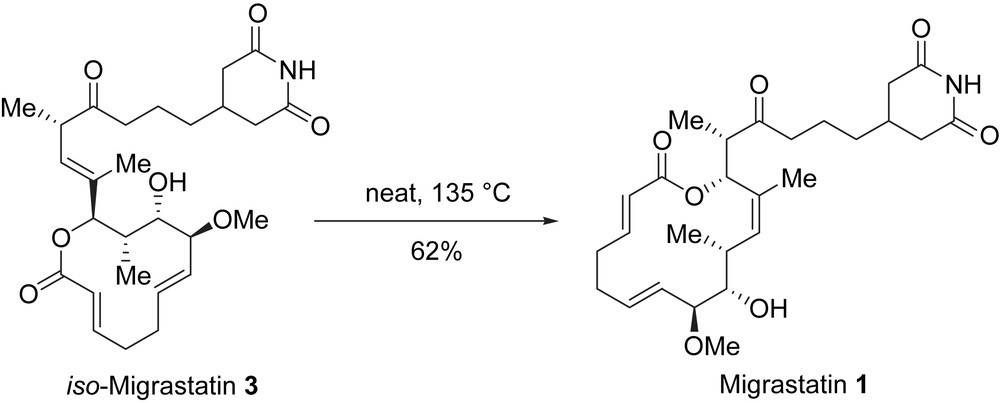
Shen et al. demonstrated that migrastatin and the structurally related dorrigocins were actually shunt metabolites of iso-migrastatin 3 [6], which is the primary metabolite biosynthesized by S. platensis. Iso-migrastatin 3 is not stable in aqueous solutions and undergoes isomerization to a mixture of 1, 4 and 5. More recently, Shen et al. showed that the thermal rearrangement of iso-migrastatin 3 was able to produce the natural product migrastatin 1 regio- and enantiospecifically in 62% yield (Scheme 8) [17]. They suggested that this rearrangement was based on a [3,3]-sigmatropic process. A series of iso-migrastatin analogues were also transformed to rearranged migrastatin analogues.
4.2 Synthesis of migrastatin analogues
4.2.1 Synthesis of iso-migrastatin 3 [27]
Recently, Danishesfy et al. reported the first total synthesis of iso-migrastatin 3. They anticipated the unstability of 3 postulating that it was due to the presence of two (E)-disubstituted double bonds in the 12-membered ring macrolactone. They decided to introduce the (E)-double bond at C2–C3 in the last stages of the synthesis.
Starting from dihydropyranone 32, which is also a precursor of migrastatin, epoxide 56 was prepared in three steps (35% yield). Then, the reductive opening of 56 furnished a diol, for which the differentiation of the primary versus the secondary hydroxyl group was achieved by using a three-step acetylation/protection/methanolysis sequence that gave rise to primary alcohol 57 in 69% yield (four steps). Alcohol 57 was treated under Swern conditions, and the obtained aldehyde was reacted with Wittig reagent 58 to produce enone 59 in 83% yield (from 57). In order to introduce the stereogenic center present at C14, an SN2′ opening of the epoxide was envisaged under Marshall conditions [28]. The stereoselective reduction of the enone carbonyl group was achieved by using a Corey reduction [(S)-Me-CBS, BH3·Me2S; dr = 10/1], and the obtained alcohol was protected with TBSCl. The SN2′ opening of the epoxide with lithium cyanomethylcuprate provided 60 in 55% yield (from 59). During this step, the methyl group at C14 was installed with the expected configuration (anti relative to the silyl ether at C15), and the (E)-trisubstituted double bond at C11–C12 was established (Scheme 9).
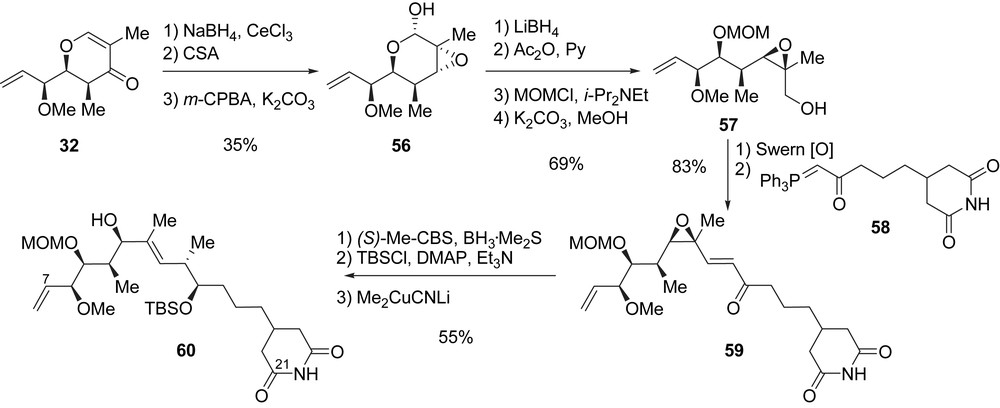
With synthon 60 (C7–C21 fragment) in hand, the completion of the synthesis required the construction of the macrolactone core of iso-migrastatin. Acylation of 60 at C11 with the racemic seleno-acid 61 afforded diene 62 with the α-selenoester moiety as the masked C2–C3 double bond. After cleavage of the silyl ether at C15 with HF·Py complex, oxidation to the corresponding ketone, and MOM deprotection, diene 63 was obtained in 68% yield (three steps). When diene 63 was treated with catalyst [Ru]-I, the RCM afforded a mixture of two macrolactone isomers with a (Z)- and (E)-double bond at C6–C7 in a 1.6/1 ratio in favour of the (Z)-isomer. After separation by chromatography, the (E)-isomer 64 was obtained in 21% yield. The final oxidative deselenylation provided 3 in 93% yield (Scheme 10). Iso-migrastatin 3 was obtained after 23 steps from tartrate 30 with a global yield of 0.7%.
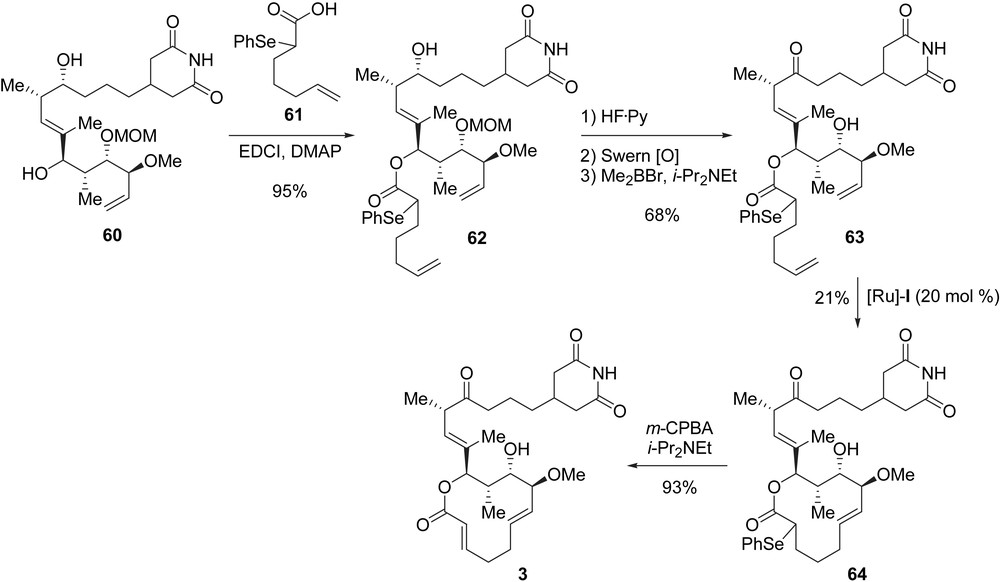
4.2.2 Synthesis of the macrolactone core 8
The macrolide core 8 was the first migrastatin analogue reported. It possesses the highest activity against the migration of tumor cells but suffers from its plasma instability that results in lactone cleavage.
The first synthesis of this analogue was reported by Danishefsky and Gaul [29], and an optimized synthesis was later reported by this group [10]. The synthesis starts with a ten-step transformation of tartrate 30 into allylic alcohol 34 (Scheme 2). After Yamaguchi acylation of 34 with the mixed anhydride 41, diene 65 was obtained in 48% yield. The RCM of diene 65 was achieved by treatment with [Ru]-I, and the final deprotection of the silyl ether with HF·Py complex yielded the macrolactone core of migrastatin 8 (36% yield for the last two steps) (Scheme 11). Analogue 8 was obtained in 13 steps from 30 in 4% global yield.
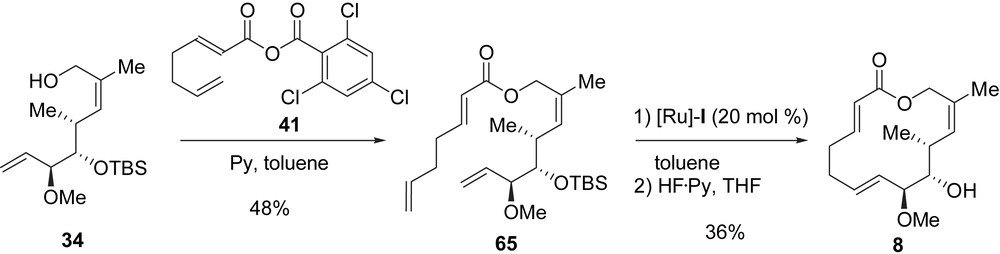
During their total synthesis of migrastatin, Cossy and Reymond reported the synthesis of macrolide 8 [16b]. To begin, ester 44 was transformed into the α,β-unsaturated ester 50 in ten steps with a global yield of 37%. After reduction of 50 with DIBAL-H (94% yield), the corresponding allylic alcohol was obtained and acylated with 41 providing ester 66 in 67% yield. The latter was converted into diene 65 in three steps: a selective cleavage of the primary TBDPS ether, a Dess–Martin oxidation of the resulting alcohol, and a Takai methylenation of the aldehyde in 49% yield (three steps). In a manner similar to Danishefsky et al., 65 was transformed into 8 after two steps and in 31% yield. Macrolactone 8 was obtained in 17 steps from 44 in 4% global yield (Scheme 12).
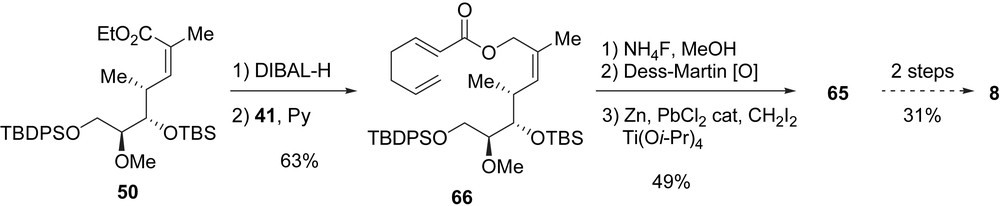
Das et al. also reported a synthesis of macrolactone 8 [30]. Their synthesis started from acrolein 67 which was reacted with Evans oxazolidinone ent-35, and after cleavage of the chiral auxiliary with LiBH4, allylic alcohol 68 was obtained in 81% yield (two steps) and excellent stereoselectivity. The reaction of diol 68 with p-methoxybenzaldehyde dimethyl acetal (69) afforded an acetonide which was selectively opened using DIBAL-H, and the obtained primary alcohol was protected as a TBS ether to yield 70 (58% for the three steps). The oxidative cleavage of the double bond in 70 provided an aldehyde which was treated with vinylmagnesium bromide in the presence of MgBr2·OEt2 to yield the syn–syn stereotriad 71 in a 7/1 ratio of stereoisomers. After the transformation of alcohol 71 into the corresponding methyl ether, the removal of the TBS group afforded primary alcohol 72 in 58% yield (two steps). This alcohol was first oxidized under Dess–Martin conditions to an aldehyde, which was treated with Ando phosphonate 73 to establish the (Z)-trisubstituted double bond (one isomer formed) [31], and finally, the reduction of the ethyl ester gave rise to allylic alcohol 74 (49% yield, three steps). To complete the synthesis of 8, alcohol 74 was acylated with 41, the RCM of the diene was achieved with [Ru]-I and the MPM ether was cleaved using DDQ (23%, three steps) (Scheme 13). In summary, macrolide 8 was prepared form acrolein in 15 steps and 2% global yield.
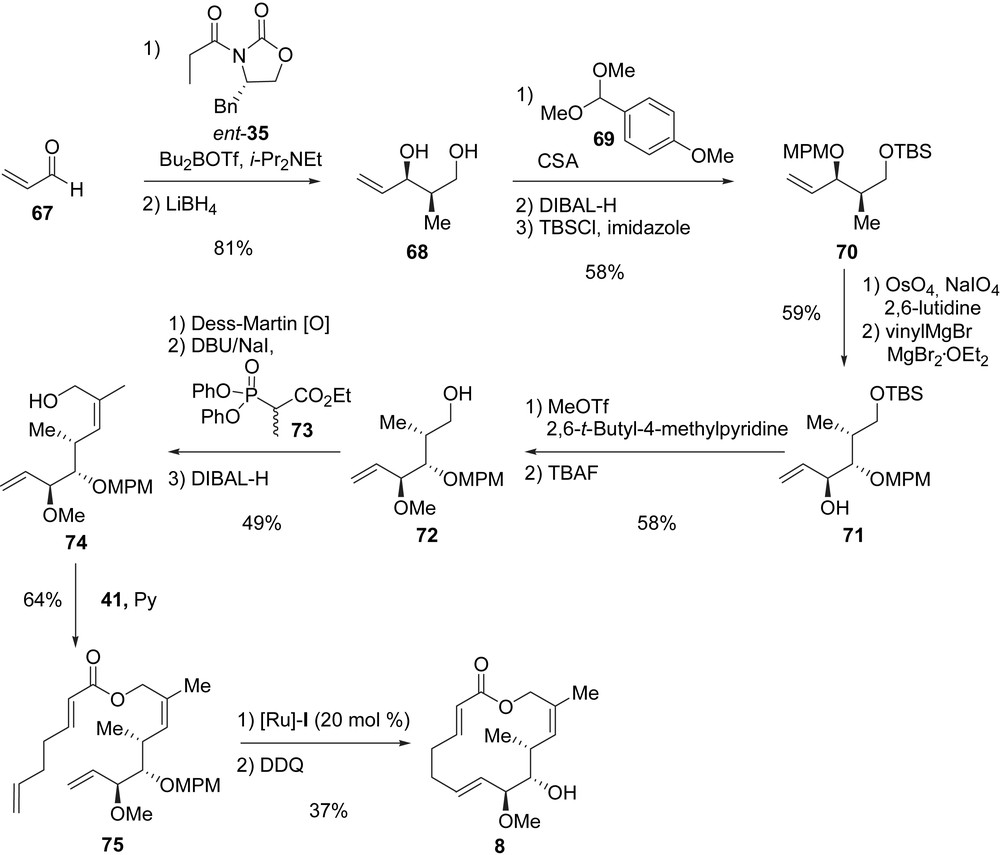
4.2.3 Synthesis of analogues 9–21[10]
Starting from allylic alcohol 34, Danishefsky et al. designed the synthesis of analogues 9–21. Upon treatment of 34 with 6-heptenoyl chloride 76 in the presence of DMAP, diene 77 was obtained in 82% yield. The RCM of diene 77 was then achieved by using [Ru]-I (20 mol%), and the removal of the TBS group produced macrolactone 9 in good yield (71%, two steps). From 9, three new analogues were prepared (Scheme 14). Acetate 12 was obtained after treatment of 9 with acetyl chloride in the presence of DMAP (76% yield), and then the Dess–Martin oxidation of 9 gave rise to ketone 13 (72%). Hydroxy-acid 14, the opened form of lactone 9, was obtained in 77% yield when 9 was subjected to NaOH in methanol.
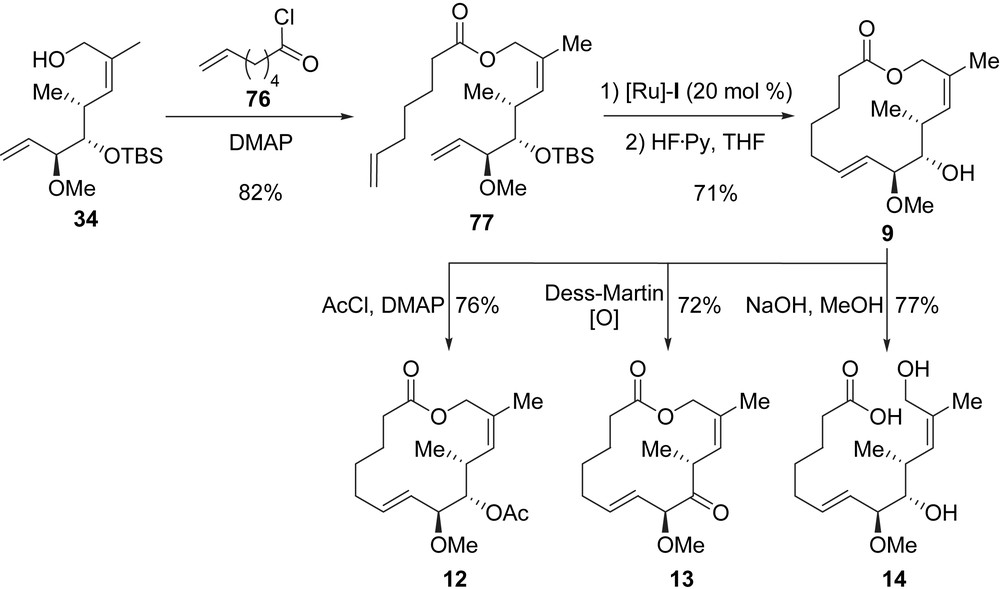
Two other macrolactones, 15 and 16, both featuring an additional stereogenic center, were prepared from 34 in five steps (Scheme 15). The Dess–Martin oxidation of 34 followed by the treatment of the resulting aldehyde with iso-propylmagnesium chloride furnished two separable diastereomeric alcohols 82 and 83 in a 2/3 ratio (86% yield for the mixture). The Yamaguchi acylation of 82 with 84 led to the formation of a diene which was cyclized by a RCM promoted by [Ru]-I, and the final cleavage of the TBS ether afforded macrolactone 15 in 46% yield (three steps). By applying the same three-step sequence to alcohol 83, macrolactone 16 was obtained in 49% yield.
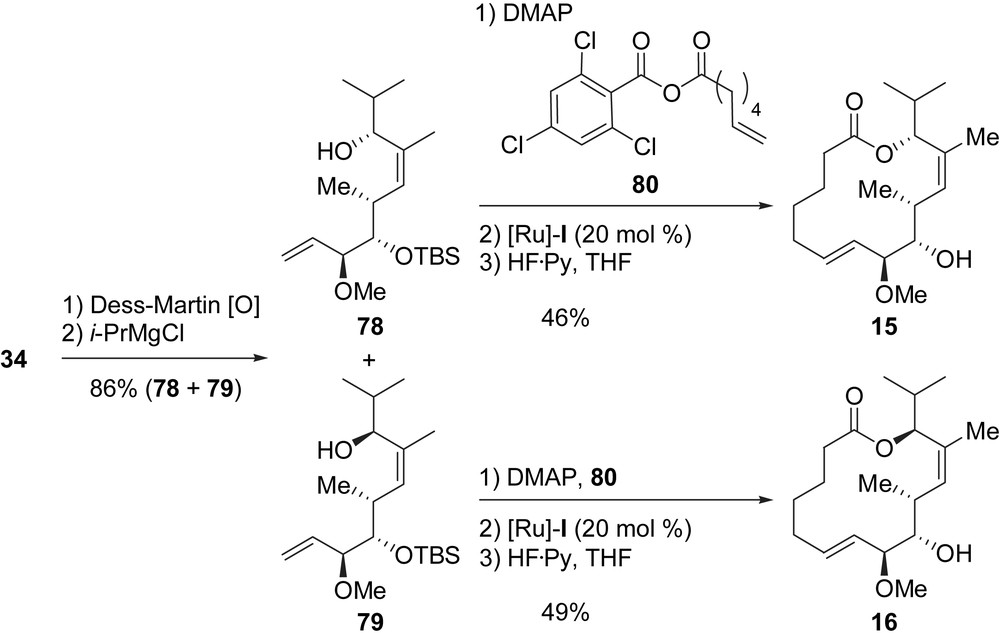
A macrolactam, 10, was synthesized in five steps from 34 (Scheme 16). Azide 81 was first prepared by using the DBU/diphenylphosphoryl azide (DPPA) method in 87% yield [32]. Azide 81 was then subjected to a Staudinger reduction to afford an amine which was directly coupled with 6-heptenoic acid (82) by using EDC to yield dienic amide 83 (92%, two steps). Finally, the RCM/TBS deprotection endgame furnished lactam 10 in 54% yield.
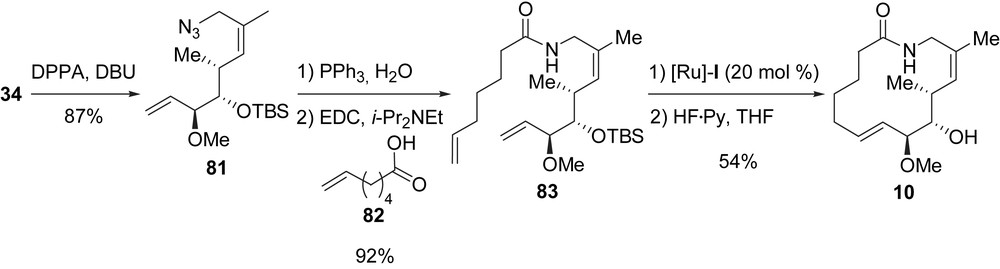
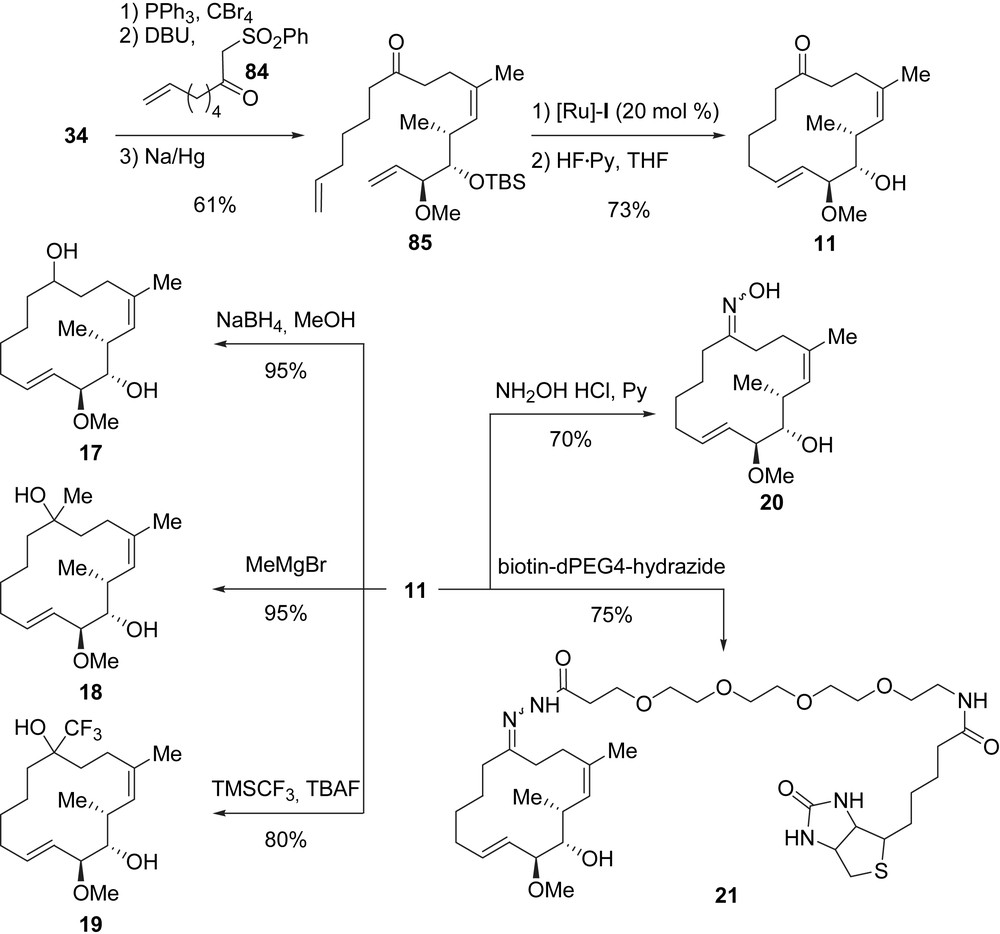
A synthesis for the preparation of macroketone analogue 11 was also designed. The synthesis of 11 started with the transformation of allylic alcohol 34 into the corresponding allylic bromide, which was displaced by treatment with β-ketosulfoxide 84 in the presence of DBU. After Na/Hg desulfonylation, dienic ketone 85 was obtained in 61% yield (three steps). The RCM/TBS deprotection endgame afforded macroketone 11 in 73% yield (Scheme 17). Starting from this ketone, five additional analogues 17–21 were prepared. Alcohol 17 (diastereomeric mixture, 95% yield) was obtained after NaBH4 reduction of the carbonyl group in 11. The addition of methylmagnesium bromide to 11 yielded tertiary alcohol 18 (95%). Reaction of 11 with TMSCF3 in the presence of TBAF allowed the formation of 19 bearing a trifluoromethyl group. Finally, 11 was converted to oxime 20 in 70% yield, and the biotin-labeled acyl-hydrazone 21 was obtained in 75% yield after treatment of ketone 11 with biotin-dPEG4-hydrazide.
4.2.4 Synthesis of analogues 22–25 [14]
Recently, Murphy et al. reported their efforts in the preparation of migrastatin and dorrigocin A analogues. Compared to the analogues described previously, these analogues have no methyl group on the double bond at C12, and at C10, a hydroxyl group is present instead of a methyl group. These analogues were prepared from tri-O-acetyl-d-glucal (86) [14a].
After a methanolysis of the acetate groups in 86, the three hydroxyl groups were protected as TBS ethers by using TBSCl. Then, the selective cleavage of the primary TBS ether with HF·Py complex yielded primary alcohol 87 (68%, three steps). The oxidation of the latter, under Swern conditions, furnished an aldehyde which was treated with vinylmagnesium bromide to afford a 1/1 mixture of separable diastereoisomers 88 and 89 (78% yield for the mixture). Both diastereoisomers were used for the preparation of migrastatin analogues. Starting from alcohol 88, the methylation of the free hydroxyl group was performed with NaH and MeI, and the two TBS ethers were exchanged with acetates after TBAF deprotection and acylation with acetic anhydride to provide 90 in 52% yield. Water-mediated allylic rearrangement of 90 then furnished a lactol which was reduced with LiBH4 to afford allylic alcohol 91 in 68% yield (two steps). The same transformation was applied to 89 thus leading to the diastereomeric allylic alcohol 92 in 34% yield (Scheme 18).
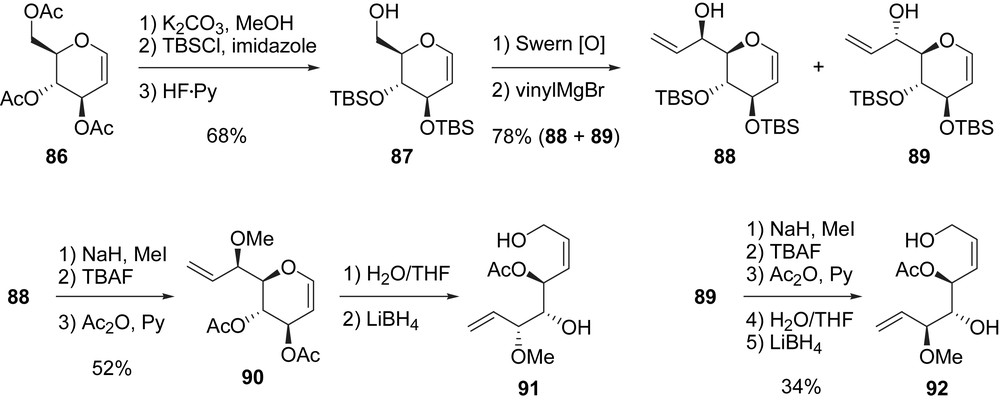
With the key building-blocks 91 and 92 in hand, Murphy et al. prepared four analogues, compounds 22–25. After selective acylation of 91 with 76 followed by RCM of the obtained diene, macrolactone 22 was obtained in 27% yield. It is noteworthy that the same sequence (acylation then RCM), when applied to 92, did not lead to the expected macrolactone but gave homodimerization and unidentified products. Thus, 92 was transformed to 93 after removal of the acetate, protection of the obtained triol with TBSOTf and selective cleavage of the primary TBS ether (59%, three steps). A Mitsunobu reaction of 93 with 6-heptenoic acid (82) allowed the formation of a diene which was treated with [Ru]-I, thus affording macrolactone 23 in 55% yield after final deprotection of the TBS groups (Scheme 19) [14a].
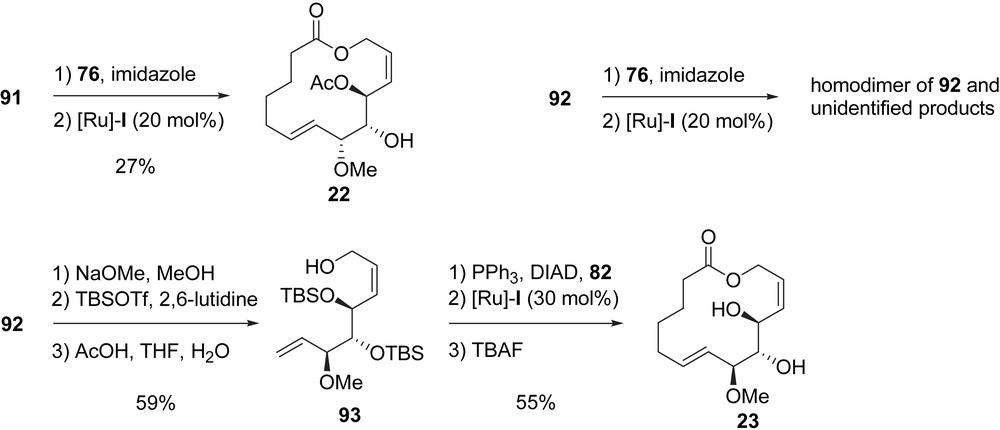
In order to synthesize macrolactam 24, allylic alcohol 93 was transformed into an allylic azide 94 by using Mitsunobu conditions with DPPA. After a Staudinger reduction of the azide and EDC coupling of the obtained amine with 82, amide 95 was obtained in 60% yield. The RCM of 95 with [Ru]-I followed by the cleavage of the TBS ethers afforded macrolactam 24 in 35% yield (Scheme 20) [14b].
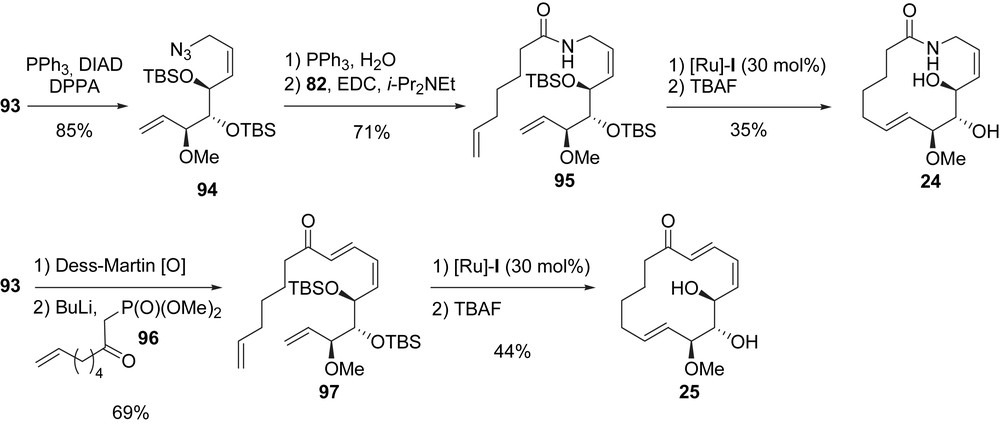
Finally, the macrocyclic ketone 25 was prepared from allylic alcohol 93. After Dess–Martin oxidation of 93, the resulting aldehyde was treated with HWE phosphonate 96 to afford enone 97 (69%, two steps). This compound was then treated with [Ru]-I and the obtained macrocyclic ketone was deprotected to provide migrastatin analogue 25 in 44% yield (Scheme 20) [14b].
5 Conclusion
Since the discovery of migrastatin and its unusual anti-metastatic activity, synthetic organic chemists have designed a diverse array of migrastatin analogues. Some of these analogues appeared to have excellent in vitro activities, such as the migrastatin macrolactone core 8. However, as this macrolactone suffers from plasma instability, other analogues such as macrolactam 10 or macroketone 11 were synthesized and have shown to display nanomolar activities and plasma stability. These promising analogues will probably stimulate the design and synthesis of new migrastatin analogues for biological testing and better comprehension of the biological mode of action of this family of compounds.