

1 Introduction
In 1984, Schmitz isolated a novel compound, tedanolide (1) from the Caribbean sponge Tedania ignis [1]. The crystalline nature of the compound enabled determination of the structure by X-ray crystallography. Five other related natural products forming the tedanolide family have been isolated from various organisms, namely tedanolide (1), 13-deoxytedanolide (2), tedanolide C (3) and candidaspongiolides (4, 5) (Fig. 1).

The family of tedanolides.
The tedanolides' complex architecture, in addition to their high cytotoxicity, attracted considerable interest within the synthetic community. Initial biological evaluation showed an arrest of P338 tumor cell growth in S-phase [1]. Seven years after, Fusetani et al. reported the isolation of a deoxygenated analogue, 13-deoxytedanolide (2), from the sponge Mycale adhaerens [2]. The amount isolated was sufficient for only limited studies on the biological profile in the course of which a unique binding to the 60S ribosome subunit [3b,4] was demonstrated. As a result of this binding, peptide elongation in eukaryotic cells was efficiently inhibited. While several compounds exhibiting specificity for prokaryotes were identified, it is noteworthy that 13-deoxytedanolide (2) is the first known macrolide to be selective for eukaryotes [3]. This selectivity for eukaryotes and archaea was rationalized by Moore et al. who successfully co-crystallized 13-deoxytedanolide (2) with the 50S ribosome subunit of Haloarcula marismortui [4]. The binding location was identified as the E site of the ribosome. More recently, Ireland isolated tedanolide C (3) from the marine sponge Ircinia sp. [5] in 2005 and McKee et al. isolated the group of candidaspongiolides (4, 5) from the sponge of the genus Candidaspongia in 2007 [6]. Degradation studies were also performed on 13-deoxytedanolide by Fusetani to probe the structure–activity relationships [3]. The first functionality targeted was the epoxide. Unfortunately, attempts to remove it only resulted in a skeleton rearrangement and the formation of a furan (Scheme 1), and thus, left open the question of its significance for the biological activity.
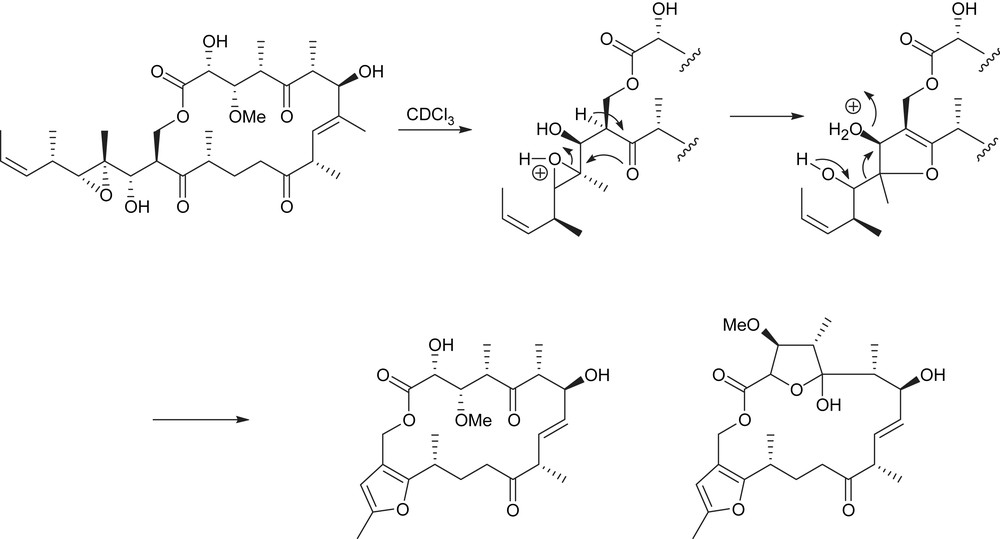
Fragmentation of 13-deoxytedanolide under acidic conditions.
One striking feature of this family resides in their structure, raising some questions on their biosynthesis. Indeed, to the best of our knowledge, a macrolactone with a primary linkage was unprecedented. Thus, the hypothesis of a different lactone as the direct PKS product was envisioned. On the synthesis perspective, tedanolide is prone to retro-aldol degradations due to the numerous β-hydroxyketone moieties. In addition, the epoxide containing side chain is acid sensitive. Since the initial isolation report, tremendous efforts were devoted to the synthesis of tedanolide. These efforts culminated with the first total synthesis of 13-deoxytedanolide (2) by Smith et al. [7] in 2004, followed three years later by the synthesis of tedanolide (1). Their strategy was designed to give access to both compounds in a unified approach. In 2005, a second synthesis of 13-deoxytedanolide was reported by the Roush group [8]. The first total synthesis of tedanolide was achieved by the Kalesse group [9a] in 2006. Fragment syntheses that lead to valuable insight into the subtle synthetic problems of this class of compounds will be covered as well.
2 Results and discussion
2.1 The common approach to the tedanolides by Smith
As already stated, Smith developed a unified strategy to synthetize both tedanolide (1) and 13-deoxytedanolide (2) from a common late stage intermediate. Retrosynthetic disconnections at the macrolactone linkage and at the C11–C12 bond gave rise to the northern and the southern fragments. While the northern fragment is identical for both compounds, simple variation of the southern fragment was required. Coupling by opening an epoxide or by displacement of an iodide would generate tedanolide (1) or 13-deoxytedanolide (2), respectively (Fig. 2). The use of a dithiane anion reversed the reactivity of the aldehyde for the coupling to proceed and protected the carbonyl from possible hemiketal formation. This hemiketal was indeed detrimental as it prevented Roush from completing the synthesis of tedanolide. Two different routes were reported by Smith for the southern fragment. The first generation [6] disclosed for the synthesis of 13-deoxytedanolide (2) was linear and closely similar to the one developed by Roush for his synthesis [8]. The second generation [7c] developed for tedanolide (1) was more convergent and more efficient (Scheme 2).

Common strategy for the synthesis of tedanolide and 13-deoxytedanolide.

First-generation approach of the synthesis of the southern fragment of 13-deoxytedanolide.
One of the main challenges encountered during the syntheses was the protecting group strategy. The change from DEIPS to SEM for C17 allylic alcohol between the two routes is noteworthy. The SEM was easily deprotected selectively in presence of a TIPS and TBS and was also more stable than the DEIPS.
2.2 Synthesis of the first-generation southern fragment
Starting from alcohol 6 standard transformations generated acetate 8. Next the terminal double bond was selectively oxidized and subjected to a Roush crotylation which proved to be more appropriate for this transformation than the Brown protocol. Indeed, the boronate was more labile and thus provided higher yields in the hydrolytic work up. After protecting group manipulations and oxidation, the Z-alkene could be introduced via Wittig olefination. Finally, saponification and iodination provided the southern segment.
2.3 Synthesis of the second-generation southern fragment
The second-generation synthesis disconnected the southern fragment at the C17–C18 bond. The coupling proceeded accordingly to Yonemitsu's study of this transformation. Starting from the protected Roche aldehyde, an alkyne was generated via the Corey–Fuchs protocol. Regioselective E-vinyl iodide formation was achieved by stannyl cupration to provide 13. Several conditions were investigated in order to obtain only the desired isomer (Schemes 3 and 4).

Second-generation approach of the synthesis of the southern fragment of tedanolide.

Synthesis of the northern fragment of tedanolide.
The C12–C17 aldehyde synthesis featured a Brown crotylation and an Evans aldol condensation. The subsequent coupling directed by the inherent properties of the aldehyde yielded 14 in a 6:1 diastereoselectivity. The southern fragment 17 was then completed by a Z-selective Wittig olefination and a Fraser-Reid epoxide formation.
2.4 Synthesis of the northern fragment
In a similar fashion to the southern fragment, the northern fragment 24 [7] was assembled by the addition of a vinyl iodide to an aldehyde. The vinyl iodide 21 was generated as previously from an alkyne and a hydrostannylation. Iterative Evans aldol reactions provided the C1–C7 aldehyde intermediate 22.
2.5 Coupling of the northern and southern segments for the tedanolides
The endgame strategy for 13-deoxytedanolide (Scheme 5) and tedanolide (Scheme 6) are closely related and only one will be described herein. The dianion of the northern fragment 24 (dithiane and free primary alcohol) was used for the key coupling which proved to be high yielding (80% BORSM). As previously stated, the dithiane was seen as a protecting group of the carbonyl to prevent hemiketalization. The downside of this strategy was the dithiane sensitivity to common oxidation methods. This issue was addressed by the development of Tishchenko oxidation. This specific oxidation promoted by samariumII relied on an internaloxidation–reduction in the presence of a β-hydroxyketone (26). The resulting ester 27 was hydrolyzed and subsequent macrolactonization of the triol (C15, C17 and C29) selectively closed on the less hindered primary alcohol to provide 28. Diastereoselective epoxidation of 30 directed by the C17 allylic alcohol (Henbest effect) and global deprotection finalized the synthesis. The longer linear sequence was of 31 steps with an overall yield of 0.31% (Scheme 7).
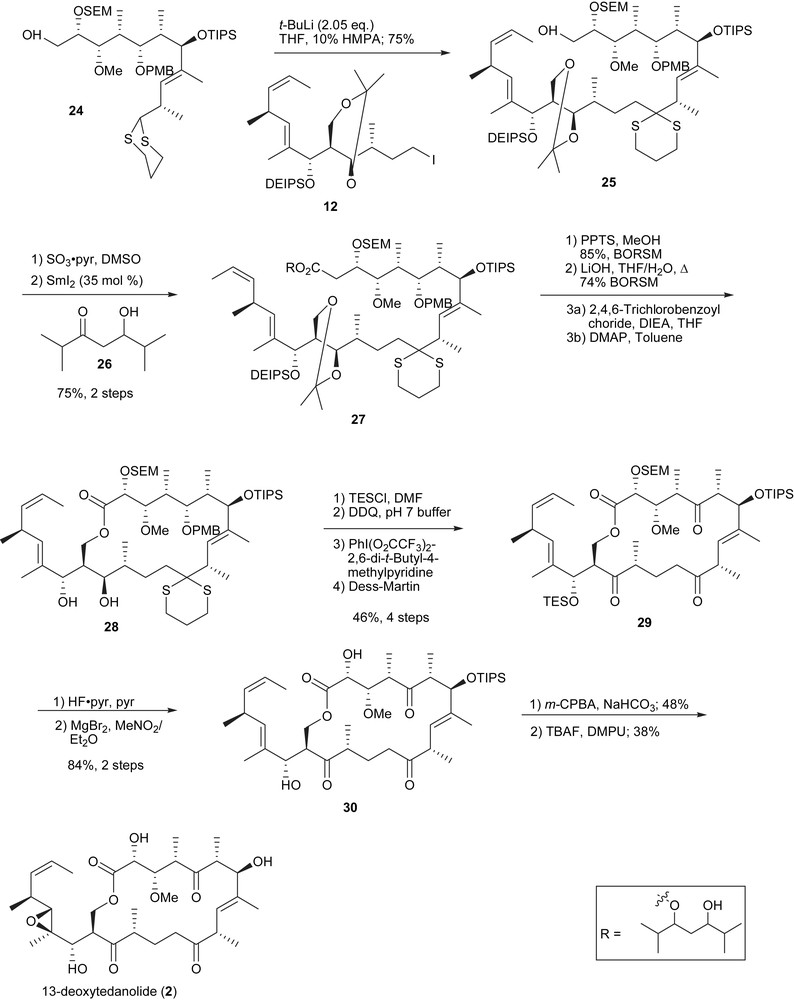
Endgame of the Smith's 13-deoxytedanolide synthesis.
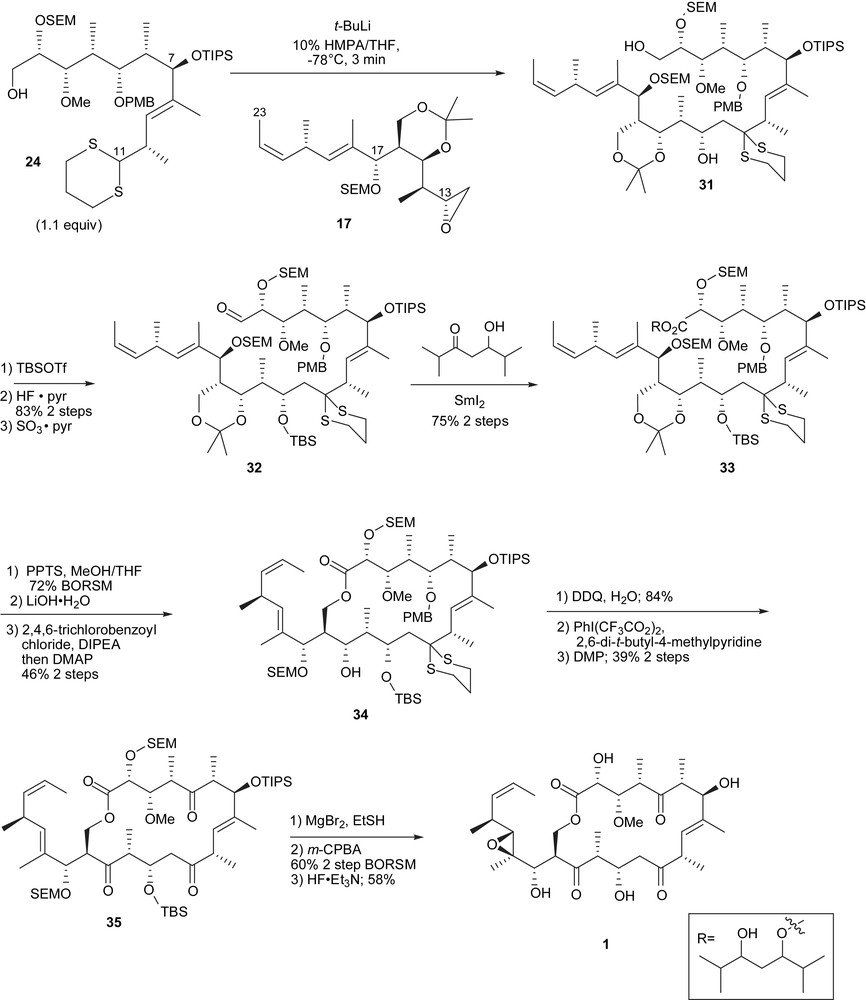
Endgame of Smith's tedanolide synthesis.

Synthesis of the C1–C12 segment of tedanolide.
2.6 The Roush synthesis of 13-deoxytedanolide
Roush envisioned a disconnection between C12 and C13 (Fig. 3). This approach should allow access to both tedanolide and 13-deoxytedanolide from the same precursor [8]. The configuration at C15 was chosen to be anti with respect to the configuration at C14 in order to maximize the Felkin enhancing effect of both stereocenters. Additionally, they decided to carry the functionality at C5 as the ketone through the synthesis. Having recognized that mild deprotection conditions were needed for the ester moiety, they employed an allyl group and the Alloc protecting group for the C29 primary alcohol. Thus, both could be removed simultaneously (Fig. 4).
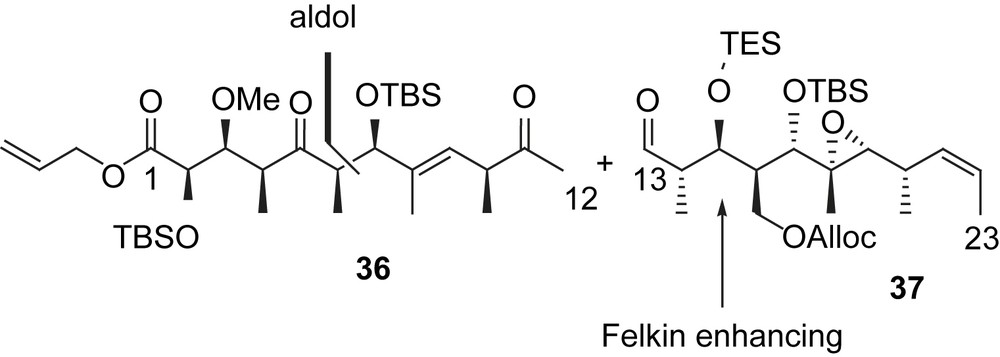
Retrosynthetic disconnection of tedanolide in the Roush synthesis.

Retrosynthesis of tedanolide.
2.7 Synthesis of the C1–C12 segment
The Roush synthesis of the C1–C12 segment began with a Noyori reduction of keto ester 38 and subsequent methylation. A sequence of reduction, acetylation and regioselective reductive acetal opening provided after oxidation compound 42. Allyl stannylation, methylation and oxidative cleavage of the double bond provided aldehyde 44 which was transformed to allyl ester 45 through Pinnack oxidation, Mitsunobu esterification and oxidation of C11 to the ketone. The key aldol coupling proceeded in high yield to provide the northern fragment 36.
2.8 Synthesis of the C13–C23 segment
One of the key issues in this segment synthesis was the appropriate introduction of the protecting groups. In particular the introduction of the Alloc group was achieved at a relative late stage since it would interfere with oxidative transformation employed in this segment synthesis. Therefore, the BEC group was exploited as a transient protecting group for the primary alcohol.
Starting from the commercially available Roche ester (Scheme 8), Wittig olefination followed by Dibal-H reduction generated the allylic alcohol 6 required for the stereoselective introduction of the epoxide. An Evans aldol reaction using (S)-56 established olefin 51 which could be transformed to the double acetate protected intermediate 53. Introduction of the C12–C22 double bond was performed with standard conditions. Finally, the pivotal introduction of the Alloc group was achieved by treatment of 54 with allyl alcohol and MeMgBr. This simultaneously liberated diol was then transformed to the corresponding aldehyde through Pd(OAc)4 cleavage.

Synthesis of the C13–C23 segment.
2.9 Endgame of the synthesis
The aldol coupling was effectively achieved using LiHMDS in THF (Scheme 9). Upon deprotection of the C15 hydroxyl group, intermediate 59 spontaneously cyclized to form an undesired and stable hemiketal. The deoxygenation of C13 allowed the equilibrium with the acyclic ketone to exist, and thus, to be further processed. However, this prevented the synthesis of tedanolide (1). Oxidation of the open form drove the equilibrating mixture to the triketone 60. Simultaneous allyl ester and Alloc deprotection to provide the seco acid which was followed by a modified Yamaguchi macrolactonization yielded 61 in 31% over two steps. Finally, global sillyl deprotection provided (+)-13-deoxytedanolide (2).
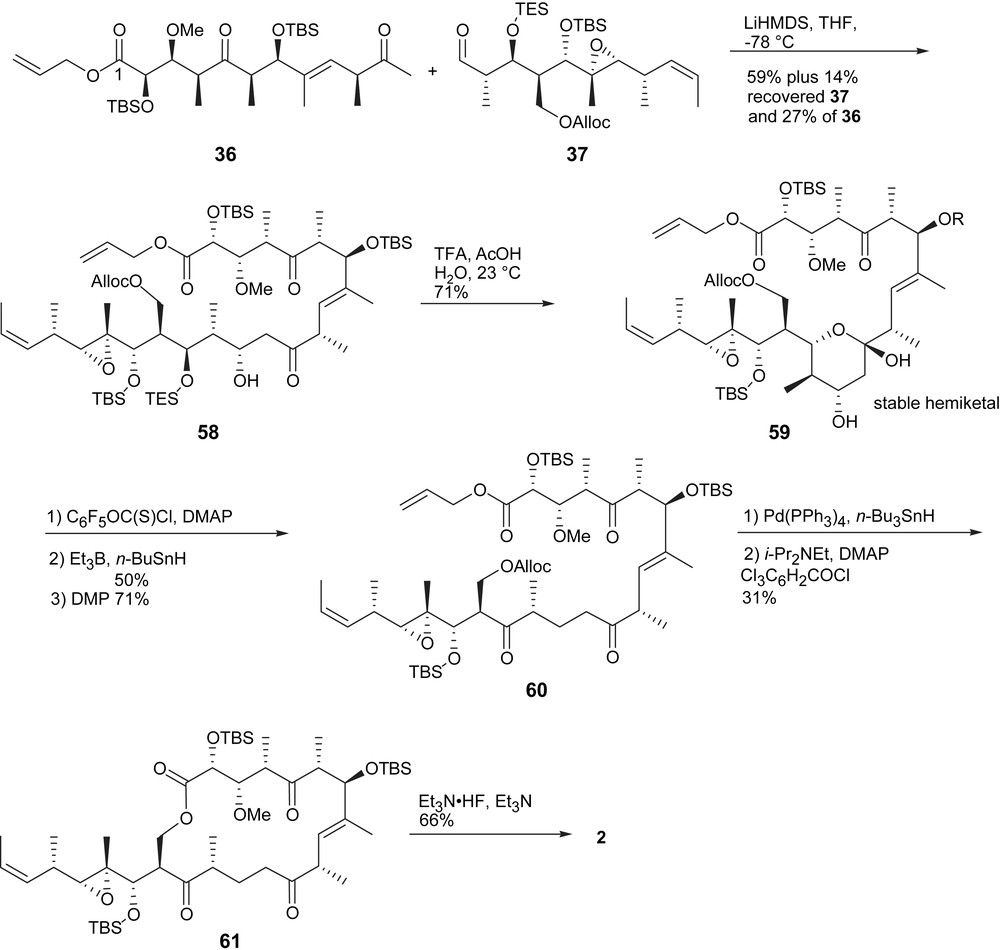
Endgame of Roush's synthesis of 13-deoxytedanolide.
2.10 The Kalesse synthesis of tedanolide
As a consequence of the labile epoxide moiety in tedanolide (1) the Kalesse group decided to introduce this functional group at the last step of the synthesis.
As in all other syntheses of the tedanolides an essential issue was the appropriate choice of orthogonal protecting groups for the C29 alcohol and the carboxylate. After substantial variations, the monomethoxytrityl group (MMTr) was identified to be an ideal choice for C29 hydroxyl since its installation and chemical stability were compatible with the operations employed. Additionally, the very mild conditions for its removal, namely treatment with hexafluoro isopropanol allowed removal even in the presence of unprotected β-hydroxyketones.
2.11 Synthesis of aldehyde 65
The originally published route had its drawbacks due to a relatively greater number of steps and occasionally uncontrolled migration of protecting groups. These disadvantages led to an improved synthesis of the C13–C23 aldehyde 65, using a more convergent aldol coupling between C16 and C17 that paralleled the fragment synthesis proposed by Loh. The synthesis commenced with trityl protected Roche aldehyde 66 and subsequent olefination to yield alkene 67. The acid catalyzed cleavage of trityl ether and subsequent Swern oxidation provided an aldehyde which was directly subjected to Wittig olefination providing the unsaturated ester 68. Dibal-H reduction and MnO2 oxidation provided aldehyde 69 (Scheme 10).
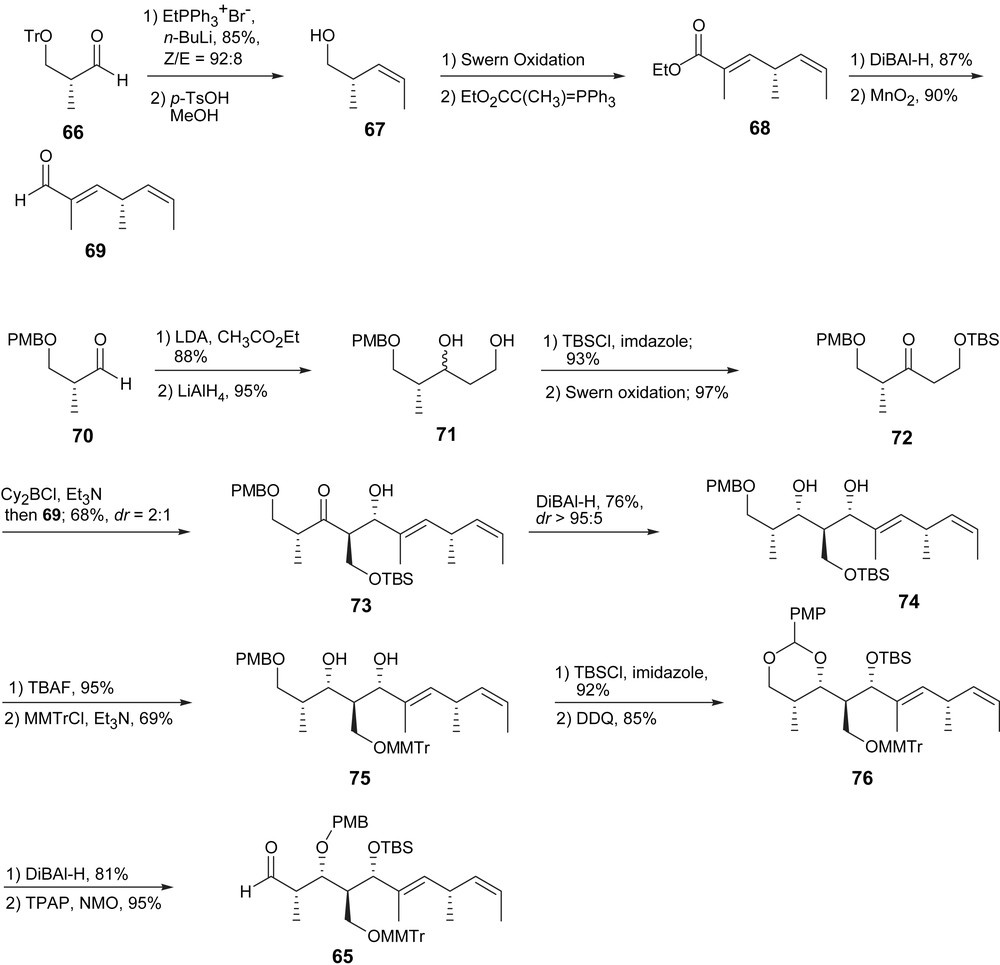
Synthesis of fragment 65.
Ketone 72 was synthesized in four steps according to the route described by Loh. An anti-selective aldol addition between 72 and 69 set the desired anti-relation between the C17 hydroxy and the C16 hydroxymethyl group. The diastereomeric ratio, however, was only of 2:1 in favor of the desired isomer 73. Dibal-H reduction provided exclusively syn-diol 74 (dr > 95:5). The TBS group was removed with TBAF. The three alcohol moieties were selectively protected with an MMTr group on the primary alcohol, a TBS group on the allylic alcohol and the closure of the PMP acetal on the third. Reductive opening of this acetal with Dibal-H followed by oxidation with TPAP/NMO provided the southern fragment 65 (Scheme 10).
Aldol reaction. Screening of different bases for the enolization of 64 was needed for the pivotal aldol coupling with aldehyde 65. KHMDS was identified to provide the highest selectivities for obtaining the Felkin product 77 (Scheme 11). To liberate the seco acid the monomethoxytrityl group was removed with hexafluoro isopropanol and the allyl group by palladium. The Yamaguchi protocol which was previously employed in both syntheses of 13-deoxytedanolide (2) provided 10–35% of the undesired 14-membered macrolactone 79 [9e].

Aldol coupling and lactonization.
To avoid the undesired lactonization the secondary alcohol at C13 was protected with TBS triflate. The moderate yield of 55% in the conversion of 77–80 could be attributed to considerable retro aldol of 77. After cleavage of the allyl ester, the free carboxylic acid was subjected to different lactonization methods. The Mitsunobu protocol led to the desired macrolactone 81 in good yields (Scheme 12).

Cyclization of hydroxy acid 80.
The oxidation state manipulation required deprotection of the PMB and TES groups followed by oxidation. Global silyl deprotection with 3HF·Et3N provided 82 after five days (Scheme 12). The final epoxidation could be achieved with substoichiometric amounts of m-CPBA at −45 °C, taking full advantage of the directing Henbest effect.
2.12 The Yonemitsu approach
Early contributions by Yonemitsu were based on computational analyses of the seco acid 84 and unraveled the cyclic acetals to induce the desired conformation for the lactonization. The aldol reaction between 86 and 87 was an additional example of how subtle differences can dominate the outcome of synthetic strategies. Indeed, the aldol reaction using LiHMDS provided a 1:1.2–1:1.9 mixture of diastereoisomers in favor of the undesired isomer. Fortunately, the isomers could be separated by chromatography and macrolactonization was achieved by using the Yamaguchi protocol in 87% yield (Fig. 5).
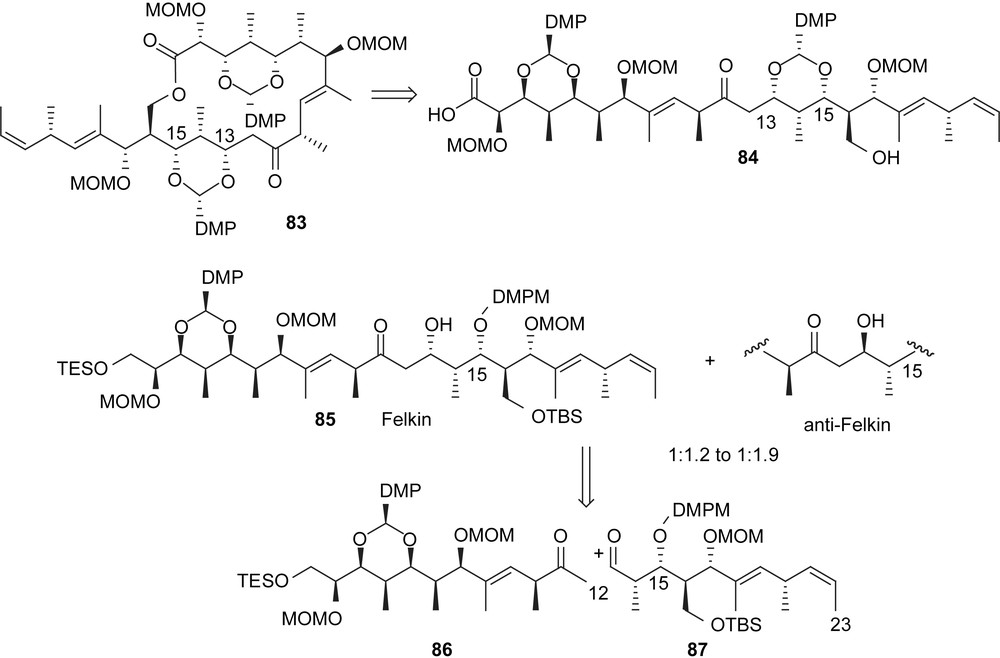
Lactonization by Yonemitsu.
Different disconnections can be envisioned to generate 86. Initially, 86 was generated from the coupling of known vinyl iodide 89 and aldehyde 88 (Fig. 6). The coupling, however, suffered from low selectivity and irreproducibility. The synthesis of the northern fragment was then reviewed, this time highlighted by a tin triflate based syn-aldol pioneered by Paterson between 90 and 91.

Retrosynthetic disconnection of tedanolide according to Yonemitsu.
The pivotal Paterson aldol provided the adduct in 80% yield with a diastereoselectivity greater than 20:1 (Scheme 13). After protecting group manipulations and oxidation of 93, a Wittig homologation provided the carbon skeleton of the C1–C11 fragment. Subsequent dihydroxylation followed by DDQ treatment provided the first acetal unit. Finally, the methyl ketone was introduced through methyl lithium addition and oxidation.
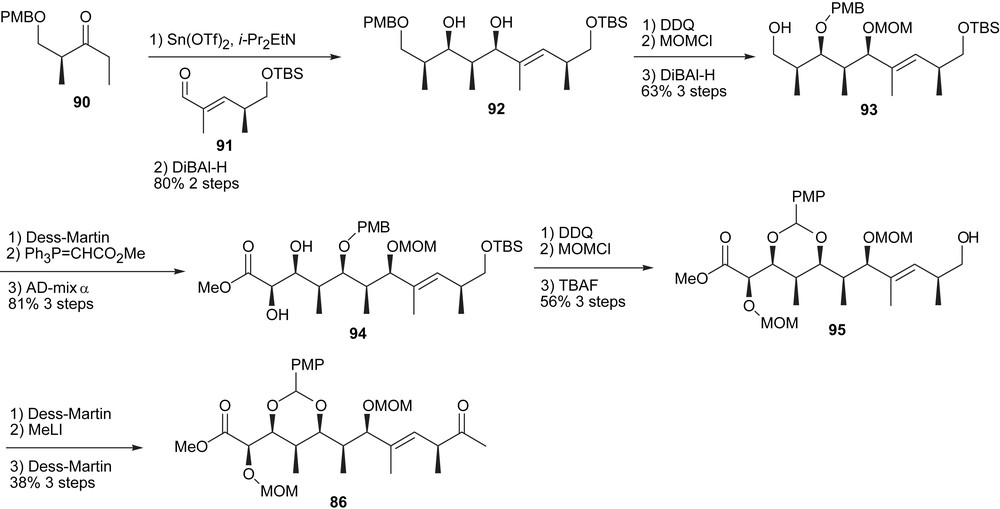
Synthesis of the C1–C12 segment.
The C13–C23 fragment was further disconnected at the C17–C18 bond with the addition of vinyl lithium 97 to aldehyde 96 (Scheme 14). The synthesis of 96 began with a TiCl4-mediated addition of oxazolidinone (R)-56 to the Roche ester-derived aldehyde 98. Reduction and acetalization provided 100. Reductive opening of the acetal followed by TBSCl protection and oxidative cleavage of the double bond provided aldehyde 96.

Synthesis of the C13–C23 segment.
In contrast to the previously discussed syntheses Jung, Miyashita and Loh mainly focused on providing segment syntheses and new approaches toward tedanolide (1). The groups of Jung and Miyashita synthesized the northern and southern fragments, respectively. Loh et al. reported the synthesis of the northern and the southern hemispheres as well as the fragment coupling via an aldol reaction to provide the corresponding seco acid 126.
Additionally, the approaches of Jung and Miyashita also aimed on developing an efficient synthetic route flexible enough to provide access to 13-deoxytedanolide (2) and tedanolide (1).
2.13 The Miyashita approach
Miyashita et al. published the stereoselective synthesis of the C13–C23 segment in 2005 [11a]. Their retrosynthetic analysis (Fig. 7) paralleled the one presented by Roush and Lane [8], Hassfeld and Kalesse [9] and Yonemitsu et al. [10]. The first retrosynthetic disconnection was also made at the macrolactone linkage. Further disconnection divided the seco acid into the C13–C23 fragment 102, already containing the labile epoxide functionality, and the C1–C12 segment 103. Both segments were envisioned to be assembled by an aldol under Felkin–Anh control [11a].

Retrosynthetic analysis of tedanolide by Miyashita et al.
Miyashita et al. designed a synthetic route for the C13–C23 segment 102 starting from the chiral unsaturated ester 105 in 23 linear steps. The main drawback was the linearity of the synthesis. However, it is interesting to note that the stereoselective synthesis was achieved without the use of any aldol reaction. Instead, the strategy is highlighted by two stereoselective epoxidation reactions of trisubstituted olefins (epoxidation/epoxide opening) and a stereospecific SN2′ methylation reaction of a trans-γ,δ-epoxy-cis-α,β-unsaturated ester 104 (Scheme 15).

Synthesis of the C13–C23 fragment of tedanolide by Miyashita et al.
The C13–C21 portion was constructed from the chiral unsaturated ester 105 via Sharpless asymmetric epoxidation followed by epoxide opening using vinylmagnesium chloride and copper(I)bromide dimethyl sulphide complex. The E-configured double bond of the C18–C19 moiety was installed via Wittig reaction. To introduce an α-secondary methyl group at the C20 position stereoselectively, the use of the stereospecific SN2′ methylation reaction developed by Miyashita et al. was applied [11b]. For this purpose, the requisite ester 104 was synthesized via the Horner–Wadsworth–Emmons reaction with the Ando reagent (Scheme 15). This reaction not only installed the methyl group at the C20 but also opened the epoxide at the same time to obtain the C17 hydroxyl stereoselectively.
To obtain the southern fragment 102, the C13–C21 polypropionate chain was extended via a Z-selective Wittig reaction. After several protecting group manipulations and stereoselective epoxidation of the C18–C19 alkene with m-CPBA, a protection–oxidation sequence yielded 102.
2.14 The Loh approach
Loh and Feng [12] reported on the synthesis of the northern hemisphere 114 and the southern hemisphere 113 of tedanolide (1) as well as the fragment coupling via aldol reaction to complete the carbon backbone. However, the successful total synthesis of tedanolide (1) has not been reported yet.
The retrosynthetic analysis of tedanolide (1) is outlined in Fig. 8. Disconnection at the C29 lactone and the C12, C13 aldol units created the two subunits 113 C13–C23 and 114 C1–C12.

Retrosynthesis of tedanolide by Loh et al.
The key step of the southern fragment 113 [12] synthesis displayed a boron-mediated aldol reaction developed by Paterson and Tillyer [13] between the α,β-unsaturated aldehyde 115 and ketone 72 in very good yield and selectivity (95% yield, major:minor = 85:15). It is interesting to note that Kalesse et al. followed the same strategy using the same ketone 72 to assemble the carbon backbone of the C13–C23 fragment. However, in the case of the Kalesse synthesis, another aldehyde was used and lower yields and stereoselectivity were obtained (68%, major:minor = 2:1).
Ketone 72 could be easily synthesized from the commercially available Roche ester via aldol reaction with methyl acetate and several oxidation and protecting group manipulations. Aldehyde 115 was synthesized from the readily available (S)-methyl-3-hydroxy-2-methylpropionate as reported by Paterson and Tillyer [13] (Scheme 16).

Boron-mediated aldol reaction.
Further elaboration of fragment C13–C23 was effectuated with a Wittig reaction to install the C21–C22 Z-olefin. After protecting group transformations, the C18–C19 epoxide was generated with m-CPBA and 121 was obtained in 54% as a single diastereomer (Scheme 17). It is the first example of a late stage epoxidation of these substrates. Indeed, it was initially expected to give a mixture of diastereomers or even the undesired isomer [19]. Gratifyingly, the desired product was formed exclusively.

Epoxidation.
The C1–C12 subunit [12] was generated from the intermediate 122 which could be obtained from the coupling of the two precursors 117 and 118 (Scheme 18). The key feature also presents a syn–syn boron-mediated aldol reaction developed by Paterson and Tillyer [13].

Boron-mediated aldol reaction.
Ketone 117 was synthesized from the Roche ester via Wittig reaction and Sharpless asymmetric dihydroxylation to incorporate the alcohol functionalities at C2 and C3. The t-butyl ester was selected due to its stability toward nucleophilic attack (Scheme 19).

Fragments coupling by Loh et al.
The coupling of the northern and southern fragments was achieved via an aldol reaction. Optimization of this pivotal transformation identified (c-hex)2BCl as a Lewis acid to provide the highest yields and diastereoselectivities.
2.15 The Jung approach
Jung et al. proposed in their approach of tedanolides 1 and 2 the preparation of the C1–C12 fragment [14] via a stereoselective non-aldol aldol process, which was developed in their laboratories. The reaction involves the internal hydride migration from an alcohol or silyl protected alcohol to open an epoxide with inversion of configuration at the epoxide center. As the product is already protected during the course of this transformation, further modifications are not required in contrast to standard aldol reactions.
The retrosynthetic disconnection of tedanolide's backbone began with the cleavage at the lactone and scission at the C12–C13 bond to generate the common precursors 126 C1–C12 fragment and 125 C13–C23 fragment (Fig. 9). These intermediates should have been coupled either by an aldol reaction of the aldehyde 125 (for tedanolide (1)) or an alkylation of the tosylate derived from 125 (for 13-deoxytedanolide (2)).

Retrosynthesis of the tedanolides by Jung et al.
The northern fragment 126 [14] was generated from the aldol coupling of the two intermediates 127 and 128 with TiCl4 and Hunig's base (Scheme 20). It is worth mentioning that only the desired Felkin product was obtained in good yield. Other coupling reagents were unsuccessful.

Aldol coupling of 127 and 128 with TiCl4 and Hunig's base.
After protection of the free hydroxyl group and reduction of the ketone by l-Selectride compound 132 was obtained containing all the required chiral centers of the northern fragment of the tedanolides 1 and 2 (Scheme 21).
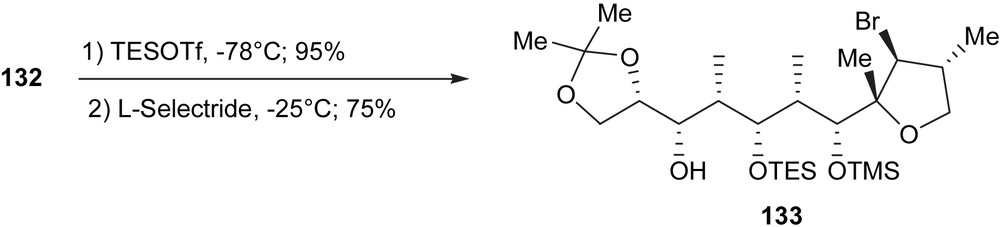
Protection of the hydroxyl group and reduction with l-Selectride.
The precursor of 127, namely ester 129 was prepared from commercially available l-ascorbic acid in three steps via diol protection with 2,2 dimethoxy-propane, cleavage of the alkene and esterification with ethyl iodide [14]. The desired ketone 127 was obtained through reduction, epoxidation and regioselective epoxide opening with methyl magnesium bromide and copper iodide (Scheme 22).

Synthesis of ketone 127.
(Scheme 23) The synthesis of aldehyde 128, already containing the required stereogenic centers from C5 to C11, started from the commercially available Roche ester 131 and was completed in 13 steps. E-selective Wittig reaction was applied to install the C18–C19 olefin. NBS-promoted cyclization of the free homoallylic alcohol to the double bond generated the bromotetrahydrofuran 136 (which demonstrated the utility of this protecting group as a masked homoallylic ketone). The protection of the alkene functionality was necessary to permit the application of the non-aldol aldol process. Thereby problems associated with allylic epoxide rearrangements were avoided. A second E-selective Wittig olefination, reduction to the allylic alcohol and Sharpless asymmetric epoxidation led to intermediate 130. The crucial non-aldol aldol rearrangement of the epoxide provided the aldol product 137 as a single diastereomer.
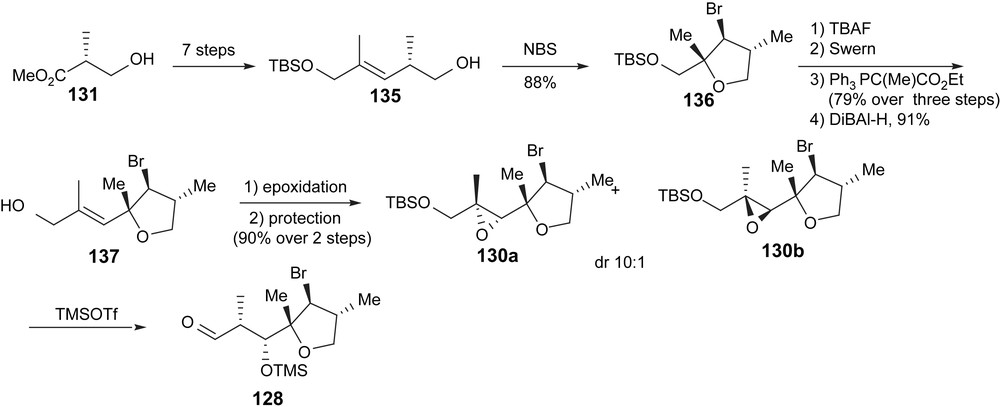
Synthesis of aldehyde 128.
2.16 The Iqbal approach
A different synthetic plan was presented by Iqbal et al. [15] who reported a general approach for the synthesis of the C3–C16 segment of tedanolide (1) and 13-deoxytedanolide (2). The central idea was to introduce a C12–C13 double bond of 138 through metathesis and to transform it depending on the natural product to be synthesized, either to saturated hydrocarbon 139 for 13-deoxytedanolide (2) or the corresponding epoxide 140 in the case of tedanolide (Fig. 10).

The Iqbal approach.
The synthesis started with the reductive transformation of 141 and subsequent acetalization. Reductive opening, oxidation and Wittig olefination provided the corresponding extended ester 144. A second reduction/oxidation sequence finally provided the corresponding aldehyde which was subjected to an Evans aldol reaction (Scheme 24).

Synthesis of 145.
Liberation of the aldehyde 146 and a second Evans aldol reaction provided stereoselectively the intermediate 147 (Scheme 25). A sequence of PMP-acetalization/Dibal-H opening provided the precursor 149 for the pivotal cross metathesis reaction. During the course of their investigations it appeared that the free C5 allylic alcohol was necessary for the reaction. The final transformations for the different intermediates could be achieved through either Dess–Martin oxidation and NiCl2-catalyzed reduction (139) or epoxidation of the allylic alcohol (140).
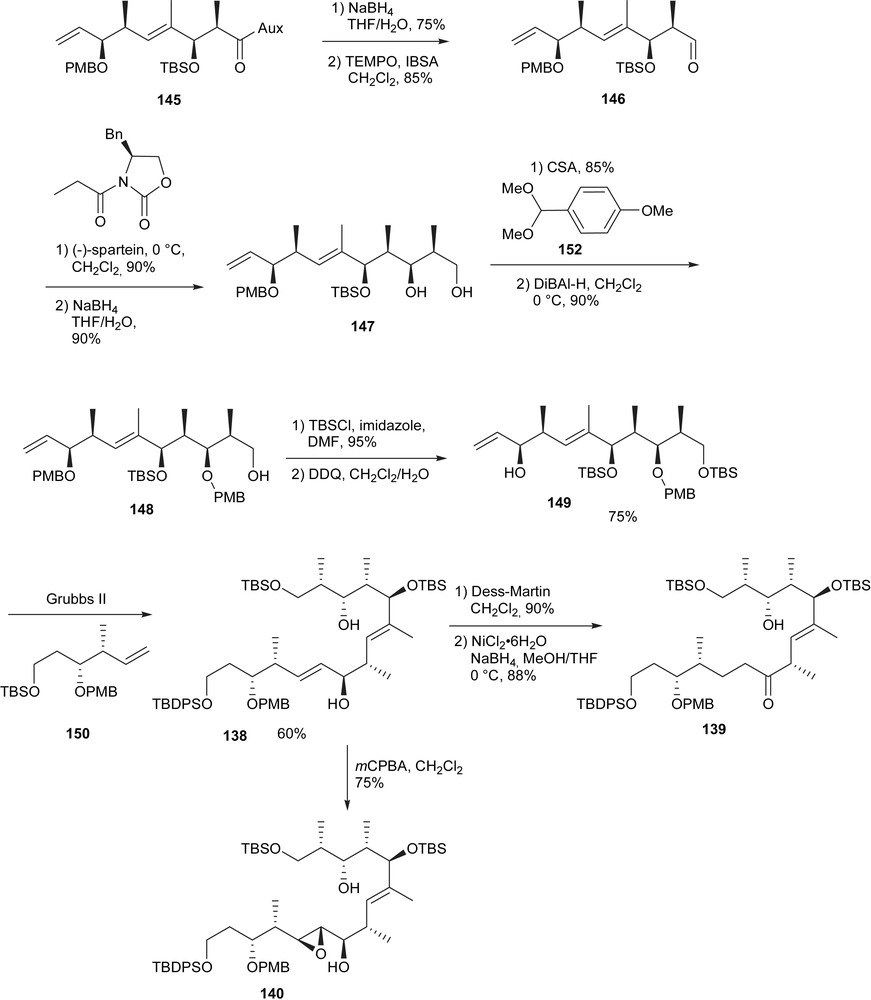
Synthesis of 139 and 140.
2.17 Myriaporones
Ten years after the discovery of the first tedanolide (1), Rinehart isolated from Myriapora truncata a closely related family of natural products, the myriaporones [16]. It appears that myriaporone 1 (151) and 2 (152) are isolation by-products of the natural compound, myriaporone 3/4 (153 and 154). Myriaporone 2 (152) arises from the opening of the epoxide by the C7 carbonyl in myriaporone 1 (151). Myriaporone 3/4 exists as a dynamic equilibrium between a closed and an open form due to the lability of the hemiketal moiety. The small amount isolated did not allow the determination of the stereochemistry at C5 and C6 at the time and they were elucidated by total synthesis. Their interesting biological activity (IC50 value of 100 ng/mL against L-1210 murine leukemia cells) as well as the similarities of their structure to the C10–C23 portion of tedanolide (1) suggests that the myriaporones are in fact naturally occurring analogues. The myriaporones would then share the same mode of action as tedanolide and their simpler structure renders them more attractive as drug candidates. Recently, a study established that like tedanolide, myriaporone 3/4 is a potent protein synthesis inhibitor selective for eukaryotes [17]. Surprisingly, despite the numerous methodologies developed for the synthesis of tedanolide, few groups applied them for the synthesis of myriaporone (Fig. 11).

The family of myriaporones.
2.18 The Yonemitsu approach
As previously mentioned, the stereochemistry at C5 and C6 was not assigned at the time of the isolation. In light of the structural similarities between myriaporone and tedanolide, one could have assumed the stereochemistry of these two centers to be identical to the corresponding carbons of tedanolide (C13 and C14). Yonemitsu, however, hypothesized the opposite configuration for C6 (C14 of tedanolide) [10i]. He also altered his route to the tedanolide and developed a new synthesis toward myriaporone 3/4. His retrosynthesis was based on the coupling of known vinyl iodide 157 and aldehyde 156. The C5–C9 aldehyde was generated from intermediate 158 which was developed for his synthesis of erythronolide A [10i]. The starting material for this synthesis was d-glucose (Fig. 12).

The Yonemitsu approach of myriaporone 3/4.
The key methodology applied in this study was the regioselective opening of MP acetal 160 with MgBr2 and SnBu3H [10i]. The opening selectivity is governed by a five-membered ring chelation of the substrate, and thus opening occurs at the more hindered carbon. This is a good complement to the Dibal-H opening of acetals (Scheme 26).
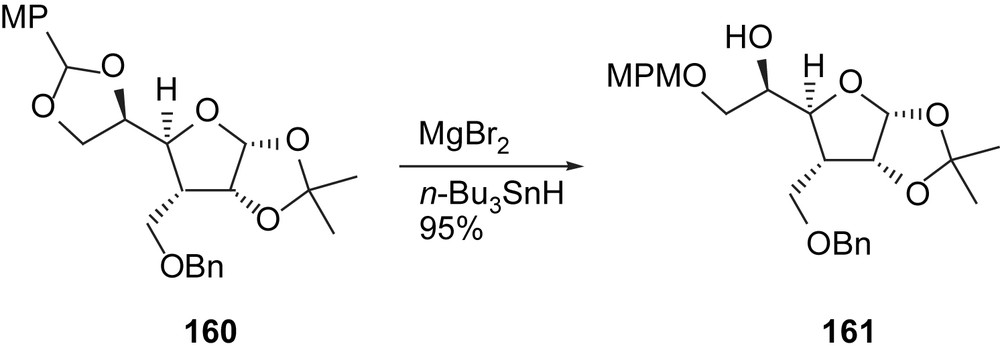
Opening of MP acetal 160.
The total synthesis was not completed and only the intermediate 155 was generated. As it is now proven that his assumption of the stereochemistry was not correct at C6, his synthesis will need to be revised to generate the natural product.
2.19 The Taylor approach
The first approach envisioned for the introduction of C5–C9 hydroxypropionate unit was two successive stereocontrolled allylations (Fig. 13) [17a]. The known intermediate 162 was in turn generated via an alternative approach using a thionyl chloride rearrangement (Scheme 27). The epoxide was chosen to be introduced prior to C9 installation even though such an early epoxidation was expected to render the synthetic intermediates more sensitive. This choice was motivated by the report depicting anti-selectivity for epoxidation of secondary allylic alcohols by Sharpless et al. [18].

The Taylor approach.
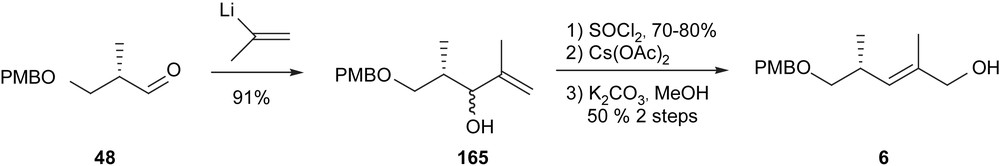
Thionyl chloride rearrangement.
Although the thionyl chloride rearrangement mechanism was already known, its application in synthesis was not common [17b]. The key reaction depended upon an intramolecular cyclic rearrangement to provide the primary allylic chloride selectively. The zirconium-mediated allylation, however, was not selective due to the steric hindrance generated by the α-methyl of the aldehyde.
Despite the lower activity of myriaporone 1 (151) compared to myriaporone 3/4, Taylor also decided to target the former [17c]. With C6 being sp2 hybridized in myriaporone 1, only the stereochemistry at C5 remained unknown, and thus, only two diastereomers needed to be synthesized (Fig. 14).

Retrosynthesis of myriaporone 1.
The C5–C7 β-hydroxyketone was masked as an isoxazoline. Formation of the C6–C7 bond was envisioned to be made by a homoallenylation.
The main drawback of this route was the sensitivity of aldehyde 167. It was generated via an Evans aldol reaction using a chiral auxiliary which proved to be selective and high yielding. C6–C7 bond formation was attempted using Nozaki–Hiyama–Kishi coupling and several homoallenylation conditions. The best results were obtained using Brown conditions giving only a moderate yield. The isoxazoline in 166 was then introduced in a 3:1 selectivity via an inverse dipolar cycloaddition.
The amount of material generated was not sufficient to finish the synthesis. Myriaporone 1 was unexpectedly generated during the synthesis of myriaporone 3/4 (vide infra) [17d] (Fig. 15).
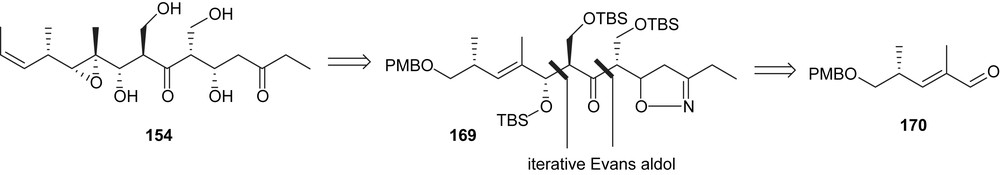
Retrosynthesis of myriaporone 3/4.
Due to the excessive sensitivity of the epoxyaldehydes and encouraged by the promising epoxidation of Loh [12a], the synthetic plan for myriaporone 3/4 was revised as well as the timing of epoxidation.
The C5–C9 hydroxypropionate unit of 171 was installed by two iterative Evans aldol reactions. The isoxazoline was then introduced without any diastereoselectivity. At the time of the synthesis, the stereochemistry at C5 was still unknown and both isomers were needed for spectroscopic comparison with the natural product. In later work, the selectivity was increased to 3:1 toward the desired isomer. The Z-olefin was installed via selective Wittig reaction to provide 173 and after protecting group manipulations, the isoxazoline was reductively cleaved to unmask the β-hydroxyketone. The final key epoxidation was highly selective according to Yonemitsu's precedence. Myriaporone 3/4 (153/154) was thus completed in 2.1% over 25 linear steps.
When acetate-protecting groups were used instead of TBS, the final deprotection led to an elimination reaction, thus providing myriaporone 1 (151) (Schemes 28–30).

Toward myriaporone 1.
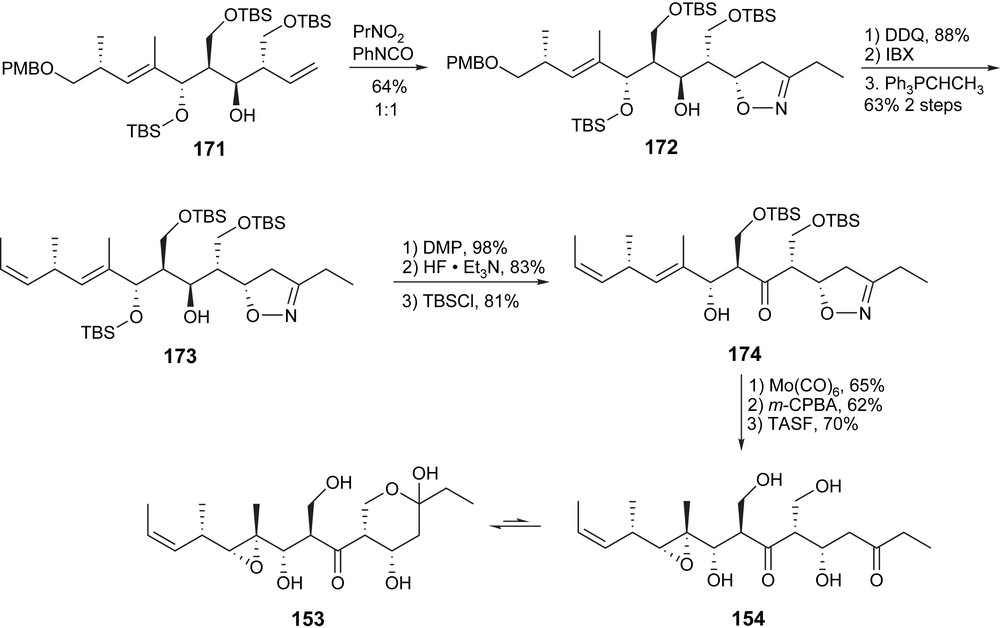
Taylor synthesis of myriaporone 3/4.
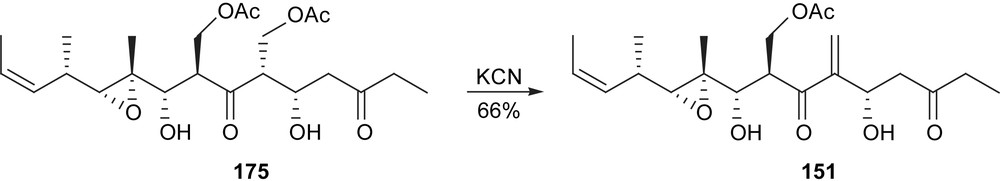
Taylor synthesis of myriaporone 1.
2.20 The Cuevas approach
Concomitantly with the Taylor synthesis, Cuevas et al. published their own synthesis in the same journal issue [19]. The synthesis developed was also linear and its beginning was similar to the Taylor synthesis. The main difference resided in the choice of an early epoxidation and the installation of the C1–C5 β-hydroxyketone moiety (Scheme 31).
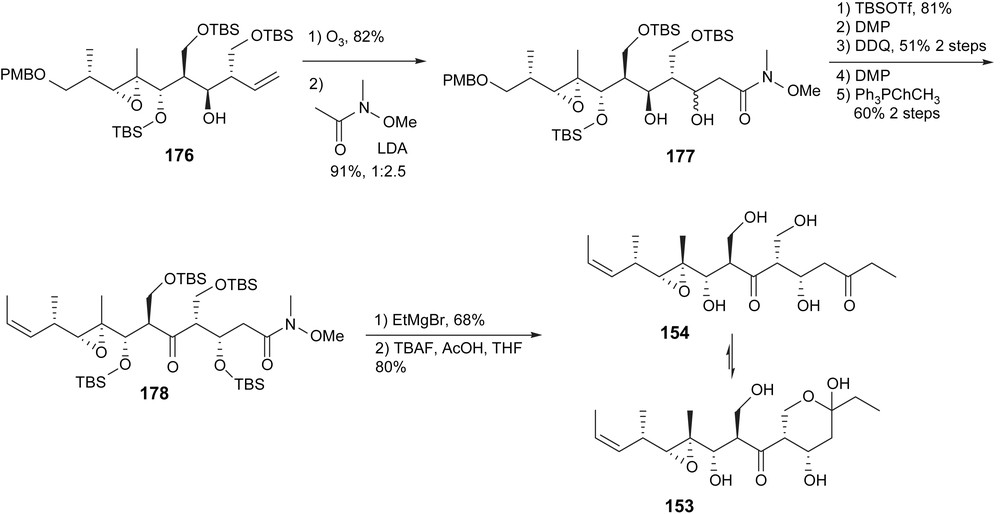
Cuevas synthesis of myriaporone 3/4.
The aldol reaction to form the C4–C5 bond gave 177 with a selectivity of 1:2.5 in favor of the unnatural isomer at C5. This is thus not a valuable selectivity a posteriori. Selective monoprotection discriminating between two secondary alcohols was performed to subsequently oxidize only C7. The carbon backbone was then completed via an Z-selective Wittig olefination to provide 178 and a Grignard addition to the Weinreb amide. Global deprotection gave myriaporone 3/4. The synthesis was completed in 24 linear steps (one step shorter than the Taylor synthesis) but with only 0.3% yield. Partial dehydration on silica gel (not observed by the Taylor group) and subsequent acetate protection of the dehydrated by-product provided them with myriaporone 1. The facility of elimination tends to prove that myriaporone 1 is indeed an isolation by-product of myriaporone 3/4.
3 Conclusion
In summary, the tedanolides are fascinating compounds due to their unprecedented biosynthetic origin, complex architecture and promising biological activity. For more than 20 years, a variety of new strategies to address the challenges offered by tedanolide were developed, completing the established set of tools available for polyketide synthesis.
Additionally, completing the total synthesis of tedanolide opens the door to structure–activity relationship studies, providing analogues that cannot be accessed from natural sources or degradation experiments. A fortiori, myriaporone 3/4 appears to be a simpler version of tedanolide that retains the latter's potency, thus suggesting that myriaporone 3/4 is a naturally occurring analogue of tedanolide. Both families represent exciting new leads for therapeutic development which will attract the attention of medicinal chemistry in the future.