

1 Introduction
Semiconducting organic materials have the potential to be used in a variety of emerging technologies such as organic thin-film transistors (TFTs) [1], organic light-emitting diodes (OLEDs) [2], and nonlinear optical media [3]. The prospect of easily forming such devices from organic molecules, oligomers, and polymers has subsequently led to a significant effort in exploratory research in these areas [4–12]. The allure of organic materials often lies in the ability to synthetically tune electronic and optical properties merely by systematically changing either the size or the constitution of the molecular skeleton. To this end, one approach toward optimizing organic materials is through the formation and study of a series of structurally related oligomers, in which the degree of conjugation is controlled by the number of repeat units. Using this so-called oligomer approach [13], structure–function trends can be elucidated and correlated with, for example, the electronic make-up of the molecules (e.g., HOMO–LUMO gap) [10,11,14]. This strategy has been applied to many, many different structural motifs, including, for example, acetylenic systems such as polydiacetylenes [15–17], polytriacetylenes [10,17,18], and poly(phenylene ethynylene)s (Fig. 1) [6,19–24]. Interestingly, the least amount of information has been reported for a series of oligomers that, arguably, represent the most efficient way to achieve the highest degree of conjugation in an organic material, namely, polyynes [25–27].
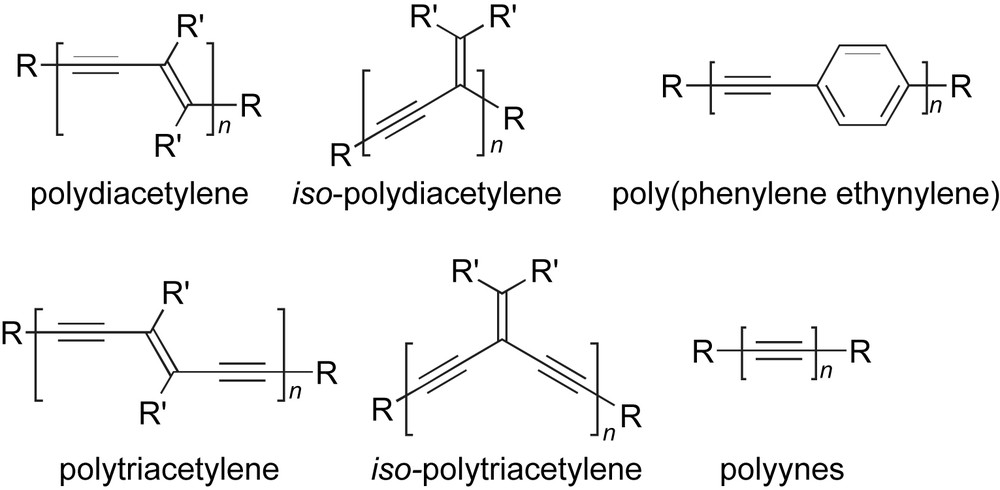
Common oligo- and/or polymeric systems based on acetylenic building blocks.
In the realm of all-carbon and carbon-rich materials, one hardly needs to be reminded of the significance of diamond and graphite and the impact they have made on society [28–31]. While certainly obvious, it is worth noting that these two very important allotropes are both composed almost entirely of carbon, yet they show inherently different materials' properties due to the difference in their hybridization, i.e., diamond is derived from sp3-hybridized carbon while graphite (as well as graphene, fullerenes, and carbon nanotubes) is made up of sp2-hybridized carbon. Extension of this series to the next logical allotrope, comprised of sp-hybridized carbon, would give carbyne (Fig. 2), the existence of which has been a controversial topic for decades [32–36]. While naturally occurring carbyne may or may not exist, defined length oligomers (polyynes) based on the contiguous connection of acetylenic units have become desirable synthetic targets over the past decade [25–27,37]. The current renaissance in polyyne chemistry is not, however, the first era in which exploration of both the synthesis and characterization of polyynes has been popular. Already in the 1950s, 60s, and 70s the study of such molecules attracted the interest of pioneers such as Jones and Thaller [38,39], Bohlmann [40–42], Walton and coworkers [43–45], and Nakagawa et al [46]. Whether a half-century ago or this year, the study of extended polyynes provides scientists with an unprecedented opportunity to gain insight into the properties of carbyne, as well as compare it to other allotropic forms of carbon.
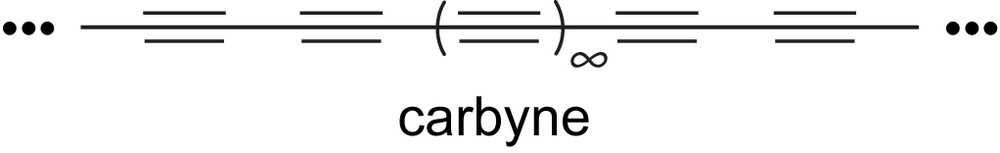
The structure of carbyne.
2 Polyyne synthesis
There are a number of common techniques currently used to synthesize polyynes. Many rely on the homo- or heterocoupling of preformed acetylenic precursors [47], while others utilize an elimination event to form the polyyne in the last step. A handful of methods have also been developed whereby the extrusion of a molecular subunit can be exploited to assemble the polyyne framework. The intent of this account is by no means to enumerate all of the variations of these methods. Rather, highlighted examples of each synthetic strategy will be presented, concentrating primarily on reports of polyynes with a length of at least 10 carbons (i.e., pentaynes). The account will conclude with a more thorough description of known polyynes with a length of at least 16 carbons, given that there are relatively few such molecules.
2.1 General synthetic considerations
The synthesis of polyynes arguably dates back to 1869 when Carl Glaser observed that copper(I) phenylacetylide undergoes oxidative dimerization in the presence of air to produce diphenylbutadiyne (Scheme 1a) [48].
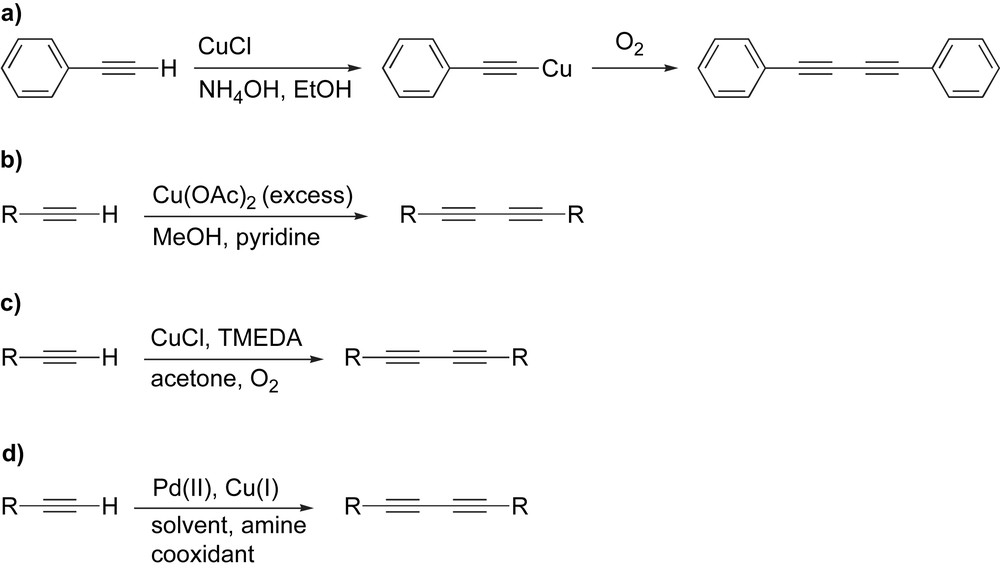
Commonly used homocoupling conditions.
Since then a variety of improvements have been made on the copper-catalyzed homocoupling method that do not require the isolation of a potentially explosive copper(I) acetylide. An example of this is the oxidative coupling of acetylenes using Cu(OAc)2 in the presence of MeOH and pyridine reported in 1956 by Eglinton and Galbraith (Scheme 1b) [49]. A breakthrough in oxidative dimerization of acetylenes has been reported by Hay in 1962, who showed that CuCl in the presence of TMEDA and O2 is an exceedingly effective system (Scheme 1c) [50]. This reaction is commonly done in either acetone or CH2Cl2, and it benefits substantially from the enhanced solubility of the Cu catalyst as a result of the TMEDA ligand. Further refinement of the typical homocoupling reactions has recently invoked the use of Pd catalysis instead of the traditional Cu(I)/Cu(II) system (Scheme 1d) [47,51]. The use of Pd catalysis can often lead to excellent yields of desired product when Cu catalysis fails in this regard [52,53]. Recently, it has also been reported that Pd-catalyzed homocoupling of acetylenes can also occur under aerobic conditions where oxygen in the air acts as the cooxidant to regenerate the active Pd(II) catalyst [54].
A common feature of Cu(I)/Cu(II)-catalyzed reactions is that they are typically not suitable for the formation of unsymmetrical derivatives such as, for example, triynes. This fact is nicely demonstrated by the work of Bohlmann and coworkers (Scheme 2) [55], where the reaction of phenylbutadiyne with pent-2-ene-4-yn-1-ol produced almost exclusively the α,ω-tetrayne, with little of the unsymmetrical triyne obtained in the reaction. This has been explained through a mechanistic investigation that shows that the relative acidity of the acetylenic precursors plays a defining role in the reaction rate. Thus, the more acidic diyne reacts first, leading to the tetrayne product.
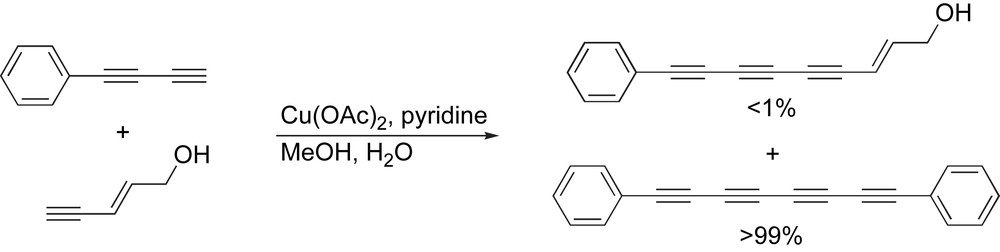
Bohlmann's attempted cross-coupling under Eglinton conditions.
The problem of forming unsymmetrical diynes, triynes and in some cases tetraynes has been addressed through the cross-coupling method developed by Cadiot and Chodkiewicz (Scheme 3) [47,56–58]. In this case, a terminal alkyne (or polyyne) is reacted with a bromo-, iodo-, or chloroalkyne derivative, leading to the unsymmetrical product. This reaction can, however, be complicated by the competing homocoupling reaction of the alkynyl halide, leading to a product mixture that is often difficult to separate. Pd-catalyzed versions of this protocol have also recently been developed [52,59].
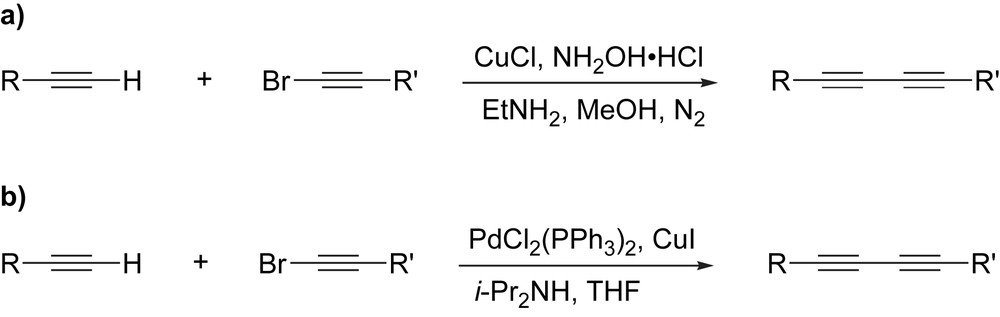
Cadiot–Chodkiewicz cross-coupling.
Finally, a more recent development in the formation of polyynes uses the Fritsch–Buttenberg–Wiechell (FBW) rearrangement [60–65] in which an alkylidene carbene/carbenoid is generated in situ followed by a 1,2-migration of a pendent alkynyl group to generate a carbon–carbon triple bond (Scheme 4). While the conditions of the original FBW rearrangement are quite harsh, a fact that renders it incompatible with many functional groups, milder variations of the FBW rearrangement have subsequently been developed and provide a means to access symmetrical and unsymmetrical polyynes [26,66–72].
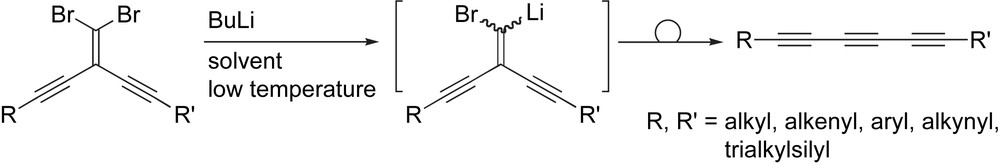
The FBW rearrangement.
2.2 Pentaynes
As a result of their odd number of acetylene units, pentaynes present a rather unique synthetic challenge. In some cases, the formation of a pentayne skeleton can be achieved by an exhaustive elimination reaction [73]. On the other hand, more “traditional” methods can be used such as the Cadiot–Chodkiewicz reaction [74] or a mixed Hay coupling reaction [45,75,76]. To circumvent the need for potentially unstable triyne precursors, however, several innovative synthetic techniques have been recently developed and deserve comment within the confines of this Account.
Tobe and coworkers reported an interesting procedure for the synthesis of symmetrical tri- and pentaynes utilizing a [2 + 1] chelotropic fragmentation (Scheme 5) [77]. In this case, a suitably functionalized enyne is irradiated with high energy UV light (254 nm), which induces a sequence of electrocyclic ring closure followed by chelotropic extrusion of indane that results in the formation of a free alkylidene carbene intermediate. A Fritsch–Buttenberg–Wiechell rearrangement via migration of one of the pendent butadiynyl groups then affords the desired pentayne. Tobe and coworkers have also used this method to form cyclic polyynes, e.g., the cyclo-Cn series [77].
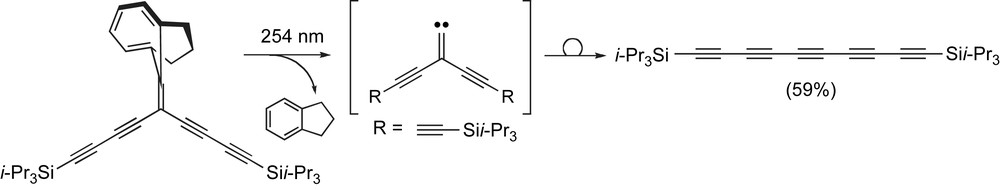
Tobe and coworkers' [2 + 1] chelotropic fragmentation – alkylidene carbene rearrangement.
Diederich and coworkers have addressed the problem of pentayne formation (as well as a hexayne) through development of a solution-spray flash-vacuum-pyrolysis (SS-FVP) technique (Scheme 6) [78]. In this case, readily available 3,4-dialkynyl-3-cyclobutene-l,2-diones can be converted to the desired polyynes on a multi-gram scale, as demonstrated by formation of the diphenyl- and bis(triisopropylsilyl)-terminated pentaynes.
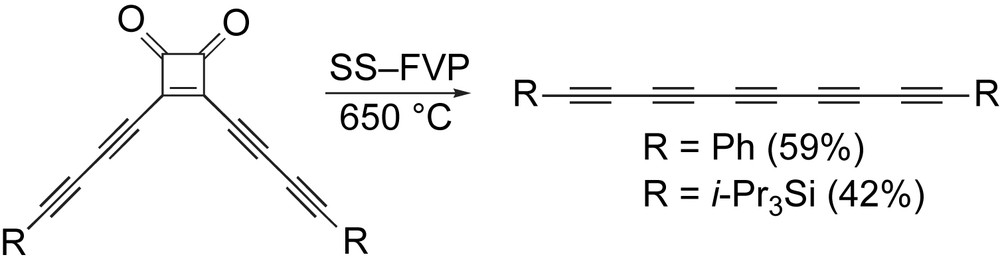
Diederich and coworkers' SS-FVP route to pentaynes.
Negishi and coworkers have applied their pair selective protocol to the formation of polyyne derivatives (Scheme 7) [79]. In this case, a Zn-acetylide, formed in situ from the corresponding E-1-chloro-1-hexen-3,5-hexadiyne precursor (not shown), is coupled with a 4-aryl-1,1-dibromo-1-buten-3-yne partner, followed by elimination to the desired pentaynes.
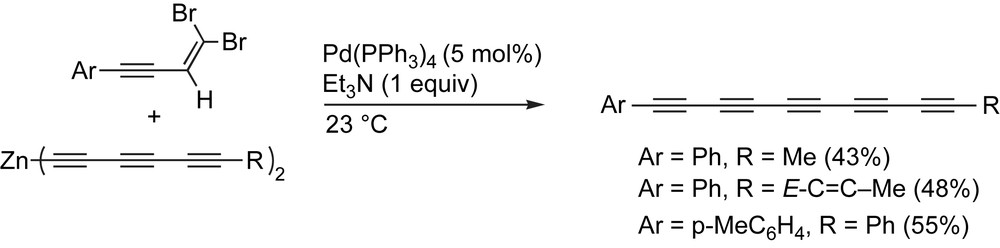
Negishi and coworkers' pair selective protocol.
Pentaynes can be formed via the FBW rearrangement starting from the appropriate dibromoolefinic precursor. In the case shown in Scheme 8, a two-fold rearrangement leads to the polyyne product in 45% yield [67].
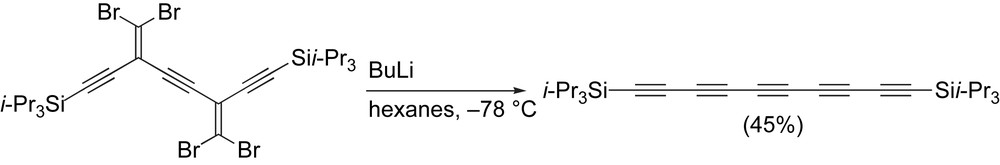
Tykwinski and coworkers' carbenoid route to a pentayne.
Finally, polyynes have been formed by Cataldo through the vaporization of graphite by electric arc and by the hydrolysis of calcium carbide (CaC2) in an aqueous solution of NH4Cl containing Cu(I)/Cu(II) salts [80–82]. Both methods lead to a mixture of hydrogen terminated polyynes that can be separated by HPLC using a C-18 column. The pentayne H–(CC)5–H is typically the longest of the series to be formed in a reasonable quantity in this manner.
2.3 Hexaynes
There are many, many examples of hexayne syntheses and most involve the initial formation a trialkylsilyl-protected triyne precursor, which is then deprotected and subsequently subjected to an oxidative homocoupling reaction. While the stability of the terminal triyne precursor can be a determining factor in the success of such reactions, if kept in solution following removal of the silyl protecting group, reactions to form hexaynes typically proceed in reasonable to good yields, such as the examples shown in Scheme 9 [68,69]. It is worth noting as well that recent work by Bryce and coworkers has also demonstrated that terminal triynes, and even tetraynes, can show surprising stability in the solid state through the use of a large, sterically demanding endgroup that effectively separates the reactive polyyne groups such that intermolecular reactions are kinetically disfavored [83–85].
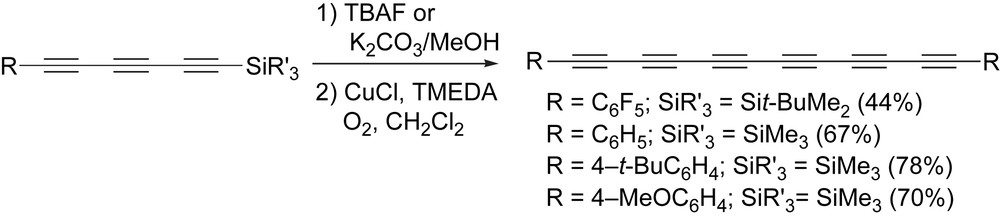
Formation of diarylhexaynes via oxidative homocoupling.
The need for an intermediate triyne can be circumvented through formation of the complete carbon framework prior to introducing the final acetylene units. As with pentaynes, this has been done with both the SS-FVP technique of Diederich [78] and the FBW rearrangement (Scheme 10) [26,67]. In both cases, a two-fold process is accomplished, i.e., dual FBW rearrangements or the pyrolytic openings of two cyclobutenediones followed by loss of carbon monoxide. The hexaynes have been obtained in moderate to good yields.
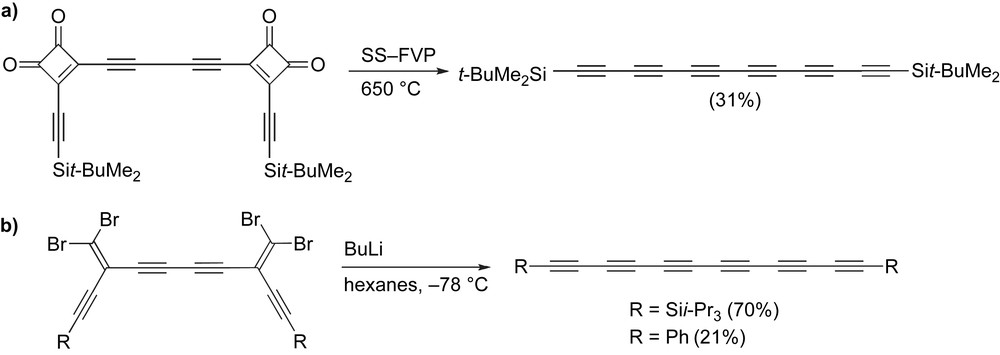
Synthesis of hexaynes through (a) SS-FVP and (b) two-fold FBW rearrangement.
An alternative route to hexayne formation using an FBW rearrangement utilizes a one-pot procedure (Scheme 11) [70]. In this case, formation of a Li-acetylide from a previously desilylated dibromoolefinic precursor is accomplished through reaction with two equivalents of BuLi in toluene. To this mixture is added CuBr and TMEDA to give an intermediate Cu-acetylide [86], and oxygen gas is then bubbled into the mixture to ultimately afford the desired hexayne in good yield.
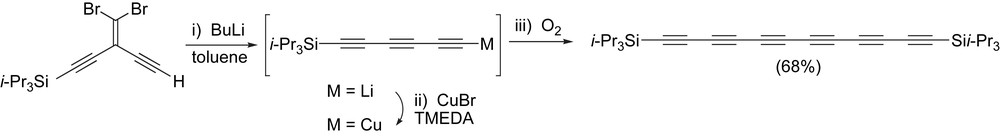
A one-pot approach to hexayne formation based on an FBW rearrangement.
The synthesis of precursors that do not require oxidative homocoupling as the final step can be used for polyyne formation, such as the example reported by Cox and coworkers [87]. This route establishes the 12 carbon framework of the nascent polyyne via an oxidative homocoupling of the two enediyne “halves”, followed by a fluoride-catalyzed elimination reaction to form the acetylenic skeleton (Scheme 12).
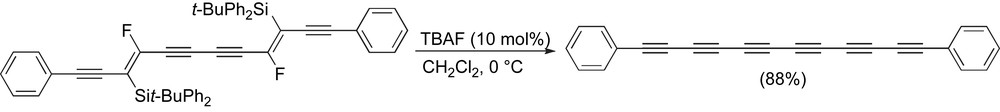
Cox and coworkers' synthesis of diphenyl hexayne.
Mori and coworkers have pioneered the concept of an oxidative acetylenic homocoupling directly from a trialkylsilyl-protected acetylene, thus forming a diyne without the need for isolation of the terminal alkyne precursor [88]. This concept has also been applied to polyynes [89]. For TES-protected triyne derivatives, a one-pot reaction has been achieved using a sequence of TBAF mediated desilylation, in situ quenching with ClSiMe3 (used as a fluoride scavenger), and Hay homocoupling to yield the desired hexayne [75]. An alternative strategy toward hexaynes involves an in situ deprotection/homocoupling protocol of a TIPS-protected triyne through the use of TBAF in the presence of a Cu(OAc)2 catalyst [90].
A number of efforts have been made to provide a more stable polyyne through insulating the polyyne core with an alkyl chain that is tethered to the two termini. For example, Hirsch and coworkers have used phenyloxy endgroups that are connected via a flexible alkyl chain. The tethered triynes, once desilylated, are subjected to Hay conditions to form the hexayne core (Scheme 13a) [91]. Gladysz and coworkers, pioneers in this area, were able to use a similar insulation strategy for hexaynes by means of a long-chain alkyl diphosphine ligand that spans the two Pt endgroups (Scheme 13b) [92,93]. The bridging tether could be introduced in one of two ways, either through ligand exchange at the platinum centers using a bidentate diphosphine ligand or through the use of ring-closing metathesis with the Grubbs I catalyst to link the two halves of the tether, followed by reduction of the resulting alkene.
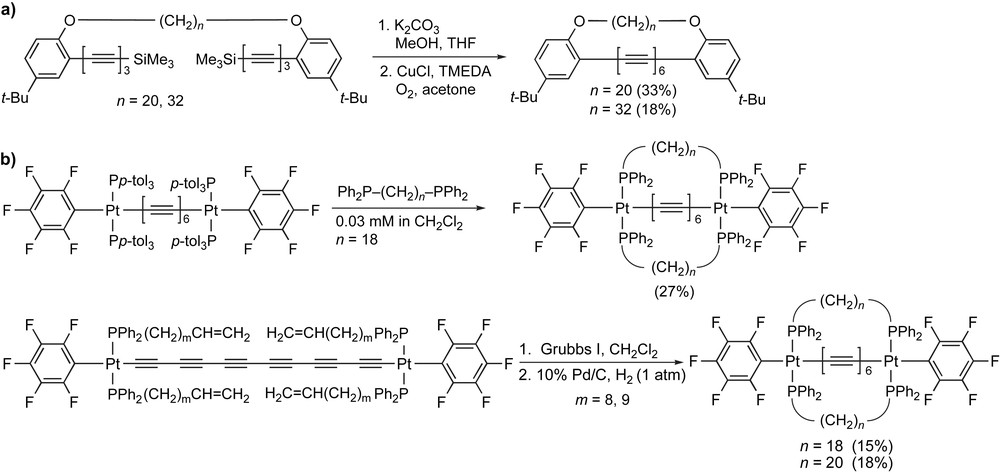
The formation of insulated hexaynes by (a) Hirsch and coworkers and (b) Gladysz and coworkers.
Finally, Tomita and Sugiyama have approached the problem of hexayne stabilization through rotaxane formation using cyclodextrins (CDs). They observed, using time-course UV–vis spectroscopic studies, that there is substantial stabilization of a bis(phenanthryl)hexayne while encapsulated within one or two CD molecules, as compared to the free hexayne [94].
2.4 Heptaynes
Even more so than pentaynes, heptaynes are quite formidable synthetic targets because they cannot be assembled by a simple dimerization reaction. Furthermore, beyond hexaynes, kinetic stability of the product becomes a more serious issue as well. The first heptayne to be produced and characterized has been by Bohlmann, based on the exhaustive elimination of a suitably functionalized tert-butyl endcapped precursor (Scheme 14) [95]. The product heptayne is reported to be crystallized from a mixture of petroleum ether and MeOH to give a yellow crystalline solid that slowly turns black upon heating to ca. 150 °C.
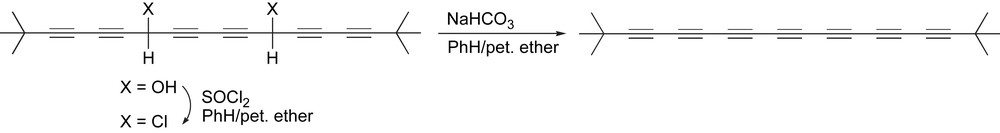
Bohlmann's synthesis of the first isolated heptayne.
Walton and coworkers have used a Hay coupling to extend a hexayne framework by one acetylene unit through a reaction with excess Et3SiC≡CH, providing the triethylsilyl endcapped heptayne (Scheme 15a) [45]. Although no yield has been given, this product could be characterized by UV–vis spectroscopy and then desilylated to give either the mono- or bisdeprotected polyyne product. Gladysz and coworkers have used a conceptually analogous chain length extension protocol toward a Pt endcapped heptayne. In this one-pot reaction, desilylation of the pentayne precursor with TBAF is followed by the addition of trimethylchlorosilane which acts as a fluoride scavenger (Scheme 15b) [75,76]. The resulting deprotected polyyne is then subjected to Hay coupling with triethylsilylbutadiyne to yield the unsymmetrical heptayne in a modest 20% yield as a red solid after chromatographic purification.
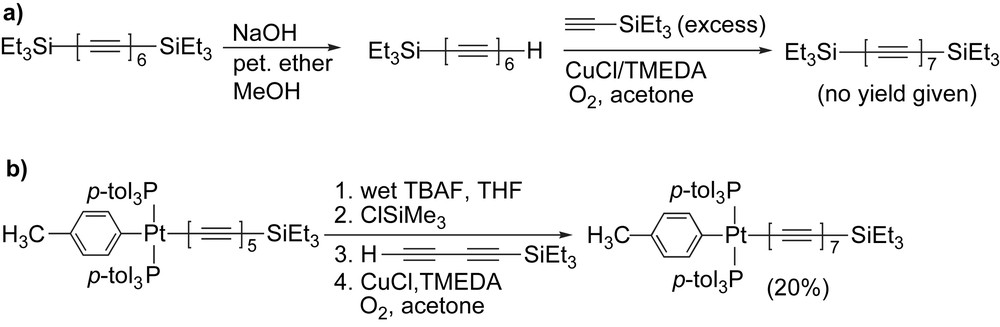
Heptayne synthesis via chain length extension by (a) Walton and coworkers and (b) Gladysz and coworkers.
As a part of their effort to encase polyynes within an insulating sheath provided by an alkyl tether (as also described in the previous section), Hirsch and coworkers reported a rather unique mode of heptayne formation (Scheme 16). Subjecting a tethered triyne to Pd-catalyzed Sonogashira coupling conditions in the presence of diiodoacetylene provided the desired heptayne in 11% yield as an orange solid that could be characterized by 1H NMR and UV–vis spectroscopies, as well as FAB MS analysis [91].
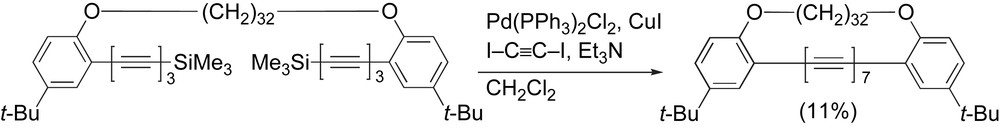
Hirsch's heptayne synthesis.
Bruce and coworkers have elegantly developed the chemistry of gold-acetylides toward the formation of both Ru and Co endcapped heptaynes (Scheme 17) [96,97]. In both cases, the Pd-catalyzed reaction of the Au-diyne with I–(CC)3–I led to the desired heptayne product, and in the case of the cobalt species a remarkable 86% yield has been obtained. Both species have been characterized crystallographically, and these are the only X-ray structures reported to date for heptaynes [96].
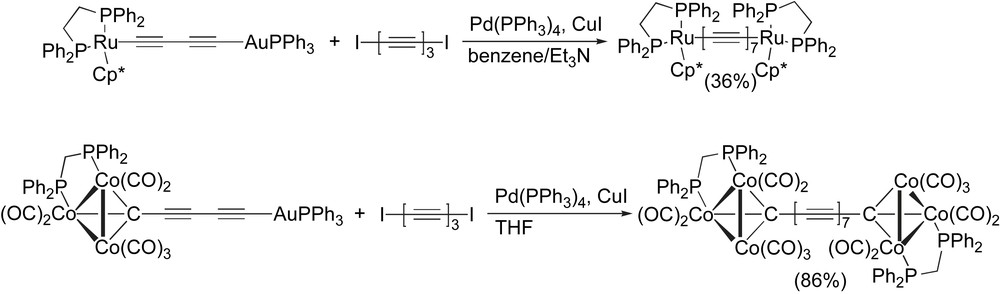
Heptayne syntheses from Bruce and coworkers.
In addition to those described above, the parent heptayne, H–(CC)7–H [45,82,98], has been formed by a number of methods, and both H–(CC)7–CN [82] and NC–(CC)7–CN [99,100] have also been synthesized and studied. In most cases, the heptayne is formed as part of a mixture of polyynes with varying length and is subsequently separated chromatographically.
2.5 Octaynes
Octaynes are much less common than their hexayne counterparts, and they typically show considerably lower kinetic stability under ambient conditions. Considering the rather substantial amount of work over the past 40 years in this area, few polyynes of 16 carbons or longer have been formed and characterized (Table 1). Given some of the difficulties encountered in the synthesis of octaynes and even longer polyynes, stabilization of the reactive polyyne framework has become an obvious goal in order to provide macroscopic quantities of the product for subsequent characterization [25,26]. To this end, there is substantial empirical evidence that supports the premise that extended polyynes can be stabilized by using bulky substituents on the polyyne termini. Hirsch and coworkers have demonstrated that the use of large dendrimeric endgroups provided bench-stable octaynes (Table 1, entry 8) [101]. Gladysz and coworkers have successfully used a bulky Pt endgroup to achieve the stability needed to isolate these materials (Table 1, entries 16 and 18) [75]. In both cases, the final octayne product is formed through Cu-catalyzed oxidative dimerization of the tetrayne “halves”. Other notable octayne systems formed through oxidative dimerization include the stable ferrocene derivative (Table 1, entry 12), and a porphyrin endcapped species (Table 1, entry 13).
Known, characterized 1,3,5,7,9,11,13,15-hexadecaoctaynes.
| Entry | Octayne | Characterizationa | Reference(s) |
| 1 | UV–vis | [45,82,98] | |
| 2 | UV–vis, EA, DP | [102] | |
| 3 | IR, UV–vis, EI-MS | [99,100] | |
| 4 | 1H and 13C NMR, IR, UV–vis, EA, DSC, NLO | [69,103] | |
| 5 | IR, UV–vis, EA, DP | [43] | |
| 6 | 19F and 13C NMR, IR, MALDI-TOF HRMS, DP | [68] | |
| 7 | 1H and 13C NMR, IR, Raman, UV–vis, FAB MS, CV | [101] | |
| 8 | b | 1H and 13C NMR, Raman, UV–vis, FAB MS | [101] |
| 9 | UV–vis, FAB MS | [91] | |
| 10 | UV–vis | [45] | |
| 11 | 1H and 13C NMR, IR, UV–vis, EI HRMS, DSC, X-ray, NLO | [67] | |
| 12 | IR, UV–vis, DP | [104] | |
| 13 | 1H NMR, IR, UV–vis, MALDI-TOF MS | [105] | |
| 14 | 1H and 31P NMR, IR, ESI MS, EA | [97] | |
| 15 | 1H, 13C, and 31P NMR, IR, Raman, UV–vis, FAB MS, CV, DSC, EA | [74,106] | |
| 16 | 1H, 13C, and 31P NMR, IR, UV–vis, FAB MS, CV, EA, TGA, DP, X-ray | [107] | |
| 17 | 1H, 13C, and 31P NMR, IR, UV–vis, MSc, EA, DP | [75,76] | |
| 18 | 1H, 13C, and 31P NMR, IR, UV–vis, CV, MSc, EA, DSC, TGA, NLO | [75,76,108] | |
| 19 | 1H and 13C NMR, IR, [α]D, UV–vis, ESI HRMS, EA, DP | [109] |
a CV = cyclic voltammetry, DP = decomposition point, DSC = differential scanning calorimetry, EA = elemental analysis, NLO = nonlinear optical analysis, TGA = thermal gravimetric analysis, X-ray = X-ray crystallographic analysis.
b Ar = Fréchet's aromatic polyether [G-3] dendrimer.
c Ionization method not reported.
Hirsch and coworkers have extended efforts toward encapsulation of an octayne. Unlike their experience with heptayne synthesis using a –O(CH2)32O– tether (Scheme 16), however, only a trace of the desired octayne product could be isolated using a slightly longer –O(CH2)40O– tether linker (Table 1, entry 9) [91].
A two-fold FBW rearrangement has been used to synthesize TIPS and the phenyl endcapped octaynes, giving the desired products in 10% and 13% yields, respectively (Scheme 18 and Table 1, entries 4 and 11) [26,67,69]. The TIPS endcapped octayne can be stored as a solid indefinitely under refrigeration without appreciable decomposition. When crystallized from ether/EtOH, Armitage reported that the phenyl endcapped octayne has been isolated as an unstable copper colored solid that tended to explode upon heating [103]. When crystallized from THF, however, the resulting rust-colored solid shows surprisingly enhanced stability, surviving years if kept under refrigeration [110]. It is unclear whether the improved stability derives from a more stable polymorph or from increased purity [69].
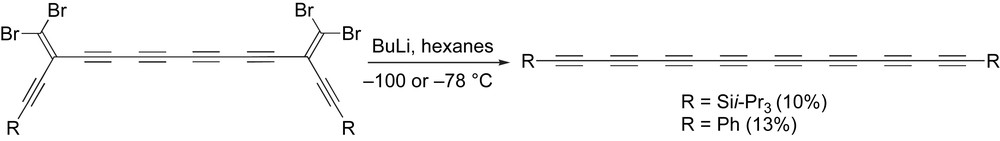
The FBW approach to octaynes.
Recently, Frauenrath and Hoheisel reported a new Negishi protocol toward the synthesis of glycosylated polyynes (Scheme 19 and Table 1, entry 19) [109]. The formation of the Zn-acetylide was carried out by monodesilylation using MeLi·LiBr followed by treatment with ZnCl2. This intermediate was subjected to Pd-catalyzed heterocoupling conditions with the brominated diyne coupling partner to generate the unsymmetrical glycosylated tetrayne in a 45% yield. Desilylation using silver nitrate followed by oxidative dimerization under Hay conditions led to the glycosylated octayne in 58% yield over two steps.
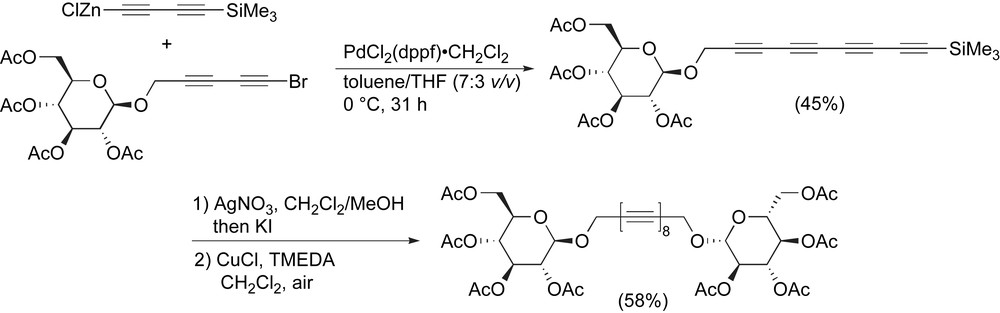
Frauenrath and coworkers' Negishi protocol toward octaynes.
It is worth noting that X-ray crystallographic analyses of octaynes are scarce, and only two structures have been reported to date. These include the TIPS endcapped species (Table 1, entry 11) and the Pt endcapped species (Table 1, entry 16).
2.6 Nonaynes
Nonaynes are rare. The only two intended syntheses of a nonayne, to our knowledge, come form the early work of Eastmond in which the monodeprotected octayne is obtained through a carefully controlled kinetic desilylation followed by a cross-coupling reaction with excess triethylsilylacetylene to obtain the bis(TES)nonayne (Scheme 20) [45]. The protected nonayne has also been desilylated by Eastmond to generate H–(CC)9–H, which is a compound that has also been observed as a product of electric arc between graphite electrodes [82]. Three other nonaynes have been reported as byproducts of the Hay coupling reaction toward decaynes, albeit in these cases characterization is limited to MS and/or UV–vis spectroscopic analysis [67,101].

Eastmond's synthesis of a nonayne.
2.7 Decaynes, and beyond
Decaynes (Table 2) have been realized as early as 1960 but their isolation and characterization have proved difficult [102]. For most series of polyynes, this length of 20 carbons seems to typically be the upper limit of polyyne stability. This is particularly true for early work, including t-Bu and Ph derivatives (Table 2, entries 3 and 5). Likewise, the decayne has been formed for the Et3Si series (entry 9), as well as longer derivatives (vide infra), but even the octayne is already unstable to isolate in its neat form. Nevertheless, there have been a few recent examples that have overcome the issue of stability and have provided decaynes in macroscopic quantities for study. Of particular note, both the Gladysz and Hirsch groups have successfully formed and isolated stable decaynes terminated with either metal-acetylide (Re and Pt) or dendrimeric endgroups, respectively (Table 2, entries 14, 15, 17 and entry 8, respectively) [75,101].
Known, characterized polyynes with ≥10 acetylene units.
| Entry | Polyyne | Characterizationa | Reference(s) |
| 1 | UV–vis | [45] | |
| 2 | UV–vis | [45] | |
| 3 | UV–vis, EA, DP | [43,102] | |
| 4 | UV–vis, DP | [43] | |
| 5 | UV–vis, IR, DP | [43] | |
| 6 | UV–vis, MALDI-TOF MS | [112] | |
| 7 | FAB MS | [101] | |
| 8 | 1H and 13C NMR, Raman, UV–vis, FAB MS, CV | [101] | |
| 9 | UV–vis | [45] | |
| 10 | UV–vis | [45] | |
| 11 | UV–visc | [45] | |
| 12 | 1H and 13C NMR, IR, UV–vis, MALDI MS, DSC, NLO | [67] | |
| 13 | UV–vis | [75,76] | |
| 14 | 1H, 13C, and 31P NMR, IR, Raman, UV–vis, FAB MS, CV, DSC | [106] | |
| 15 | 1H and 31P NMRc, IRc, UV–vis, FAB MSc | [107] | |
| 16 | 1H and 31P NMRc, IRc, UV–vis | [107] | |
| 17 | 1H, 13C, and 31P NMR, IR, UV–vis, CV, MSd, EA, DSC, TGA, NLO | [75,76,108] | |
| 18 | 1H, 13C, and 31P NMR, IR, UV–vis, MSd, EA, DSC, TGA, NLO | [75,76,108] | |
| 19 | 1H, 13C, and 31P NMR, IR, UV–vis, MSd, EA, DSC, TGA | [75,76] | |
| 20 | 1H and 31P NMR, IR, ESI MS | [97] |
a CV = cyclic voltammetry, DP = decomposition point, DSC = differential scanning calorimetry, EA = elemental analysis, NLO = nonlinear optical analysis, TGA = thermal gravimetric analysis.
c Analysis of an impure sample.
d Ionization method not reported.
One of the most studied series of polyynes is that with terminal TIPS groups, which include polyynes up to the decayne (Table 2, entry 12). An attempt at making the bis(TIPS)decayne utilizing a four-fold FBW rearrangement proved to be futile, with no desired product being isolated (Scheme 21) [26,67]. The decayne has been synthesized, however, through first implementing a two-fold FBW rearrangement on tetrabromoolefin to produce the unsymmetrical pentayne (Scheme 21). The pentayne is then subjected to standard protodesilylation conditions to give the unstable deprotected pentayne, which is subjected directly to oxidative homocoupling conditions yielding the decayne in an overall yield of 30%. The third-order nonlinear optical characteristics of this series of polyynes, including the decayne, have been explored and show an impressive increase in molecular second hyperpolarizability as a function of increasing conjugation length [111].
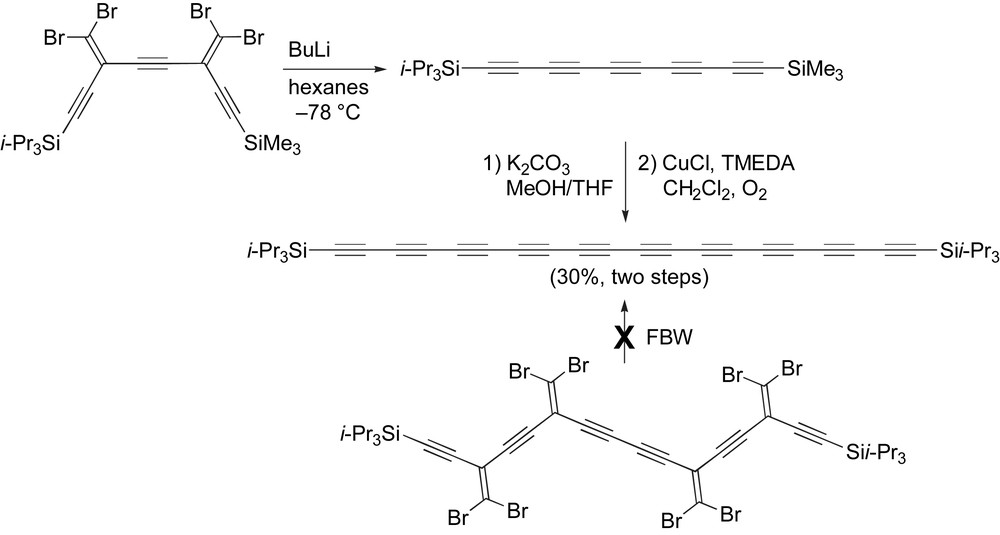
Synthesis of a triisopropylsilyl endcapped decayne.
The synthesis of polyynes beyond 20 carbons in length has been nothing less then a monumental synthetic challenge, one for which there still remains no easy or general answer. There has been evidence of the formation of dodecaynes within the literature as early as 1972 [43,45]. For example, in a classical “tour de force” approach, Walton and Johnson have synthesized one of the first known dodecaynes t-Bu–(CC)12–t-Bu (Scheme 22) [43]. The mixed Hay coupling of Et3Si–(CC)4–H and H–(CC)2–t-Bu gives the desired unsymmetrical hexayne, which is carefully desilylated with NaOH to give the terminal hexayne. Attempted Hay coupling of this compound resulted in a black solid from which a trace amount of the desired dodecayne could be observed spectroscopically, but not fully characterized. Thus, a MeOH solution of the terminal hexayne was subjected to Eglinton–Galbraith coupling conditions, from which again a black solid has been obtained. Extraction of this solid with CHCl3 and column chromatography provided reddish-brown needles of the product that decomposed in less than 10 min under ambient conditions. Walton and coworkers have also formed Et3Si–(CC)12–SiEt3 and characterized it by UV–vis spectroscopy (Table 2, entry 10). This same study provided evidence of Et3Si–(CC)16–SiEt3 (entry 11), although characterization was limited to UV–vis spectroscopic analysis of an impure sample.
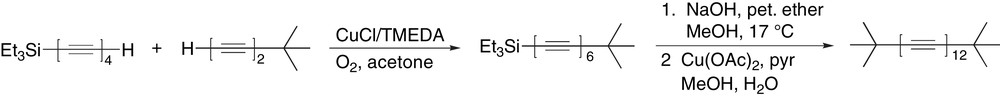
Walton's synthesis of the t-butyl endcapped dodecayne.
Cox and coworkers recently reported the synthesis of the first aryl endcapped dodecayne using a very interesting new strategy of β-chlorovinylsilane elimination (Scheme 23) [112]. In this case, the masked precursor, which contains all 24 carbons of the dodecayne skeleton, has been assembled through a series of oxidative homocoupling events using modified Eglinton–Glaser–Hay coupling conditions (Cu(OTf)2, TMEDA, CH2Cl2, O2). Final polyyne formation is accomplished through exhaustive elimination with TBAF. They have been, unfortunately, unable to isolate this precious material, but UV–vis spectroscopy and ‘matrix free’ MALDI-TOF mass spectrometry have been used to confirm its identity.
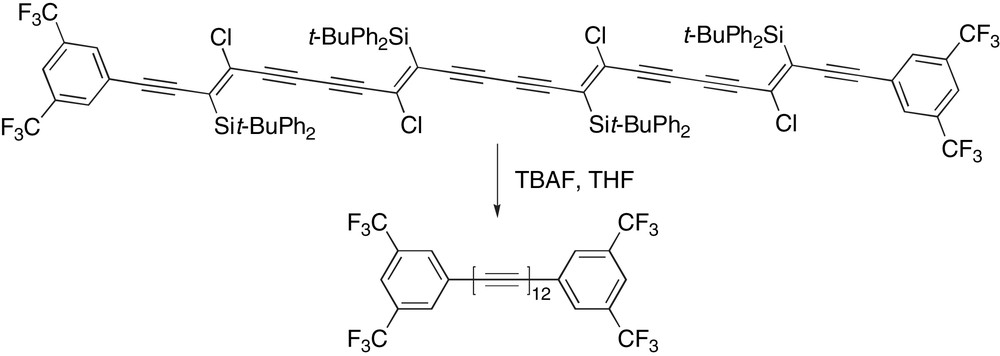
Cox's synthesis of an aryl endcapped dodecayne.
Using the same general protocol as described earlier for metal endcapped heptaynes (Scheme 17), Bruce and coworkers have very recently reported the Pd-catalyzed formation of a dodecayne terminated with tricobalt carbonyl complexes (Scheme 24). This same reaction gave a lesser amount of the corresponding octayne (Table 1, entry 14), the result of oxidative homocoupling of two equivalents of the gold-acetylide. While the dodecayne product is reported to be insufficiently soluble for 13C NMR spectroscopic analysis, the constitution of the product could be confirmed by ESI MS analysis, and it has been characterized as well by 1H NMR, 31P NMR, and IR spectroscopies.
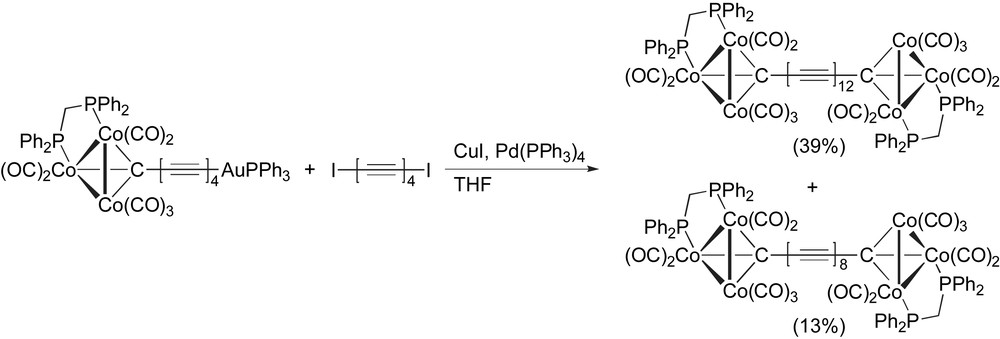
Dodecayne synthesis by Bruce and coworkers.
Using a chain length extension protocol, Gladysz and coworkers have successfully achieved the longest stable polyynes known to date [75]. This achievement is realized as a breakthrough in the field, proving that polyyne molecules of 24 and even 28 (Scheme 25) contiguous sp-hybridized carbons can be synthesized and, perhaps even more importantly, that they can be stable. Most remarkable is the fact that an entire suite of spectroscopic and physical characterizations could be obtained for this series, providing tantalizing insight into some of the possible properties of carbyne.
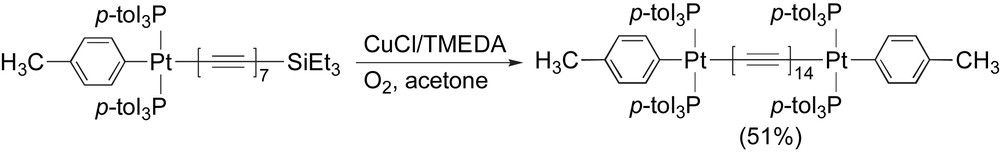
Synthesis of a C28 polyyne by Gladysz and coworkers.
3 Conclusions
Although there have been many examples of polyynes up to and including 12 carbons in length, i.e., hexaynes, there is a significant decrease in examples beyond this length. There have been clever methodologies developed for the synthesis of polyynes beyond 12 carbons but there still seems to be a limit in their stability beyond this length. Through the cunning design by a few groups using endgroups that stabilize long polyynes, it has been shown that this upper barrier of stability can be extended, allowing the synthesis of even longer molecules.
Acknowledgements
This work has been generously supported by the University of Alberta and the Natural Sciences and Engineering Research Council of Canada (NSERC) through the Discovery Grant program. W.A.C. thanks NSERC (PGS-D) and the Alberta Ingenuity Fund for scholarship support.