

1 Introduction
Shape-persistent cyclic molecular structures based on the phenylene, phenylene ethynylene or phenylene butadiynylene backbone have attracted considerable interest during the past 15 years [1]. Shape-persistence means in this context that the time or ensemble average diameter of the cycle is equal to the effective contour length of the molecule divided by π [1b]. As it holds also for linear rigid structures, shape-persistent cyclic structures have to be decorated with side groups that guarantee processability, either from solution or from the melt [2]. However, the attachment of side groups is not a disadvantage but opens the possibility to attach functionality at the inside and/or at the outside of the ring. Groups pointing outwards (extraannular side groups) can be used to control the macrocycle arrangement in two dimensions whereas functional groups that point to the inside (intraannular function groups) can be used to bind appropriate guest molecules (Fig. 1) [3,4].
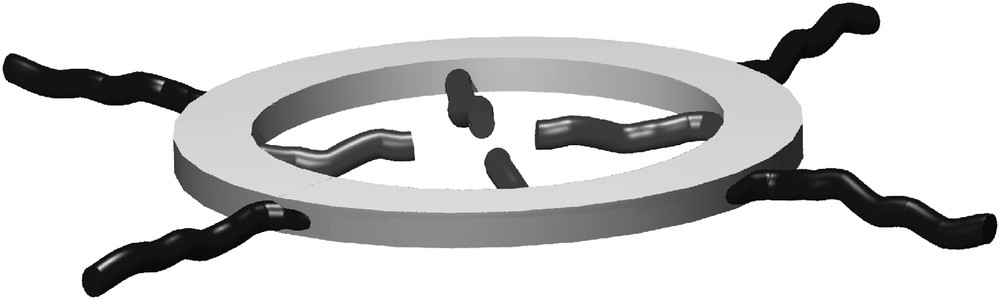
Shape-persistent macrocycle with intraannular and extraannular side groups.
The functionality can also be incorporated to the cyclic structure by an exchange of the phenylenes to heteroaromatics. The synthesis and supramolecular chemistry of several rigid heteroaromatic ethynylene macrocycles have been reported, among these are rings with thiophene, pyridine, bipyridine and terpyridine units, often in combination with other rigid building blocks. While “thiophene rings” are able to bind fullerenes [5], nitrogen containing macrocycles can bind metal ions [6,7] or be easily alkylated yielding charged structures [8]. This shows that heteroaromatic shape-persistent macrocycles are valuable compounds for the creation of complex supramolecular assemblies.
Here, we present a simple synthesis of two shape-persistent macrocycles with intraannular pyridine groups in which the heteroaromatic ring part is synthesized from a pyrylium salt. Triaryl pyrylium salts are rather easily availabe from acetophenones and benzaldehydes which can contain a variety of different functional groups, including halogens [9]. That offers the possibility to build up halogenated oligophenylenes by simple transformation of the pyrylium ring to the corresponding benzene or heterocycle without the use of protective groups.
2 Results and discussion
Scheme 1 shows the synthesis of the pyrylium salts 3 and 6 and the iodophenyl pyridine compounds 4 and 7. Compounds 3 and 4 have been described before and synthetic details are given in the literature [8]. It has to be mentioned that the transformation of 3 and 4 by reacting the pyrylium salt with sodium 4-pyridylacetate according to the procedure reported by Zimmermann and Fischer [10] offers a simple synthetic approach to macrocycle building blocks that contain aryliodides, which can undergo a variety of transition metal catalyzed coupling reactions, and pyridine rings, that are sterically not blocked so that they can be easily alkylated to charged pyridinium rings. On the other hand, triaryl pyrylinium salts can be transformed to triaryl pyridines by reaction with aqueous ammonia. Using this transformation, 7 is obtained from 6 in 81% yield.
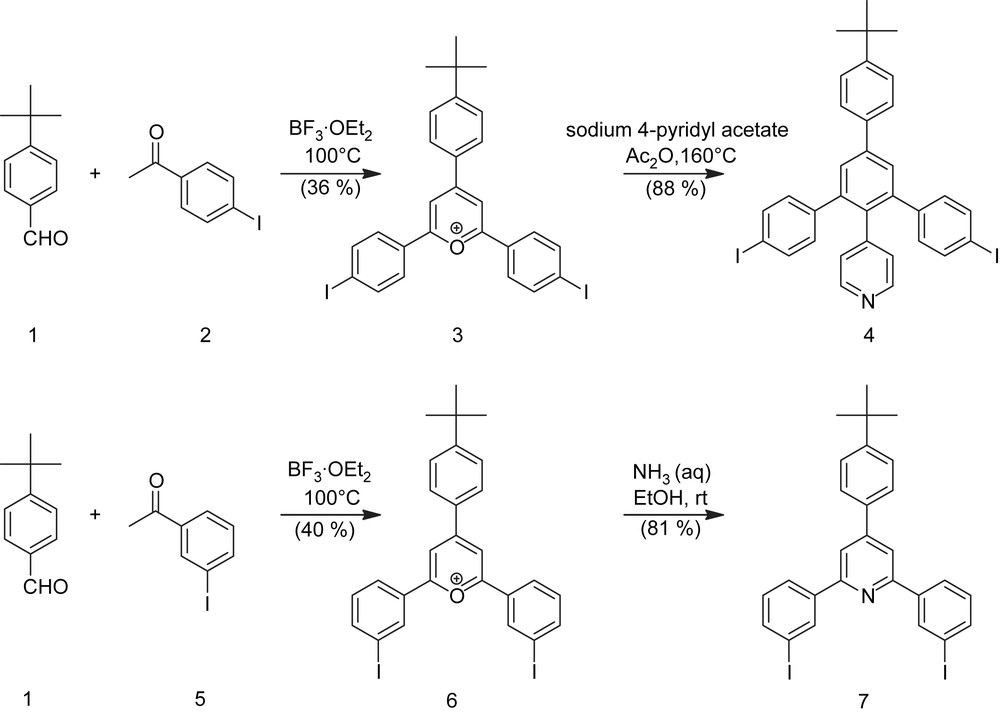
Synthesis of iodo pyridyl compounds.
Compounds 4 and 7 are used as cornerpieces in shape-persistent macrocycles. Reaction of 4 with 8 [11] and removal of the triisopropylsilyl (TIPS) groups by fluoride-induced desilylation gave the “half-ring” 10 (Scheme 2). Similarly, 7 was treated with TIPS-acetylene and subsequently desilylated with tetrabutylammonium fluoride (TBAF) to give the half-ring 13.
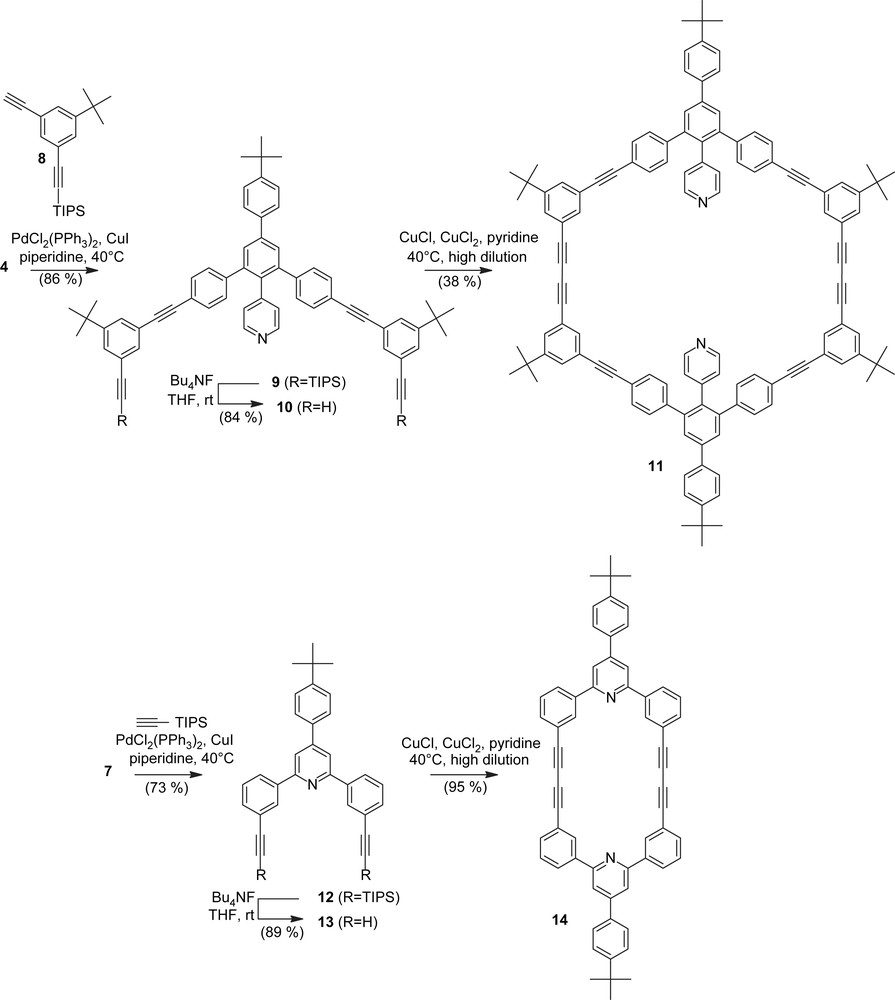
Synthesis of the macrocycles 11 and 14.
Cyclodimerization of the bisacetylenic “half-rings” 10 and 13 was performed under pseudo high-dilution conditions by slow addition of the solution of the bisacetylene in pyridine to a suspension of CuCl2/CuCl in the same solvent [12]. After workup, the cyclodimers were obtained in 38% (11) and 95% (14) yield, respectively. The nearly quantitative yield of 14 is remarkable and cyclization studies with macrocycles of a similar structure are in progress [1b].
All macrocycles were analysed by NMR, mass spectrometry and gel permeation chromatography (GPC). The latter method is necessary to estimate the purity of the compounds. While for 11 only one peak could be observed by GPC, 14 is only of >98% purity and contains a small amount of a side product with a molecular weight of approximately 700 g mol−1 (according to GPC analysis). Attempts to remove this impurity by digesting 14 with CH2Cl2 as well as THF were not successful.
Recrystallization of 11 from pyridine gave single crystals suitable for x-ray analysis. Compound 11 crystallizes as a solvate with 8 molecules pyridine per ring. The molecular structure of 11 is depicted in Fig. 2 as an ORTEP representation. The macrocyclic backbone exhibits normal thermal displacement parameters; those of the tert-butyl group and even more, of the solvent, have larger displacement parameters. The distance between opposite diacetylene bonds measures about 20.9 Å, the nitrogens N1 and N1′ are 7.4 Å apart.
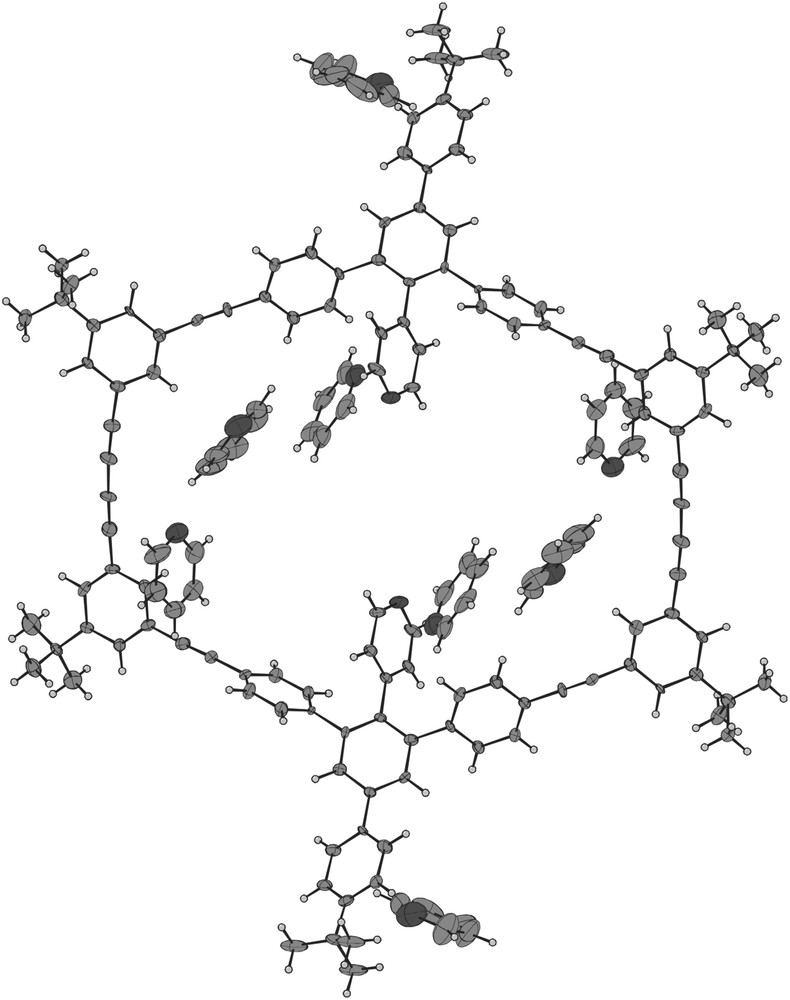
ORTEP plot of the crystal structure of 11, thermal ellipsoids are presented at a 50% probability level.
Fig. 3 shows a space filling model of 11 in the crystal (solvent molecules are not shown for clarity). The macrocycle is not planar, but adopts a cyclohexane-like chair structure with one bulky t-butyl phenyl group pointing above and one t-butyl phenyl group pointing below the ring plane, respectively. The view on two neighbouring macrocycles indicates that the t-butyl phenyl group of one ring points into the cavity of a neighbouring ring, thus filling a part of the internal ring void. The remaining free space is occupied by the solvent as shown in Fig. 4. There exist no specific interaction between the solvent molecules and the macrocycles. This, and the fact that the pyridine is not trapped in cavities but in a channel-like structure, explains the high sensitivity of the crystals when removed from the mother liquor due to the loss of solvent.
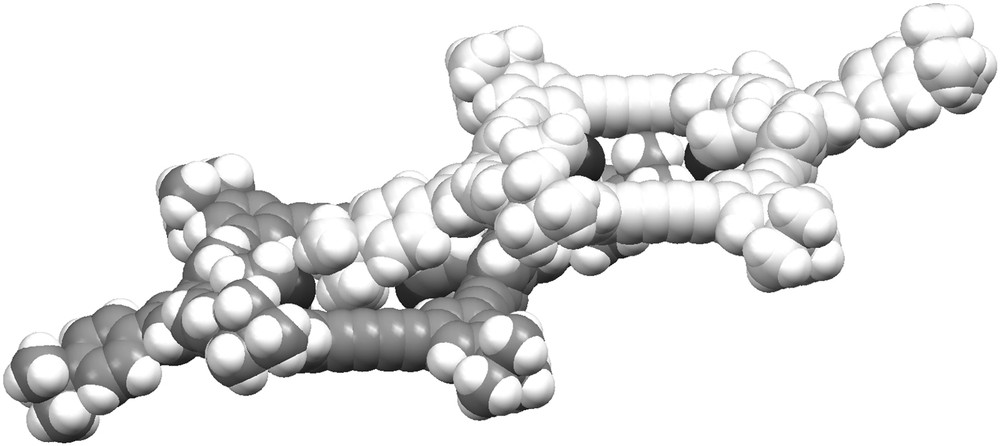
Space filling presentation of two molecules of 11 in the crystal (different shading for clarity; solvent molecules not shown).
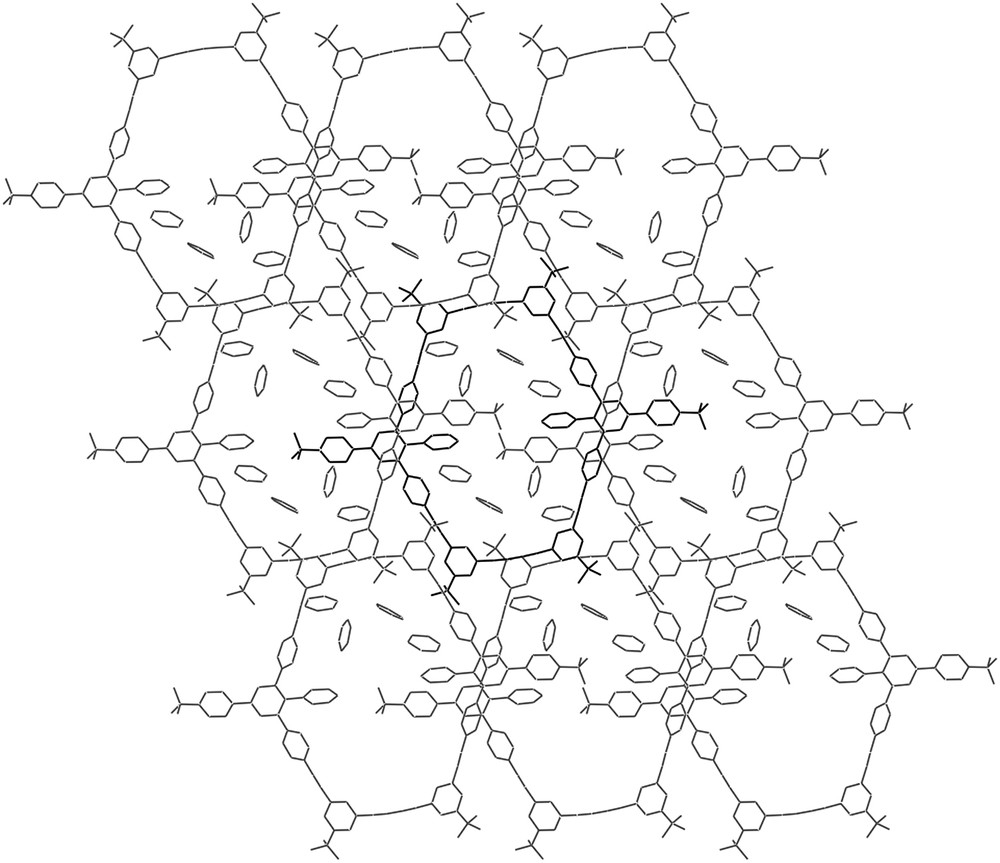
X-ray structure of 11 (view along a). The internal ring void is occupied by the tert-butyl phenyl groups of adjacent macrocycles and by the solvent (one macrocycle is shaded differently fro clarity).
In summary, we have shown that pyrylium salts are for several reasons valuable starting materials for shape-persistent macrocycles. They are accessible in large scale from commercially available starting materials, they can contain halogens necessary for subsequent transition metal catalyzed coupling reactions and they can be transformed either to functionalized aromatic or, as depicted here, to functionalized heteroaromatics in which the pyridine ring is either sterically hindered or not.
3 Experimental
3.1 General
Reactions requiring an inert gas atmosphere were conducted under argon, and the glassware was oven-dried (140 °C). THF was distilled from sodium prior to use. Piperidine and pyridine were distilled over CaH2 and stored under argon. Commercially available chemicals were used as received. Thin-layer chromatography was performed on aluminum plates precoated with Merck 5735 silica gel 60 F254. Column chromatography was performed with Merck silica gel 60 (230–400 mesh). Radial chromatography was performed on a chromatotron (Harrison research) using silica gel/gypsum including fluorescence indicator (Merck) as stationary phase. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX 400 or DRX 500 spectrometer (400 and 500 MHz for 1H). Chemical shifts are given in parts per million, referenced to residual proton resonances of the solvents. Gel permeation chromatograms were measured in THF (flow rate 1 mL min−1) at room temperature, using a combination of either two (porosity 103, 105) or four (porosity 102,103, 105, 106) styragel columns and an UV detector operating at λ = 254 nm. The molecular weight was obtained from polystyrene calibrated SEC columns. The matrix-assisted laser desorption ionization time-of-flight (MALDI TOF) mass spectroscopy measurements were carried out on a Bruker reflex spectrometer. FAB and EI mass spectra were recorded on a Finnigan MAT 90 or MAT 95 XL machine. Compounds 3, 4 and 8 were prepared according to the procedure described previously [8,11].
3.2 Compound 6
Under an argon atmosphere, BF3·OEt2 (2 mL, 16 mmol) was added to a mixture of 4-tert-butylbenzaldehyde (0.65 g, 4.0 mmol) and 3-iodoacetophenone (2.0 g, 8.1 mmol) without the use of further solvent. The mixture was stirred at 100 °C for 3 h. After cooling to room temperature, the viscous red-brown precipitate was filtered off and washed with Et2O and dried in vacuum. Compound 6 (1.13 g, 40%) was obtained as a yellow powder and used as received; 1H NMR (DMSO-d6): δ 1.39 (s, 9H), 7.57 (t, J = 7.90 Hz, 2H), 7.79 (d, J = 8.52 Hz, 2H), 8.20 (d, J = 7.84 Hz, 2H), 8.49 (d, J = 8.08 Hz, 2H), 8.56 (d, J = 8.54 Hz, 2H), 8.91 (s, 2H), 9.17 (s, 2H) ppm; 13C NMR (400 MHz, DMSO-d6) δ 30.68, 35.39, 96.06, 115.64, 126.98, 127.91, 129.76, 130.49, 131.19, 131.82, 136.75, 143.20, 159.63, 165.25, 168.46 ppm.
3.3 Compound 7
Under an argon atmosphere, an aqueous NH3 solution (25%, 5 mL) was added to a suspension of 6 (1.13 g, 1.6 mmol) in EtOH (10 mL). The mixture was stirred at rt for 5 min, and then poured into water and CH2Cl2. The organic layer was separated, washed with water, acetic acid (10%), water, and brine. After drying over MgSO4 and evaporation of the solvent, the crude product was purified by column chromatography (silica gel, CH2Cl2/petroleum ether = 1:1; Rf 0.8). Compound 7 was obtained as a foamy colorless solid (0.80 g, 81%). 1H NMR (CDCl3): δ 1.32 (s, 9H), 7.20–7.16 (m, 2H), 7.49 (d, J = 8.45 Hz, 2H), 7.61 (d, J = 8.04 Hz, 2H), 7.71 (d, J = 7.85 Hz, 2H), 7.77 (s, 2H), 8.06 (d, J = 7.88 Hz, 2H), 8.42–8.41 (m, 2H) ppm; 13C NMR (CDCl3): δ 31.36, 34.84, 94.82, 117.60, 126.22, 126.44, 126.93, 130.48, 135.60, 136.60, 138.05, 141.55, 150.54, 152.70, 156.01 ppm; MS (EI, m/z): 614.8 (M+).
3.4 Compound 9
A solution of 8 (0.87 g, 2.56 mmol) in piperidine (5 mL) was added to a solution of 4 (0.74 g, 1.07 mmol), PdCl2(PPh3)2 (50 mg), triphenyl phosphine (50 mg), and CuI (25 mg) in piperidine (20 mL) and heated to 40 °C for 2.5 h. The mixture was cooled to room temperature and diethyl ether and water were added. The organic phase was separated and washed with acetic acid (10%), water, and brine. After drying over MgSO4 and evaporation of the solvent, the crude product was purified by column chromatography (silica gel, CH2Cl2/petroleum ether = 3:5; Rf 0.4). Compound 9 (0.98 g, 83%) was obtained as a white powder. 1H NMR (CD2Cl2): δ 1.02 (s, 42H), 1.21 (s, 18H), 1.25 (s, 9H), 6.84 (d, J = 5.65 Hz, 2H), 7.13 (d, J = 8.48 Hz, 4H), 7.39 (d, J = 8.48 Hz, 4H), 7.45–7.46 (m, 4H), 7.51–7.53 (m, 4H), 7.67 (d, J = 8.27 Hz, 2H), 7.72 (s, 2H), 8.25 (d, J = 5.65 Hz, 2H) ppm; 13C NMR (CD2Cl2): δ 11.40, 18.50, 30.88, 31.13, 34.59, 34.65, 88.97, 89.52, 90.73, 100.01, 106.69, 121.61, 122.99, 123.51, 126.06, 126.67, 126.79, 128.21, 128.93, 130.04, 131.17, 132.19, 134.57, 136.89, 137.92, 141.04, 141.38, 141.68, 147.38, 149.03, 151.85 ppm; GPC (THF): single peak (Mw = 1430 g mol−1); MS (FD, m/z): 1113.7 (M+).
3.5 Compound 10
A solution of Bu4NF in THF (1.0 M; 3.0 mL, 3.0 mmol) was added to a solution of 9 (1.58 g, 1.42 mmol) in THF (25 mL). The mixture was stirred at rt for 2 h and then poured into ether and water. The organic layer was separated and washed with water and brine. After drying over MgSO4 and evaporation of the solvent the residue was purified by radial chromatography (silica gel, CH2Cl2/petroleum ether = 1:2). The compound was additionally recrystallized from CH2Cl2/petroleum (1:5) (10 mL) to give 10 as a slightly yellow solid (0.95 g, 84%) (CH2Cl2/petroleum ether = 5:3; Rf 0.64). 1H NMR (CD2Cl2): δ 1.32 (s, 18H), 1.37 (s, 9H), 3.13 (s, 2H), 6.84 (d, J = 5.58 Hz, 2H), 7.14 (d, J = 8.48 Hz, 4H), 7.39 (d, J = 8.48 Hz, 4H), 7.45 (t, J = 1.86 Hz, 2H), 7.50 (t, J = 1.86 Hz, 2H), 7.53 (d, J = 8.69 Hz, 2H), 7.55 (t, J = 1.86 Hz, 2H), 7.67 (d, J = 8.69 Hz, 2H), 7.72 (s, 2H), 8.26 (d, J = 5.58 Hz, 2H) ppm; 13C NMR (CD2Cl2): δ 30.85, 31.12, 34.58, 34.67, 77.09, 83.19, 89.10, 89.34, 121.57, 122.09, 122.97, 123.16, 126.06, 126.68, 126.79, 128.30, 128.42, 130.04, 131.18, 132.04, 135.13, 136.88, 141.05, 141.42, 141.66, 147.55, 149.02, 151.32, 151.98 ppm; GPC (THF): single peak (Mw = 1010 g mol−1); MS (FD, m/z): 800.4 (M+).
3.6 Compound 11
Under an argon atmosphere, a solution of 10 (0.77 g, 0.96 mmol) in pyridine (25 mL) was added to a suspension of CuCl (10.52 g, 106.26 mmol) and CuCl2 (2.05 g, 15.25 mmol) in pyridine (200 mL) at 40 °C over 168 h. After further stirring at rt for 24 h, methylene chloride (200 mL) and water (200 mL) were added, the organic phase was separated and washed with water, aqueous NH3 (25%) solution (until the aqueous phase stayed nearly colorless), water, acetic acid (10%), water, sodium hydroxide (10%), and brine. After drying over MgSO4 the solvent was evaporated to about 20 mL. Methanol (about 100 mL) was added and the precipitate was filtered off. The crude product was purified by radial chromatography (silica gel, CH2Cl2/CHCl3 = 1:3; Rf 0.6). Compound 11 (0.52 g, 68%) was obtained as white powder. 1H NMR (C2D2Cl4): δ 1.33 (s, 36H), 1.39 (s, 18H), 6.81 (d, J = 5.82 Hz, 4H), 7.13 (d, J = 8.32 Hz, 8H), 7.42 (d, J = 8.32 Hz, 8H), 7.51–7.54 (m, 12H), 7.62 (m, 4H), 7.67 (d, J = 8.53 Hz, 4H), 7.74 (s, 4H), 8.29 (d, J = 8.29 Hz, 4H) ppm; 13C NMR (C2D2Cl2): δ 30.94, 31.24, 34.42, 34.57, 81.36, 89.16, 89.48, 121.14, 121.44, 123.08, 125.87, 126.48, 126.71, 127.88, 128.86, 129.07, 129.77, 131.15, 133.70, 134.93, 136.47, 140.84, 140.93, 141.25, 141.43, 147.43, 148.88, 151.00, 151.77 ppm; GPC (THF): single peak (Mw = 1860 g mol−1); MS (FD, m/z): 1596.8 (M+).
3.7 Compound 12
Compound 12 was prepared analogue to 9 from 7 (44.3 mg, 0.072 mmol) and TIPS-acetylene (33 mg, 0.18 mmol), Pd(PPh3)2Cl2 (10 mg), CuI (5 mg) and triphenylphosphine (10 mg) in THF (1 mL) and piperidine (2 mL) at 40 °C for 1 h. The crude product was purified by column chromatography (silica gel, CH2Cl2/petroleum ether = 1:3; Rf 0.5). Compound 7 was obtained as a colorless solid (38 mg, 73%). 1H NMR (CDCl3): δ 1.09 (s, 42H), 1.32 (s, 9H), 7.41–7.37 (m, 2H), 7.51–7.48 (m, 4H), 7.63 (d, J = 8.46 Hz, 2H), 7.80 (s, 2H), 8.11–8.10 (m, 4H) ppm; 13C NMR (CDCl3): δ 11.43, 18.78, 31.37, 34.83, 90.89, 107.08, 117.55, 124.04, 126.19, 126.99, 127.40, 128.71, 130.45, 132.80, 135.92, 139.71, 150.40, 152.52, 156.88 ppm; MS (MALDI TOF, m/z): 724.5 (M+).
3.8 Compound 13
Compound 13 was prepared analogue to 10 from 12 (150 mg, 0.2 mmol) in THF (5 mL) and Bu4NF in THF (1.0 M; 0.5 mL, 0.5 mmol). The crude product was purified by column chromatography (silica gel, CH2Cl2/petroleum ether = 1:3; Rf 0.3). Compound 13 was obtained as a colorless solid (73 mg, 89%). 1H NMR (CDCl3): δ 1.30 (s, 9H), 3.05 (s, 2H), 7.37 (t, J = 7.73 Hz, 2H), 7.48–7.44 (m, 4H), 7.58 (d, J = 8.46 Hz, 2H), 7.78 (s, 2H), 8.11–8.08 (m, 2H), 8.20–8.19 (m, 2H) ppm; 13C NMR (CDCl3): δ 30.98, 34.43, 77.02, 83.37, 116.96, 122.21, 125.79, 126.52, 127.31, 128.45, 130.42, 132.29, 135.33, 139.34, 149.98, 152.18, 156.14 ppm; MS (EI, m/z): 411.2 (M+); GPC (THF): single peak (Mw = 560 g mol−1).
3.9 Compound 14
Compound 14 was prepared analogue to 11 from 13 (120 mg, 0.29 mmol) in pyridine (20 mL) was added to a suspension of CuCl (2.29 g, 23.3 mmol) and CuCl2 (0.58 g, 4.3 mmol) in pyridine (150 mL) at 45 °C over 48 h. The crude product was purified by column chromatography (silica gel, CH2Cl2/petroleum ether = 1:1; Rf 0.45) and subsequent digestion. Compound 11 (112 mg, 95%) was obtained as white powder. 1H NMR (C2D2Cl4): δ 1.43 (s, 18H), 7.55 (t, J = 7.77 Hz, 4H), 7.61 (d, J = 8.41 Hz, 4H), 7.71 (d, J = 6.60 Hz, 4H), 7.76 (d, J = 8.38 Hz, 4H), 8.00 (s, 4H), 8.03 (d, J = 7.93 Hz, 4H), 8.84 (t, J = 1.68 Hz, 4H) ppm; 13C NMR (C2D2Cl4): δ 30.18, 31.13, 74.29, 81.61, 117.08, 122.22, 125.36, 126.67, 127.64, 128.65, 131.54, 132.97, 135.42, 139.19, 150.24, 152.47, 155.66 ppm; MS (MALDI TOF, m/z): 818.4 (M+); GPC (THF): (Mw = 930 g mol−1; >98%).
3.10 X-ray crystallographic analysis of 11
Crystals of 11·8 pyridine were obtained by recrystallization of 11 from pyridine and embedded in inert oil prior to data collection. X-ray diffraction data of 11 were collected on a Nonius KappaCCD diffractometer, working with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structure was solved by the heavy atom method (Patterson) and refined on F with anisotropic temperature factors for all non-H atoms. H atoms were refined with fixed temperature factors in the riding mode. Selected crystallographic data: 11 × 8 pyridine (T = 120 K): triclinic, P-1, a = 9.1700(4), b = 16.9372(7), c = 22.0968(8) Å, α = 71.2253(14), β = 84.6179(12), γ = 82.1400(13)°, V = 3214.3(2) Å3, Z = 2, Dx = 1.151 g cm−3, 71301 reflections measured, 14319 unique reflections (Rint = 0.089), 8723 reflections observed (I > 3σ(I)); the structure was solved by direct methods (Shelxs) and refined by full matrix least-squares analyses on F with anisotropic temperature factors for C, O, Cl. The H atoms were included with fixed isotropic temperature factors in the riding mode. R = 0.0609, Rw = 0.0711. CCDC-689277 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223 336033; or email: deposit@ccdc.cam.ac.uk.
Acknowledgement
Financial support by the Deutsche Forschungsgemeinschaft (SFB 624) is gratefully acknowledged.