

1 Introduction
Highly unsaturated conjugated compounds have attracted substantial interest from synthetic, theoretical, and materials chemists in recent years [1]. Specifically, highly delocalized alkyne-containing systems [2] have elicited most of this attention for several reasons. First, the development of new synthetic procedures based on organometallic cross-coupling reactions [3] has provided access to a wide variety of compounds whose preparation was not previously possible. Second, many of these carbon-rich systems and macrocycles exhibit important materials properties such as nonlinear optical (NLO) activity [4], liquid crystalline behavior [5], and molecular switching [6]. Recent developments have shown that dehydrobenzoannulenes [7] and other related phenyl–acetylene macrocycles [8] are useful precursors to a number of carbon-rich polymeric systems, such as molecular tubes [9], ladder polymers [10], and novel allotropes of carbon [11]. Thus, it is essential to have easy access to these high carbon content molecules in sufficient quantities via straightforward synthetic methods if their technological potential is to be explored and eventually utilized.
We have been studying acetylene-rich dehydrobenzoannulenes (DBAs) [7,12] with the goal of examining their diverse chemical and physical properties. Our group has largely focused on the development of new or improved synthetic techniques for the assembly of such macrocycles. Our current methodologies allow us to introduce donor and/or acceptor functionalities on the phenyl rings of the DBA in a judicious manner, thus effectively “tuning” the optoelectronic properties of the annulene [12a]. To explore further DBA functionalization for detailed structure–property relationship studies, we chose to incorporate thiophene moieties onto the dehydroannulene skeleton [13]. The choice of thiophene as the fused aromatic ring was inspired by this moiety's materials applicability such as the chemical/electrochemical polymerizability of thiophene, the ability to form two-dimensional π-systems useful for electronics and photonics, the easier polarizability of thiophene, and the interaction among the individual macrocycles due to the lone pairs on the sulfur in each thiophene ring [14]. With respect to other heteroaromatics, thiophenes in general are easier to manipulate and to functionalize over pyrroles or furans, allowing easy access to tailored thiophene derivatives. Furthermore, by locking the conjugated system into planarity, thiophene-containing macrocycles [14d,e,15] and cyclic thiophene–acetylene hybrids [16–19] have the potential to be more efficient materials because of geometrically enforced π-orbital overlap with increased quinoidal character of the delocalized system and thus a decreased optical band gap [20].
Prior studies on thieno-fused dehydroannulenes such as 1 [17] and 2 [18] (Fig. 1) relied on metal-mediated intermolecular couplings for macrocycle construction. The structures of the starting materials, however, limited the resultant molecules to either Cnh- or Dnh-symmetry. In 2000 we reported the preparation of dehydrothieno[18]annulenes ([18]DTAs, e.g., 3) having lower symmetries (C2v, Cs) [13a,b]. While these macrocycles could be assembled in a systematic, stepwise manner, poor stability of the thienyldiyne intermediates necessitated exploration of other topologies. More recently, we described the synthesis and optoelectronic properties of a series of thiophene-containing [14]DBA/DTA hybrids (e.g., 4, henceforth [14]DBTAs) and the corresponding all-thiophene containing [14]DTAs (e.g., 5) [13c]. That study showed that subtle changes in molecule symmetry can “tune” the properties of annulenes. For example, the UV–vis spectra of this class of macrocycles showed that the large transitions remain essentially the same, but displayed distinct differences in the low-energy vibronic region. Expanding on our success with [15]DBAs [12a,c,e] and our further understanding of thieno-fused annulenes, this report details the synthesis and characterization (thermal, optical, electro-chemical, computational) of [15]DBTA hybrids 6–9 (Fig. 2) and [15]DPTA (dehydropyridothienoannulene) hybrids 10 [12c] and 11.
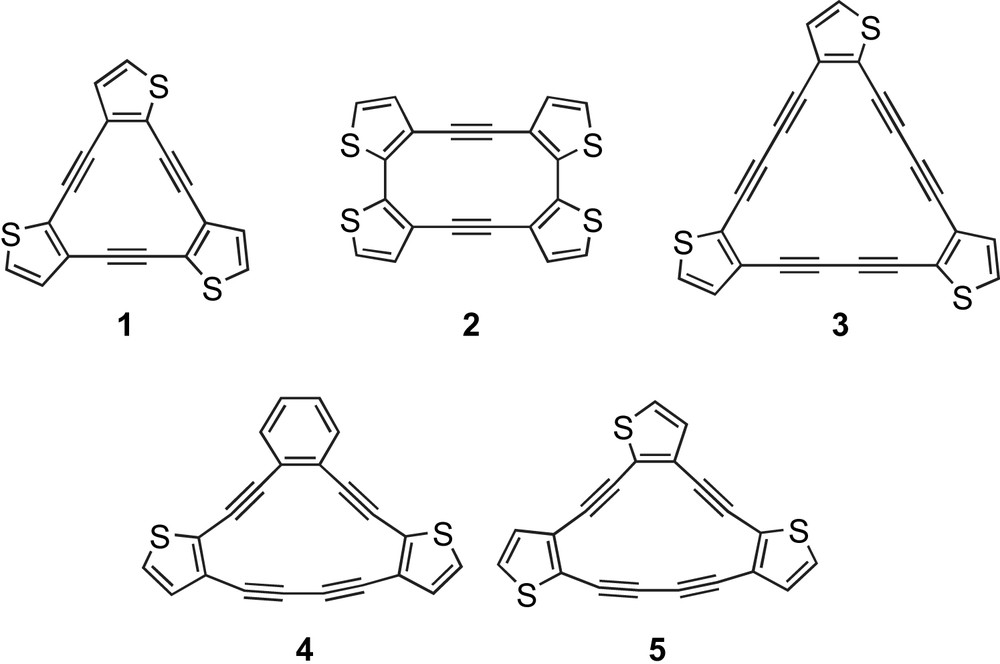
Examples of known thieno-fused dehydroannulenes.
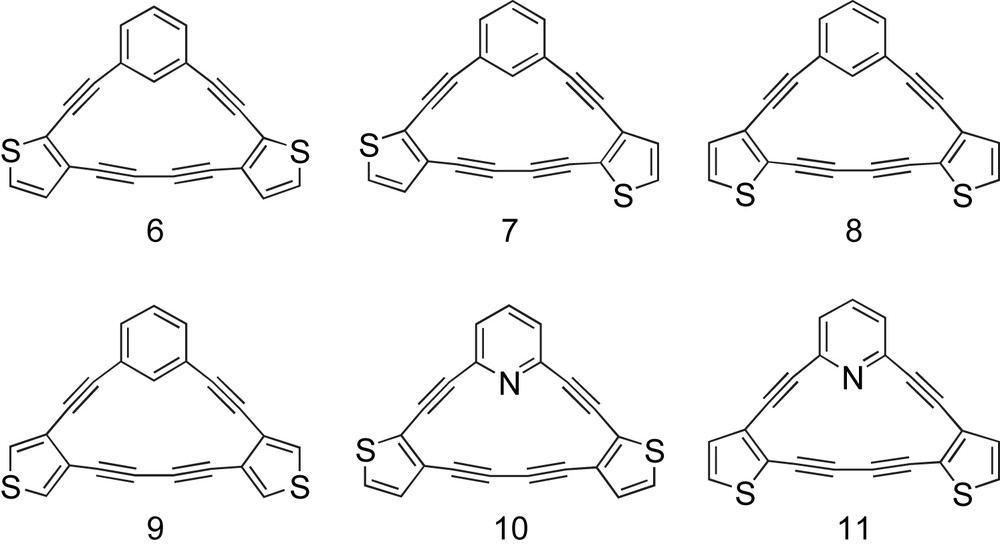
Target thieno-fused dehydro[15]annulenes 6–11.
2 Results and discussion
2.1 Macrocycle synthesis
All six macrocycles were constructed in a convergent fashion from the synthons given in Fig. 3. Dihaloarenes 12–14 are commercially available, whereas diynes 15–17 were prepared starting from the appropriate dihalothiophenes using our previously reported syntheses [12c,13c].
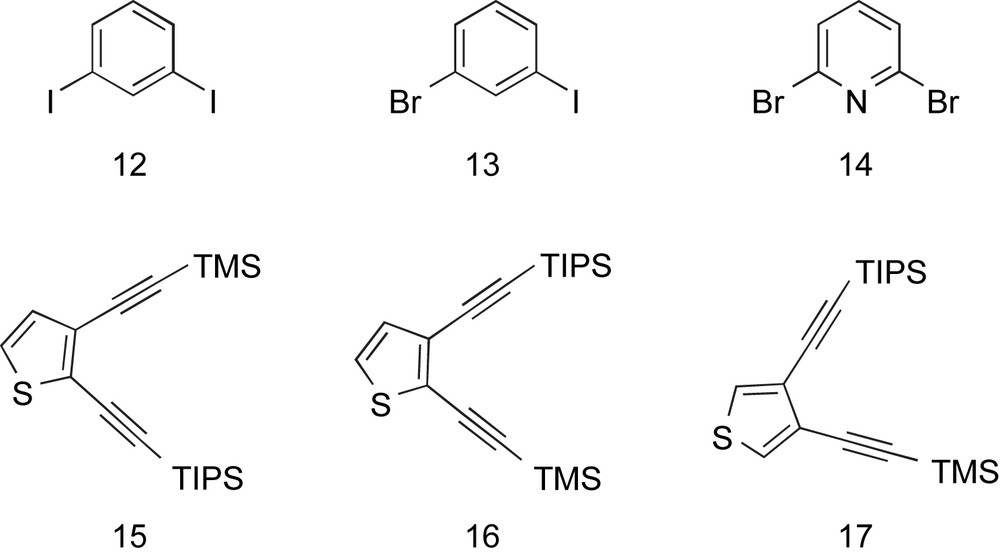
Building blocks 12–17.
An example of symmetrical macrocycle synthesis is given in Scheme 1. Selective removal of the TMS group in diyne 16 with K2CO3 in MeOH/THF followed by cross-coupling to 1,3-diiodobenzene (12) afforded precursor 18 (pre-6) in 86% yield. Desilylation with TBAF and cyclization using modified Glaser–Eglinton conditions generated hybrid 6 in 55% yield.
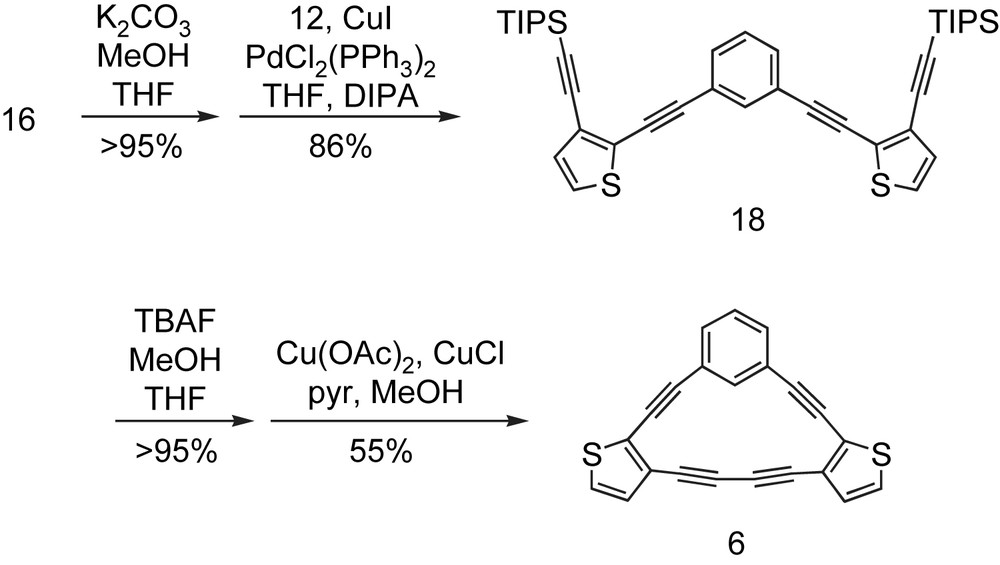
Synthesis of ‘symmetrical’ [15]DBTA 6.
Preparation of the only unsymmetrical macrocycle, 7, is shown in Scheme 2. Selective desilylation of diyne 15 as described above and then cross-coupling to 1-bromo-3-iodobenzene (13) yielded 19, which proved to be moderately sensitive to silica. After rapid purification, compound 19 was then cross-coupled to monodesilated 16 to furnish precursor 20 (pre-7), which was fully characterized. Desilylation and cyclization afforded 7 in 29% yield. A summary of the various syntheses is given in Table 1.
Synthesis of ‘unsymmetrical’ [15]DBTA 7.
Synthesis and yields of macrocycles 6–11.
| Haloarene | “Equivalents” of diyne | Precursor, yield | Annulene, yield | ||
| 15 | 16 | 17 | |||
| 12 | 2 | pre-6 (18), 86% | 6, 55% | ||
| 13 | 1 (second) | 1 (first) | pre-7 (20), 67% | 7, 29% | |
| 12 | 2 | pre-8, 47% | 8, 50% | ||
| 12 | 2 | pre-9, 79% | 9, 53% | ||
| 14 | 2 | pre-10, 74%a | 10, 73%a | ||
| 14 | 2 | pre-11, 67% | 11, 38% |
a Ref. [12c].
As with the previously reported [14]DBTAs, the thieno-fused [15]annulenes are only moderately stable during workup and are sensitive to acid, light, and silica, which accounts for the lower than normal cyclization yields. Workups were most successful when avoiding halogenated solvents and utilizing fast chromatographic methods (chromatotron or flash chromatography). NMR spectroscopic characterization of the annulenes was accomplished primarily in THF-d8 for both stability and solubility. In general, those molecules with UV–vis absorptions past 400 nm and/or where both sulfurs are oriented ‘down’ (sulfur α to the diacetylene linkage), as is the case with 8 and 11, showed the greatest sensitivity. Most could be stored in solution or in the solid state for several weeks if kept cold and dark sealed under argon. The most difficult macrocycle to work with was 9, which was found to decompose gradually over 2–3 d. The instability is likely due to the more exposed α-positions of the 3,4-fused thiophenes, which have been shown to affect the integrity of samples [13c,21].
Thermal analysis of 6–9 by DSC revealed broad exotherms with onset after 200 °C and maxima ca. 225 °C, with decomposition above 300 °C. Thermal scans of pyridine macrocycles 10 and 11, however, showed larger and sharper exotherms between 175 and 200 °C. Nonetheless, these data as a whole suggest random, disordered polymerizations that yield black amorphous materials.
2.2 Spectroscopic properties
NMR spectroscopic analysis of the [15]DBTAs shows only one significant difference between the macrocycles and their respective precursors: upon cyclization the triplet signal for the intraannular arene proton shifts from a range of 7.7–7.8 to 8.5–8.9 ppm. This large change is readily attributed to an increased anisotropic deshielding effect of the transannular triple bonds [12a,c,e,22]; otherwise, the NMR data for the [15]annulenes are unremarkable.
Compared to the [14]DBTAs, the peaks in the electronic spectra of the [15]DBTAs (Fig. 4a) are bathochromically shifted, some up to 40 nm. For example, [14]DBTA 4 has its main peak at 329 nm with two low energy absorptions at 363 and 373 nm and cut-off by 390 nm. The analogous [15]DBTA, 6, has a main peak at 337 nm and cut-off around 450 nm, with no readily discernable low energy bands. These bands do become apparent and grow in as thiophene orientation relative to the annulene ring changes from ‘up’ to ‘down’ (6 → 7 → 8), similar to the [14]DBTAs [13c]. In 8 the low energy bands are at 410 and 448 nm with a cut-off of ca. 465 nm. In comparison, the analogous bands in the ‘down’ [14]DBTA are 376 and 408 nm and a cut-off of ca. 425 nm. Compound 9 has an appreciable low energy absorption band at 349 nm and overall displays different features than 6–8. This reflects the decreased conjugation in the macrocycle due to 3,4-fusion of the two thiophene moieties. Once again, the 15-membered ring of 9 affords a band that is lower in energy than the corresponding band in the 3,4-fused [14]DBTA (339 nm), though the magnitude of this difference is smaller than the 2,3-fused systems.
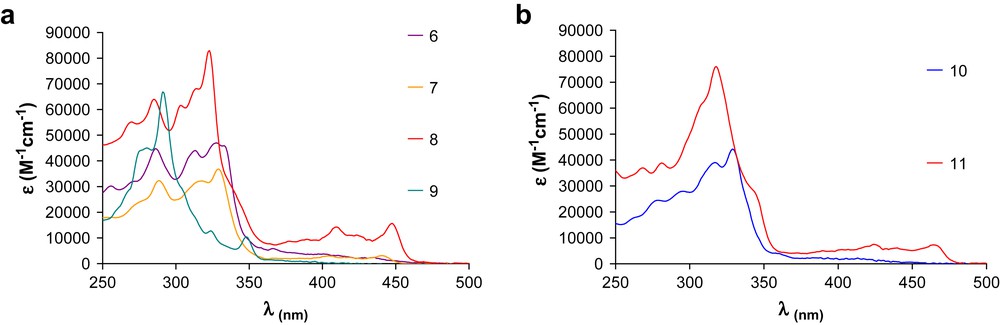
UV–vis absorption spectra of (a) 6–9 and (b) 10–11. All spectra recorded in CH2Cl2 at analyte concentrations of 15–25 μM.
Replacing the benzene with a pyridine unit (Fig. 4B) yields only minor changes to the electronic absorption data. Compounds 6 and 10 have nearly identical electronic absorption spectra and thus identical band gaps, whereas [15]DPTA 11 red shifts to 465 nm compared to 448 nm for 8. This 17 nm difference not only demonstrates that the orientation of the sulfurs plays an important role in the electronic properties of this class of compounds, but also suggests that direct conjugation of the sulfur to the benzene (or pyridine) is a driving factor in lowering the energy of the transition states. The addition of the electron-withdrawing N-atom likely allows for a more charge separated excited state (vide infra). None of the reported macrocycles, however, displayed any appreciable solvatochromism.
The [15]DBTAs and [15]DPTAs do exhibit modest emissive properties (quantum yields of 0.03–0.17), unlike the [14]DBTAs (quantum yields < 0.01). The emissive properties of cross-conjugated systems over directly conjugated systems are a phenomenon often observed elsewhere [23] and have been observed in many of our previous studies [12a,e,24]. Emission spectra and quantum yields were taken in both toluene and dichloromethane, and are summarized along with the absorption data and Stokes shifts in Table 2. [15]DPTA 11 is the only system to exhibit appreciable solvatochromism in the emission spectrum with the emission peak shifting from 471 nm to 524 nm when changing solvent from toluene to CH2Cl2.
Photophysical parameters of [15]annulenes 6–11.
| Lowest energy Abs. λmax (cut-off) [nm] | Emission energy [nm] (CH2Cl2, PhMe) | Φf (CH2Cl2, PhMe) | Stokes shift, (CH2Cl2, PhMe) [cm−1] | |
| 6 | 366 (450) | 443, 447 | 0.11, 0.17 | 4975, 5028 |
| 7 | 440 (450) | 449, 449 | 0.03, 0.03 | 507, 404 |
| 8 | 448 (460) | 456, 455 | 0.06, 0.05 | 392, 244 |
| 9 | 349 (360) | 363, 363 | 0.04, 0.05 | 1023, 1105 |
| 10 | 363 (460) | 512, 510 | 0.04, 0.05 | 8017, 7789 |
| 11 | 465 (480) | 524, 471 | 0.05, 0.06 | 2421, 228 |
2.3 Electrochemistry
Cyclic voltammograms were obtained of the [15]annulenes using a Pt disk electrode, Ag wire reference, and Pt swirl auxiliary in CH2Cl2 solutions of Bu4NPF6 and referenced to ferrocene. Electrochemical band gap values for compounds 6–8, 10, and 11 were discerned using previously described methods [13c,25]. Table 3 lists the oxidation and reduction onsets and the electrochemical band gap, along with the optically estimated and DFT calculated band gap values. Compound 9 was not tested, as it was too difficult to handle and thus garner reliable CV information. The CVs showed irreversible oxidation peaks and multiple irreversible and sometimes quasi-irreversible reduction waves. In certain instances, however, multiple oxidations were found to occur. This could be due to an increased HOMO energy, allowing more electron transfer reactions to happen outside the solvent window. As was the case with the [14]annulenes, no polymer films were deposited on the electrode. The electrochemistry results match the relative trends observed in the UV–vis absorption data. For example, sulfurs ‘up’ macrocycles have wider band gaps (e.g., 6, 2.85 eV) than sulfurs ‘down’ (e.g., 8, 2.70 eV), in agreement with the [14]DBTA series [13c]. Similarly, the optical and electrochemical band gaps of the [15]DPTAs are larger than those of the corresponding [15]DBTAs.
Electrochemical data and band gaps.
| Compound | Epa (ox) (vs. Fc) [V] | Epa (red) (vs. Fc) [V] | Electrochemical band gap [eV] | Optical band gap [eV] | DFT calculated band gap [eV] |
| 6 | 1.14 | −1.71 | 2.85 | 3.39 | 3.18 |
| 7 | 1.14 | −1.64 | 2.78 | 2.82 | 3.09 |
| 8 | 1.13 | −1.57 | 2.70 | 2.53 | 3.02 |
| 9 | – | – | – | 3.55 | 4.03 |
| 10 | 1.20 | −1.86 | 3.06 | 3.42 | 3.06 |
| 11 | 0.97 | −1.76 | 2.73 | 2.66 | 2.93 |
2.4 Computational results
DFT calculations (B3LYP/6-311G∗∗) [26] were performed on both the [15]DBTAs and [15]DPTAs. The computed HOMO/LUMO gaps are also given in Table 3, and show excellent relative agreement with the empirical trends. For instance, the largest band gap, [15]DBTA 6 (2.85–3.39 eV), corresponds well to the calculated value (3.18 eV). In most cases the DFT computed energies are larger than the experimentally determined ones, as was found for all of the [14]DBTAs and [14]DTAs [13c]. The two exceptions in the current study are sulfurs ‘up’ annulenes 6 and 10. Given their UV–vis spectra in Fig. 4, we can easily attribute this difference to unresolved low energy peaks in the data.
The computed HOMO/LUMO plots for 6–11 are given in Fig. 5. These show little if any difference in topology with respect to the sulfur orientation in [15]DBTAs 6–8, similar to what was observed in the [14]annulene series (e.g., 4–5) when 2,3-fused thiophenes were incorporated into the macrocycle [13c]. Compound 9, possessing 3,4-fusion, shows disruption in the continuity of electron delocalization around the macrocycle; however, it is not as dramatic and well-defined as in the plots of the corresponding [14]DBTA [13c]. DPTAs 10 and 11 also show remarkable similarity to DBTAs 6–8 with the exception of some slight additional electron density at C4 of the pyridine.
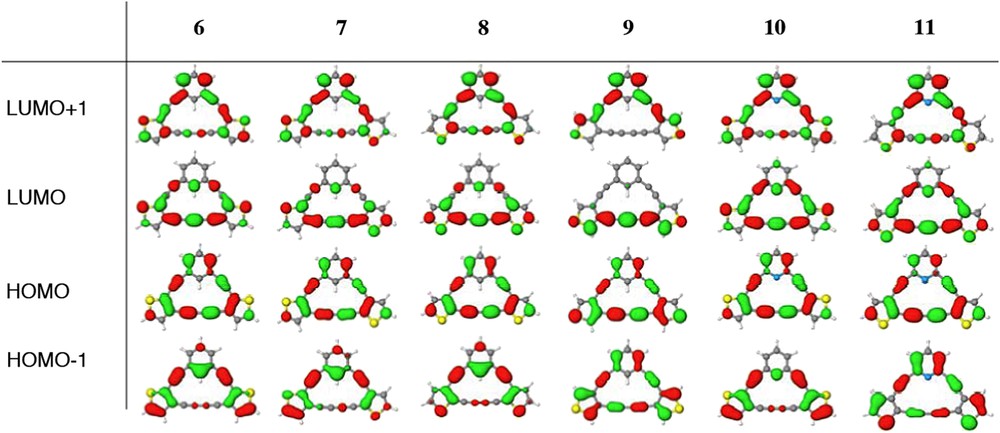
Calculated HOMO and LUMO plots of [15]DBTAs 6–9 and [15]DPTAs 10–11.
The placement of the electron density on the carbon lying between the triple bonds in the LUMOs of 6–8 and 10–11 alludes to perhaps some charge transfer effects. Furthermore it is serendipitous that the lowering of energy of the UV absorption for the pyridine, sulfurs ‘down’ DPTA 11 occurs when the sulfurs are directly in conjugation with the pyridine nitrogen atom, in contrast to 10, when the sulfur is not conjugated with the nitrogen, where no discernable change in the UV–vis spectrum is observed. This could imply that there is some level of charge transfer component to 8 and 11.
3 Conclusions
A short series of [15]DBTA and [15]DPTA hybrids composed of multiple fused thiophenes, benzenes, and/or pyridines were successfully prepared. Planarizing the thiophenes into the [15]annulene scaffold was found to increase orbital overlap even in a cross-conjugated system. Concurrently, the same relative electronic trends were observed in both the 14- and 15-membered macrocycles that are contingent on the orientation of the sulfur. When the sulfur atoms is oriented ‘down’ (as in 8), the absorption bands of the macrocycle red shift and intensify compared to when the sulfurs are oriented ‘up’ (as in 6). When 3,4-fused onto the annulene scaffold, as in 9, the net conjugation around the ring is disrupted by the increased single bond character. Incorporating pyridines in the macrocycle (DPTAs 10–11) can significantly red shift the UV–vis absorptions of the molecule, but only when the sulfurs are oriented down (11). These results as a whole are supported by DFT-B3LYP calculations of the HOMO–LUMO energy gap. Overall, the [15]annulene scaffold offers some benefits over the [14]scaffold with a wider dispersion of UV–vis absorption energies, and increased emissive properties despite the loss of the ‘aromaticity’ found with the 14-membered macrocycle. Electrochemical measurements were performed and again failed to yield polymers typical of thiophenes, most likely due to the increased ‘annulene’ character of the compounds. Future reports will focus on stabilized α-alkylated thiophene targets in order to expand the scope of syntheses of these macrocycles by solubilizing the products and protecting against unwanted degradation.
4 Experimental
4.1 General methods
These are described in Ref. [12a]. Diynes 17 [12c], 18 [13c], and 19 [13c] and annulene 10 [12c] were prepared according to known procedures.
4.2 General procedure for preparation of ‘symmetrical’ annulene precursors
To a solution of diyne (2.5 equiv.) dissolved in MeOH/THF (10:1, 0.05 M) was added K2CO3 (10 equiv.) and the mixture was stirred for 30 min. The solution was filtered and then Et2O was added. The solution was washed successively with H2O, 10% NaCl solution and water, and then dried (MgSO4). Solvent was removed in vacuo and the residue was added to a solution of THF/DIPA (1:1, 0.1 M) and dihaloarene (1 equiv.). The mixture was degassed under Ar for 30 min after which CuI (6 mol%) and Pd(PPh3)4 (3 mol%) were added. The sealed vessel was stirred at room temperature until TLC showed the reaction complete (12 h–2 d). The solvent was removed in vacuo and the crude product chromatographed on silica to afford the annulene precursor.
4.2.1 Precursor to 6 (18/pre-6)
Chromatography on silica (10:1 hexanes:CH2Cl2) furnished 18 (pre-6, 334 mg, 85%) as an orange oil. 1H NMR (300 MHz, CDCl3): δ = 1.20 (s, 42H), 7.08 (d, J = 5.3 Hz, 2H), 7.21 (d, J = 5.3 Hz, 2H), 7.33–7.39 (m, 1H), 7.48–7.54 (m, 2H), 7.69 (t, J = 1.5 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ = 11.6, 19.0, 82.9, 95.7, 96.4, 100.9, 123.5, 126.3, 127.0, 127.6, 128.7, 130.3, 131.5, 134.9; MS (APCI): m/z (%) 650.8 (MH+, 50); IR (NaCl): v = 2149, 2202, 2864, 2891, 2942 cm−1.
4.2.2 Precursor to 8 (pre-8)
Chromatography on silica (10:1 hexanes:CH2Cl2) furnished pre-8 (184 mg, 47%) as an orange oil. 1H NMR (300 MHz, CDCl3): δ = 1.17 (s, 42H), 7.09 (d, J = 5.3 Hz, 2H), 7.16 (d, J = 5.3 Hz, 2H), 7.34–7.39 (m, 1H), 7.47–7.53 (m, 2H), 7.71 (t, J = 1.4 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ = 11.3, 18.7, 84.4, 92.2, 98.2, 100.4, 132.4, 125.8, 126.7, 126.8, 128.2, 129.3, 131.2, 134.8; MS (APCI): m/z (%) 721.3 (MH+ + THF, 100); IR (NaCl): v = 2142, 2213, 2891, 2864, 2942 cm−1.
4.2.3 Precursor to 9 (pre-9)
Chromatography on silica (10:1 hexanes:CH2Cl2) furnished pre-9 (913 mg, 79%) as an orange oil. 1H NMR (300 MHz, CDCl3): δ = 1.07 (s, 42H), 7.41–7.47 (m, 5H), 7.67 (t, J = 1.4 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ = 11.3, 18.7, 83.7, 90.4, 93.2, 100.0, 123.4, 125.0, 125.3, 128.1, 128.2, 129.0, 131.2, 135.0; MS (APCI): m/z (%) 721.3 (MH+ + THF, 100), 791.3 (M+ + 2THF, 10); IR (NaCl): v = 2155, 2219, 2960, 2863, 3109 cm−1.
4.2.4 Precursor to 11 (pre-11)
Chromatography on silica (10:1 hexanes:CH2Cl2) furnished pre-11 (153 mg, 56%) as an orange oil. 1H NMR (300 MHz, CDCl3): δ = 1.34 (s, 42H), 7.11 (d, J = 5.3 Hz, 2H), 7.15 (d, J = 5.3 Hz, 2H), 7.41 (d, J = 7.6 Hz, 2H), 7.63 (t, J = 7.6 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ = 11.2, 18.7, 84.0, 91.7, 97.9, 100.8, 125.9, 126.0, 126.2, 127.9, 129.7, 136.1, 143.7; MS (APCI): m/z (%) 651.9 (MH+, 100); IR (NaCl): v = 2143, 2214, 2942, 3109 cm−1.
4.3 Procedure for preparation of ‘asymmetrical’ annulene precursor 20 (pre-7)
To a solution of diyne 15 (111 mg, 0.311 mmol) dissolved in MeOH (6 ml) and THF (0.5 ml) was added K2CO3 (106 mg, 0.78 mmol) and the mixture was stirred for 30 min. The solution was filtered and then Et2O was added. The solution was washed successively with H2O, 10% NaCl solution and water, and then dried (MgSO4). Solvent was removed in vacuo and the unstable crude material (19) was added to a solution of THF (1.5 ml), DIPA (1.5 ml) and 1-bromo-3-iodobenzene (13, 80 mg, 0.282 mmol). The mixture was degassed under Ar for 30 min after which CuI (3 mg, 0.017 mmol) and Pd(PPh3)4 (10 mg, 0.008 mmol) were added. The sealed vessel was stirred at room temperature until TLC showed the reaction complete (ca. 2 d). The solvent was removed in vacuo and the crude product chromatographed on a short plug silica to afford the halodiyne precursor. This material was redissolved in THF (1.5 ml) and DIPA (1.5 ml), and then diyne 16 (153 mg, 0.429 mmol), desilylated in the manner described above, was added. The solution was degassed under Ar for 30 min and then CuI (4 mg, 0.019 mmol), Pd(PPh3)2Cl2 (7 mg, 0.0086 mmol), and Pd(PPh3)4 (5 mg, 0.0043 mmol) were added. The reaction mixture was transferred to a sealed pressurized tube and heated at 85 °C for 18 h. The solvent was removed in vacuo and the crude product was chromatographed on silica (6:1 hexanes:CH2Cl2) to afford 20 (pre-7, 191 mg, 67%) as a dark orange oil. 1H NMR (300 MHz, CDCl3): δ = 1.14 (s, 42H), 7.04 (d, J = 5.3 Hz, 2H), 7.16 (d, J = 5.3 Hz, 1H), 7.17 (d, J = 5.3 Hz, 1H), 7.28–7.36 (m, 1H), 7.43–7.48 (m, 2H), 7.68 (t, J = 1.5 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ = 11.3, 18.7, 84.5, 84.5, 92.2, 95.4, 96.3, 98.1, 100.5, 100.6, 123.1, 123.5, 125.8, 126.0, 126.8, 126.9, 127.2, 128.3, 129.3, 130.0, 131.0, 131.2, 131.4, 134.7; MS (APCI): m/z (%) 757.4 (M+ + THF + 2H2O, 80); IR (NaCl): v = 2145, 2209, 2891, 2865, 2943 cm−1.
4.4 General procedure for annulene formation
To a solution of annulene precursor (1 equiv.) in THF (0.1 M) was added TBAF (20 equiv., 1.0 M THF). The mixture was stirred for 5 min and then Et2O was added. The solution was washed 4–6 times with brine and then water, and the organic layer was dried (MgSO4). The solution was diluted with Et2O (to ca. 50 ml total volume) and then slowly injected over 8 h into a pyridine solution (0.01 M) of CuCl (10 equiv.) and Cu(OAc)2 (10 equiv.) with stirring at 45 °C. Once TLC showed completion of the reaction, the solvent was removed under reduced pressure and the residue was run through a silica plug (1:1 hexanes:CH2Cl2). The product was further purified on a chromatotron (8:1 hexanes:EtOAc).
4.4.1 [15]DBTA 6
Obtained 6 (8 mg, 55%) as a pale yellow solid. 1H NMR (300 MHz, THF-d8): δ = 7.14 (d, J = 5.3 Hz, 2H), 7.25–7.40 (m, 3H), 7.47 (d, J = 5.3 Hz, 2H), 8.52 (t, J = 1.5 Hz, 1H); 13C NMR (75 MHz, THF-d8): δ = 79.6, 80.0, 87.5, 101.9, 125.1, 127.1, 127.9, 129.5, 129.7, 130.1, 130.4, 146.1; MS (APCI): m/z (%) 406.7 (MH+ + THF, 85); IR (KBr): v = 2105, 2198 cm−1; UV–vis (CH2Cl2) λmax (log ε): 285 (4.67), 327 (4.67), 333 (4.65), 366 (3.77) nm.
4.4.2 [15]DBTA 7
Obtained 7 (10 mg, 29%) as a yellow solid. 1H NMR (300 MHz, THF-d8): δ = 7.05 (d, J = 5.2 Hz, 1H), 7.15 (d, J = 5.2 Hz, 1H), 7.22–7.37 (m, 3H), 7.47 (d, J = 5.2 Hz, 1H), 7.52 (d, J = 5.2 Hz, 1H), 8.51 (bs, 1H); 13C NMR (75 MHz, THF-d8): δ = 78.1, 79.7, 82.4, 83.6, 87.5, 89.4, 98.9, 102.2, 125.3, 125.6, 127.2, 127.5, 128.1 (2), 129.6, 129.8, 130.2, 130.6, 132.4, 146.8; MS (APCI): m/z(%) 406.9 (MH+ + THF, 85); IR (KBr): v = 2130, 2190 cm−1; UV–vis (CH2Cl2) λmax (log ε): 288.0 (4.51), 328 (4.56), 406 (3.46), 441 (3.47) nm.
4.4.3 [15]DBTA 8
Obtained 8 (23 mg, 50%) as a yellow solid. 1H NMR (300 MHz, THF-d8): δ = 7.05 (d, J = 5.3 Hz, 2H), 7.22–7.36 (m, 3H), 7.53 (d, J = 5.3 Hz, 2H), 8.51 (t, J = 1.5 Hz, 1H); 13C NMR (75 MHz, THF-d8): δ = 79.2, 82.4, 88.2, 98.0, 124.6, 126.5, 126.8, 127.1, 128.9, 129.1, 131.4, 146.2; MS (APCI): m/z (%) 407.0 (MH+ + THF, 90); IR (KBr): v = 2173, 2205 cm−1; UV–vis (CH2Cl2) λmax (log ε): 285 (4.81), 303.0 (4.78), 314 (4.83), 323.0 (4.92), 410 (4.15), 448 (4.19) nm.
4.4.4 [15]DBTA 9
Obtained 9 (27 mg, 53%) as an unstable white solid. 1H NMR (300 MHz, THF-d8): δ = 7.27–7.36 (m, 3H), 7.58 (d, J = 2.9 Hz, 2H), 7.82 (d, J = 2.9 Hz, 2H), 8.47 (t, J = 1.5 Hz, 1H); 13C NMR (75 MHz, THF-d8): δ = 77.0, 77.8, 88.2, 94.9, 125.0, 126.2, 126.5, 127.6, 128.1, 130.0, 132.2, 145.3; MS (APCI): m/z (%) 407.0 (MH+ + THF, 90); IR (KBr): v = 2136, 2200 cm−1; UV–vis (CH2Cl2) λmax (log ε): 292 (4.83), 325 (4.16), 349 (4.08) nm.
4.4.5 [15]DBTA 11
Obtained 11 (10 mg, 38%) as a yellow solid. 1H NMR (300 MHz, THF-d8): δ = 6.99 (d, J = 5.2 Hz, 2H), 7.10 (d, J = 7.8 Hz, 2H), 7.45 (d, J = 5.2 Hz, 2H), 7.59 (t, J = 7.8 Hz, 1H); 13C NMR (75 MHz, THF-d8): δ = 78.9, 85.3, 86.3, 99.2, 123.0, 129.2, 129.3, 130.3, 131.9, 138.1, 146.0; MS (APCI): m/z (%) 337.5 (MH+, 100); IR (KBr): v = 2145, 2212 cm−1; UV–vis (CH2Cl2) λmax (log ε): 281 (4.85), 317 (4.87), 345 sh (4.27), 426 (3.87), 464 (3.86) nm.
4.5 Computational methods
Geometry optimizations were performed using the B3LYP [26] functional and 6-31G∗ basis set. HOMO–LUMO gaps and dipole moments were taken from single-point energy calculations which were carried out on the optimized structures using B3LYP/6-311G∗∗, in a solvent modeled using the Cosmo reaction field model [27] with a dielectric corresponding to methylene chloride (ε = 9.08). The set of atomic radii optimized by Stefanovich and Truong [28] was used for the solvation energy calculations. The NWChem (v4.7 and v5.0) [29] program was used for all geometry optimizations and single-point energy calculations. Building and modeling of all compounds were done using the program Ecce v3.2.4 [30].
4.6 Electrochemistry
Electrochemical measurements were taken in a CH2Cl2 solution of 100 mM Bu4NPF6 and 1 mM macrocycle using Pt disc working electrode, Pt wire auxiliary electrode, and silver wire reference. Measurements were then referenced with respect to an internal ferrocene standard.
Acknowledgements
We thank National Science Foundation (CHE-0718242) for financial support.