

In terms of development of environmentally more friendly synthetic organic processes, the search for alternative methodologies such as electrochemical synthesis constitutes interesting targets. As compared to conventional oxidative and reductive processes in organic chemistry, in which the classical chemical oxidants and reductants are often used in stoichiometric amounts, the synthesis via electrochemical methods uses electrons as clean, mild, selective, cheap and environmentally friendly reagents [1].
We have been interested in the development of reductive intramolecular cyclisations using electrochemical methods. Whereas non-reductive cyclisations of unsaturated halides have been widely reported using organometallic catalysis [2–4], conventional reductive cyclisations require the use of stoichiometric amounts of metallic reductants, such as, for example, diiodo samarium [5,6] or tin hydrides (Scheme 1) [7,8].
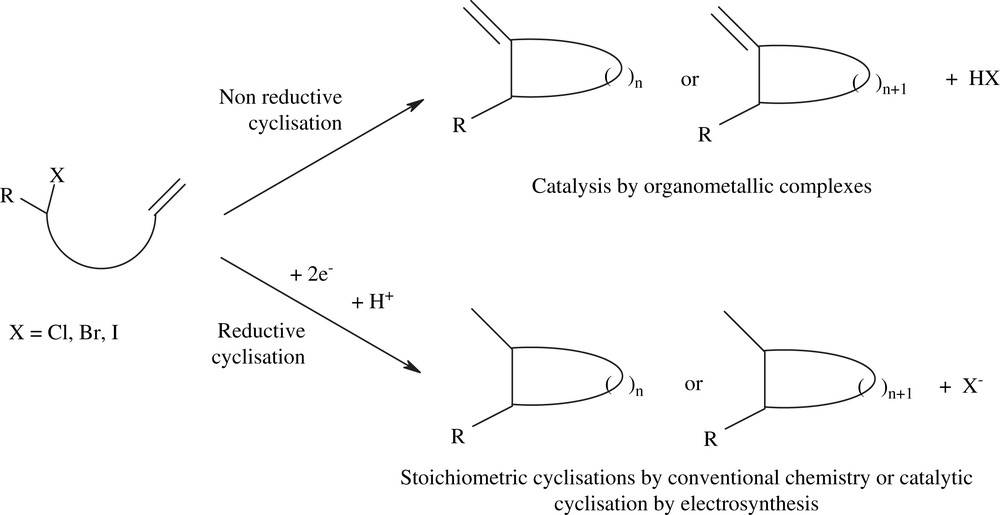
Reductive intramolecular cyclisations.
We have been working with Ni(II) complexes as catalyst precursors in view of the electrochemical in situ generation of Ni(I) species [9]. In particular, Ni(II) macrocyclic complexes have been reported to generate single-electron reduced Ni(I) species, which can further catalyse the reductive radical cyclisation of unsaturated organic halides [10–12], α-bromoacetals bearing olefinic moieties [13] and 2-haloaryl ethers containing unsaturated side chains [14]. In these electrochemical cyclisations the solvent used is an aprotic and polar solvent, generally DMF. However, DMF may present some toxicity as the solvent [15,16]. The possibility to perform reductive cyclisations in protic media has been recently reported [17]. Within a perspective aimed at cleaner and catalytic syntheses, we report here some comparative electrosyntheses run either in DMF or in ethanol and ethanol–water mixtures as the solvents for selected Ni-catalysed electroreductive intramolecular cyclisations. The examination of the comparative behavior of these systems in aprotic and protic solvents examined by cyclic voltammetry is also discussed.
We studied the compared reactivity of three different unsaturated organic bromides: the 3-propargyloxy α-bromoester, 1, the 3-allyloxy α-bromoester, 2, and the 2-bromoaryl olefinic derivative, 3 (see Eqs. (1)–(3)). The cyclisations of 1–3 were carried out under catalytic and mild conditions, at room temperature. Two stable and easily available NiII catalyst precursors, Ni(cyclam)Br2, (cyclam = 1,4,8,11-tetraazacyclotetradecane) and Ni(tmc)Br2, (tmc = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) [18] were used in 20 mol% for the cyclisations (Fig. 1).
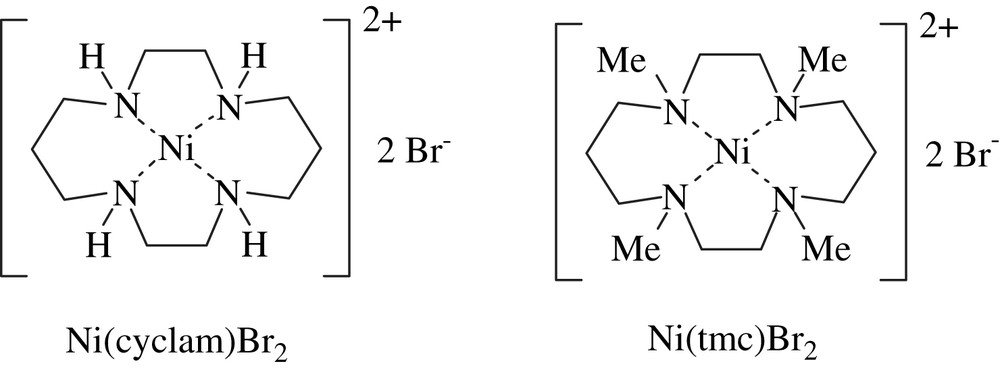
Ni(II) catalysts.
These two NiII complexes, which generate Ni(I) intermediates after a one-electron reduction [19], have been shown to be efficient in reductive processes involving aryl halides [14,20].
The cyclisations of 1–3 were run on a preparative-scale using a single-compartment cell fitted with a carbon fibre cathode and a consumable metal anode, generally a magnesium rod, and were carried out in different solvents at a constant current intensity of 30 mA (j = 0.15 A dm−2). The complete consumption of the substrates required 2–4 F mol−1.
The electroreduction of 1 (Eq. (1)) was carried out in the presence of Ni(cyclam)Br2 using Mg anode in DMF with n-Bu4NBF4 as the supporting electrolyte (6 mM), led to an overall yield of 63% cyclisation as a mixture of furan derivatives 4 and 5 in a 48:52 ratio (Table 1, entry 1). No six-membered pyran rings were formed and both 4 and 5 presented a trans-configuration, in agreement with the configuration of the starting material.
Intramolecular cyclisation of 1 (10 mM) catalysed by electrogenerated NiI complexes (20 mol%) in different solvents.
| Entry | Catalyst | Solvent/supporting electrolyte | Anode | Cyclisation products (4 + 5) (%) | Ratio 4:5 |
| 1 | Ni(cyclam)Br2 | DMF/n-Bu4NBF4 | Mg | 63 | 48:52 |
| 2 | Ni(cyclam)Br2 | DMF/n-Bu4NBF4 | Zn | 78 | 100:– |
| 3 | Ni(tmc)Br2 | DMF/n-Bu4NBF4 | Zn | 68 | 100:– |
| 4 | Ni(tmc)Br2 | EtOH/Et4NBr | Zn | 83 | 82:18 |
| 5 | Ni(tmc)Br2 | EtOH/Et4NBr | Mg | 71 | 21:79 |
| 6 | Ni(tmc)Br2 | EtOH/n-Bu4NBF4 | Mg | 87 | 40:60 |
| 7 | Ni(tmc)Br2 | n-BuOH/Et4NBr | Mg | 88 | 63:37 |
| 8 | Ni(tmc)Br2 | EtOH:H2O (9:1)/Et4NBr | Mg | 97 | 88:12 |
During electrolysis, the cathodic reaction concerns the NiII to NiI reduction, which is followed by the insertion of NiI to the C–Br bond of 1 and further reaction on the triple bond. Additional reduction and protonation by the electrolytic medium affords the furan derivative 4 as the primary expected cyclisation product. The furan moiety is an important subunit in a wide range of biologically active natural products [21], and the nickel-catalysed radical cyclisation has also been applied to the synthesis of several substituted tetrahydrofurans [22–24]. Dihydrofuran 5 is issued from the isomerisation of 4 to the more conjugated isomer.
The same reaction of 1 with Ni(cyclam)Br2 using a zinc anode led to the selective formation of 4 in 78% yield (entry 2). Using Ni(tmc)Br2 with a zinc anode, the cyclisation of 1 led to 4 in 68% yield (entry 3).
The reaction of 1 in EtOH was run in presence of Ni(tmc)Br2 and a Zn anode and afforded 4 and 5 in 83% yield and a 82:18 ratio (entry 4). It was interesting to note that the protic ethanol medium led to a more efficient cyclisation. The same observations can be done for the cyclisation of 1 using a zinc anode in DMF and EtOH as the solvents (entries 3, 4). When the same reaction was carried out with a Mg anode, the cyclisation yield was of 71% (entry 5). When the supporting electrolyte was changed from Et4NBr to n-Bu4NBF4 in EtOH, the yields of 4 and 5 raises to 87% (entry 6). The cyclisation was also efficient in n-BuOH leading to 88% of 4 and 5 and a 63:37 relative ratio (entry 7). When 1 was reacted in EtOH:H2O (9:1) as the solvent, the results were the best with an overall yield of 97% and a 4:5 relative ratio of 88:12 (Table 1, entry 8). The change of the catalyst–substrate ratio from 20 to 10 mol% led to similar results. This indicates that the ratio [RBr]/[NiII] does not interfere in reaction mechanism. Higher water ratios did not allow the complete solubility of the substrate.
The electrolyses of the allyloxy bromoester substrate 2 were also carried out with Ni(tmc)Br2 as the catalyst in DMF, EtOH and EtOH:H2O (9:1), as summarised in Table 2. In these cases, the cyclisation led selectively to a single five-membered ring tetrahydrofuran structure, 6, as a mixture of two diastereoisomers (Eq. (2)).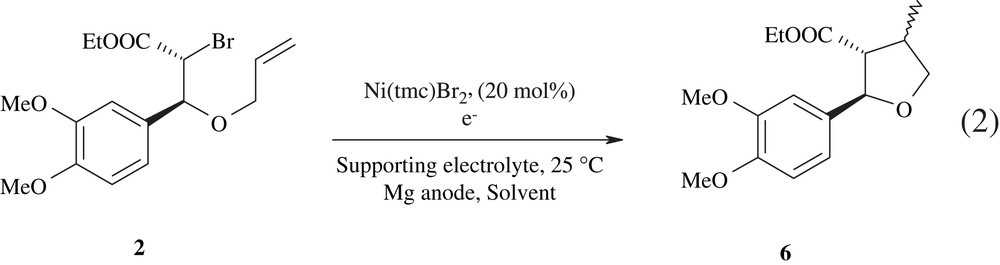
Intramolecular cyclisation of 2 catalysed by Ni(tmc)Br2 (20 mol%) in different solvents.
| Entry | Solvent | Supporting electrolyte | % of 6 (d.r.) |
| 1 | DMF | n-Bu4NBF4 | 38 (91:9) |
| 2 | EtOH | Et4NBr | 61 (93:7) |
| 3 | EtOH:H2O (9:1) | Et4NBr | 75 (93:7) |
In DMF, the cyclisation yield was of 38%. As we observed above in acetylenic series, the efficiency of the reductive cyclisation was increased using EtOH as the solvent, reaching 61% of 6, with a relative isomeric ratio of 93:7. In EtOH:H2O (9:1), an even higher 75% yield was attained with the same isomeric ratio of 93:7.
The role of EtOH as a protic solvent was also examined in the cyclisation of bromoaryl derivative 3. In this case, the comparison of the electroreductive cyclisations was done in DMF and in EtOH, with Ni(cyclam)Br2 as the catalyst and n-Bu4NBF4 as the supporting electrolyte, using an Mg anode (Eq. (3)).
The cyclisation of homoallyl ether 3 led to the indane structure 7 as the main reaction product (cis:trans), together with some dehalogenated compound 8. In DMF the yield of 7 was of 58% with a cis:trans ratio of 40:60 [25]. While in that case the use of EtOH as solvent did not lead to an improved yield of the cyclisation of 7, it lowered the amount of dehalogenation in significant manner (15% in EtOH versus 26% in DMF).
The difference of efficiency of the cyclisations of 1 and 2 in protic and aprotic media may be explained by the mechanism of the electrochemical cyclisation.
The cyclic voltammogram of 1 in DMF, in the presence of Ni(tmc)Br2 is presented in Fig. 2.
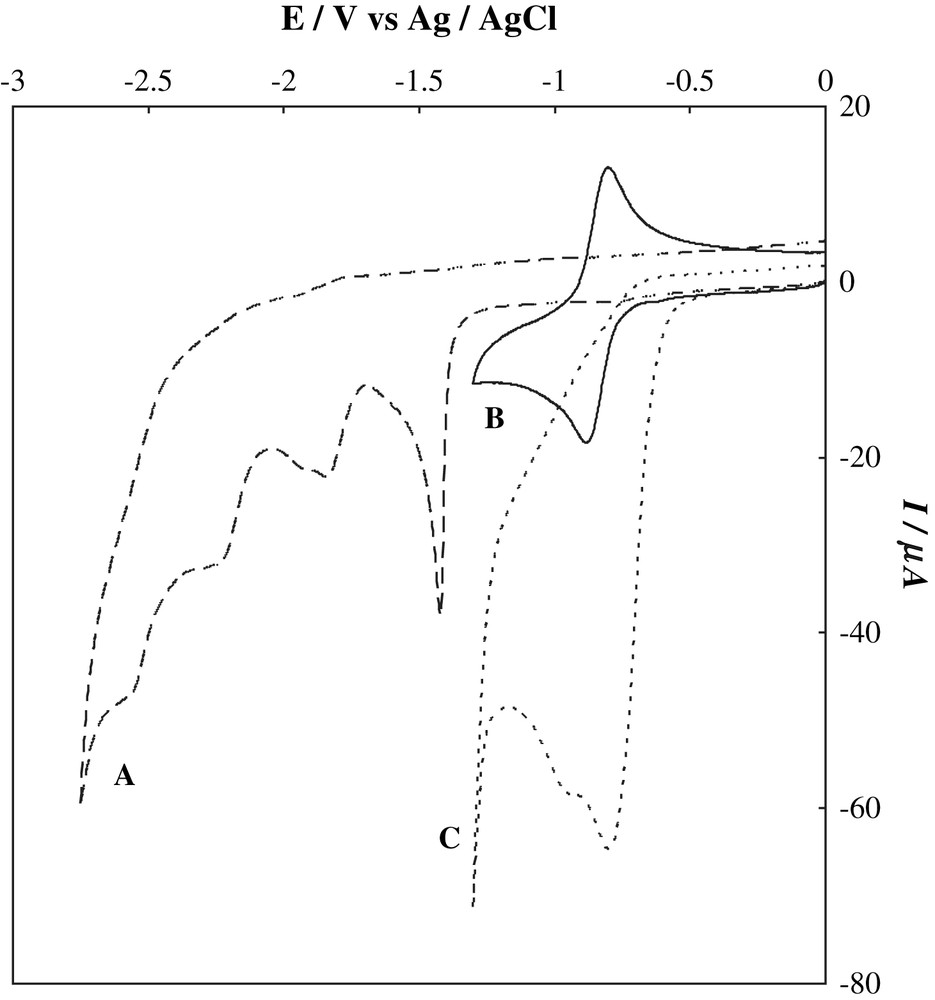
Cyclic voltammograms recorded with a glassy carbon electrode (area = 0.077 cm2) at 100 mV s−1 in DMF containing 0.1 M n-Bu4NBF4 at room temperature: (A) 1.0 mM 1; (B) 1.0 mM [Ni(tmc)]Br2; (C) 1.0 mM [Ni(tmc)]Br2 and 5.0 mM 1.
The NiII(tmc)2+ was reversibly and monoelectronically reduced at −0.92 V versus Ag/AgCl to form a NiI(tmc)+ species. The addition of 1 equiv. of 1 to the DMF solution increased the reduction peak to a two-electron wave and the peak became irreversible. The cathodic peak increased its intensity up to a NiII/1 ratio of 1/5, but upon addition of five or more equivalents of 1 versus NiII (up to 10 equiv.) no further important increase was observed. The chemical bulk cyclisation reaction was therefore a slow reaction in terms of catalyst recycling. The data obtained from these experiments are presented in Table 3. It should be noted that the substrate 1 itself was reduced beyond −1.5 V in the absence of the Ni(II) complex.
Peak-current ratios obtained from cyclic voltammograms of solutions containing Ni(tmc)Br2 (1.0 mM) and various concentrations of 1 at 100 mV s−1.
| Solvent | Ic/Ip | ||
| γ = 2 | γ = 5 | γ = 10 | |
| DMF | 3.14 | 3.74 | 4.20 |
| EtOH | 2.20 | 5.10 | 9.80 |
In contrast, the cyclic voltammetry of 1 in the presence of NiII(tmc)2+ in EtOH, indicated that the progressive addition of the substrate to the reversible NiII/NiI redox couple involved an important catalytic current at −0.85 V versus Ag/AgCl, as shown in Fig. 3.

Cyclic voltammograms recorded with a glassy carbon electrode (area = 0.07 cm2) at 100 mV s−1 in EtOH containing 0.10 M Et4NBr at room temperature: (A) 2 mM 1; (B) 1 mM [Ni(tmc)]Br2; (C) 1 mM [Ni(tmc)]Br2 and 2 mM 1; (D) 1 mM [Ni(tmc)]Br2 and 10 mM 1.
The catalytic wave observed in Fig. 3 is indicative of a faster chemical reaction, as compared to that in DMF, involving a more efficient recycling of the active catalytic species in the alcohol medium. The comparative data obtained from these experiments are summarised in Table 3.
The cyclic voltammograms of 1 and Ni(tmc)2+ in EtOH:H2O (9:1) were similar than that observed in EtOH.
To explain the efficiency of the cyclisation in protic medium as compared to DMF, a catalytic cycle is presented in Scheme 2 for compound 1.
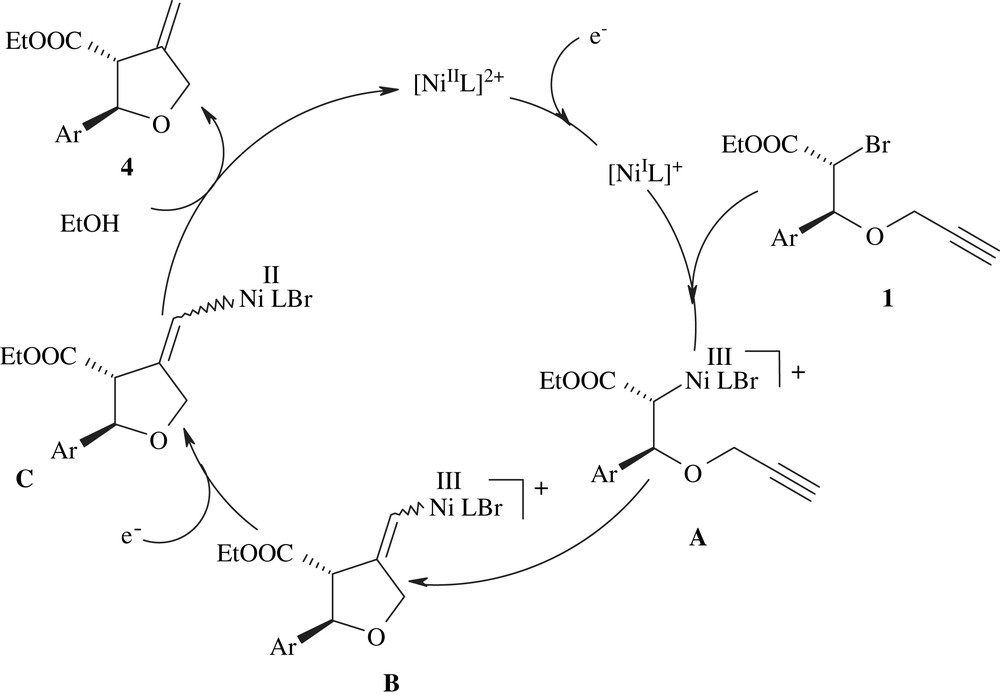
Proposed catalytic cycle for the cyclisation of 1 in EtOH.
The two first steps of NiII to NiI catalyst reduction and further oxidative addition to the C–Br bond of the substrate should be very similar in both media. The proposed organometallic NiIII species A undergoes cyclisation to form a vinyl-NiIII intermediate B that is further reduced to C and protonated, thus liberating the organic product 4 and NiII. The most important difference in the mechanism upon solvent change should lie in the last reduction/protonation step. In DMF, the protonation is a slow process, due to the absence of protons in the medium. It has been reported in other related reactions, that the supporting electrolyte can be a source of protons affording tributylamine and butene in the case of decomposition of the tetrabutylammonium salts [26]. The DMF solvent can also act as a proton source in electrochemical reactions [26]. However, these protonation reactions should be slow processes.
In the reactions run in protic media, the protonation of the NiII intermediate C is highly facilitated and can take place efficiently. As a result, the NiII catalyst is easily and rapidly recycled and accounts for the more intense catalytic wave observed in the cyclic voltammogram of Fig. 3 as compared to Fig. 2.
In conclusion, the radical-type cyclisations catalysed by NiII macrocyclic complexes can be efficiently carried out in protic solvents with increased yield when compared to the same reactions carried out in DMF. The possibility to run organic electrochemical reactions in EtOH as the solvent medium is an interesting asset from the point of view of the use of environmentally more friendly reaction media. The use of electrochemical reductive methodologies instead of the more classical redox reagents also contributes to cleaner processes that can be run with high selectivities.
Acknowledgments
MJM would like to thank Fundação para a Ciência e Tecnologia (PPDCT/55576/2004) for the partial financial support of this work.