

1 Introduction
The alkyne functionality occurs widely in natural products [1], and increasingly as a component of highly π-conjugated organic polymers [2], which are finding application in advanced materials. Moreover, since alkynes are amenable to elaboration, they are also used widely as intermediates for further synthesis. Although a number of alkyne building blocks are commercially available, de novo routes to this functionality from other starting materials are still very commonly used. In this perspective, we wish to review an elimination strategy to alkynes, which we have developed over the past five years and applied to oligoyne assembly. Our approach has required the development of a new method for synthesising β-halovinylsilanes, which act as our alkyne precursors. The elimination route to alkynes, however, has a long history and before we describe our own contribution to this field, we provide a brief overview of existing elimination approaches to alkynes, which will place our own work into its wider context.
1.1 Elimination approaches to alkynes
Elimination strategies offer a particularly attractive approach to alkynes [3]. Of the many variants that are available, the base-mediated dehydrohalogenation of vinyl halides is one of the oldest and still most widely used (Scheme 1a) [4]. The two-fold dehydrohalogenation of vicinal [5] and geminal [6] dihalides provides a useful variant (Scheme 1b), and in a related approach, a 1,4-dehydrohalogenation of 1-bromo[3]cumulenes has been used to prepare 1,3-diynes (Scheme 1c) [7].
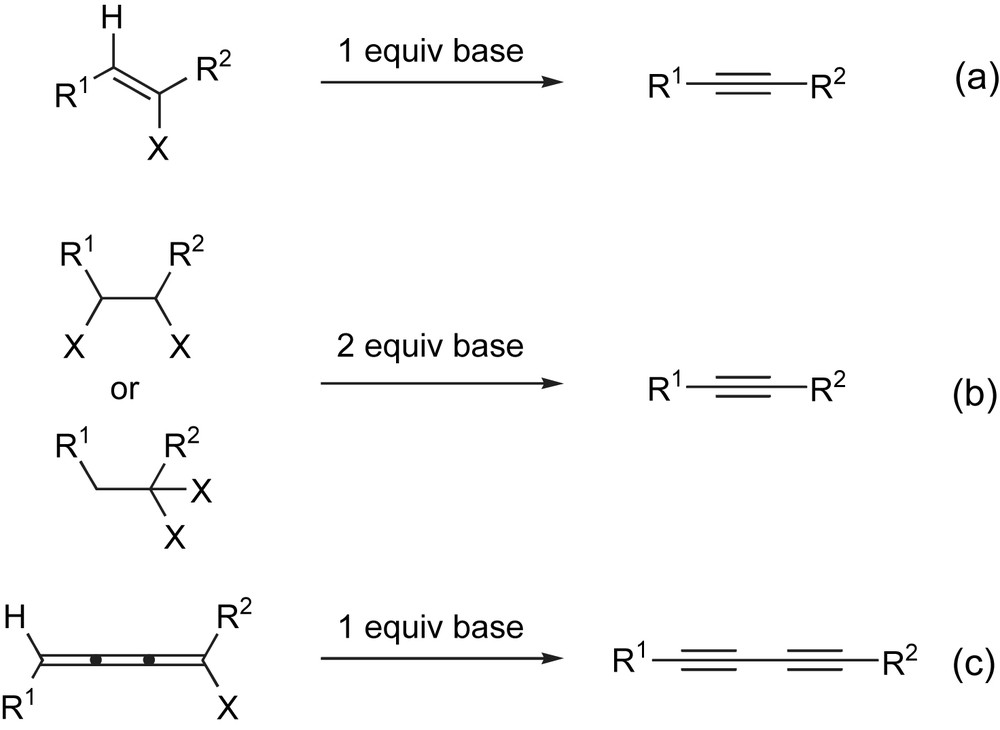
Base-mediated dehydrohalogenation provides a traditional route to alkynes.
Although halogen substituents are the most common nucleofuges in elimination precursors, a wide range of other leaving groups has been employed including sulfones [8], sulfonates [9] and stannanes [10]. Enol phosphates, such as 1 also undergo elimination to the corresponding alkyne 2 on treatment with base (Scheme 2a) [9d,f,11]. Moreover, since this alkyne precursor is generated from the corresponding ketone enolate, this approach provides a useful route to alkynes from readily obtained carbonyl compounds. In an attractive variant on this strategy, Fleming and Ramarao reported a decarboxylative elimination route to alkynes (Scheme 2b) [12]. The enol triflate precursor 3 was readily prepared from the corresponding β-ketoester 4. Subsequent acid-mediated hydrolysis of the tert-butyl ester in 3, followed by treatment with base, generated the corresponding carboxylate 5, which underwent decarboxylative elimination to provide the desired alkyne 6 in good yield. A related approach has also been reported by Zard [13]. More recently, Dudley and Tummatorn reported a related ring opening/fragmentation of dihydropyrones 7 for the synthesis of homopropargyl alcohols 8 (Scheme 2c) [14].
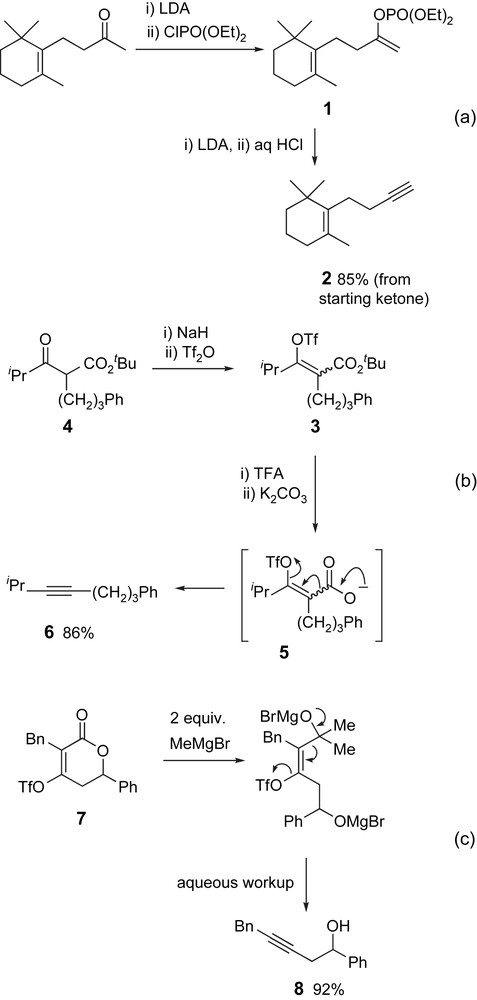
Enol phosphates and triflates, and dihydropyrones serve as useful alkyne precursors.
Reductive elimination of vinyl anions possessing a vicinal heteroatom provides another important variant on this approach to alkynes [15]. In the case of 1,2-dihaloalkene precursors, treatment with a metal, such as Zn [16], Sm [17], or SmI2 [18] likely generates an intermediate vinylmetal species1, which undergoes β-elimination to provide the corresponding alkyne (Scheme 3a). Ene-1,2-disulfides also undergo a similar reaction on exposure to lithium naphthalenide (Scheme 3b) [19].
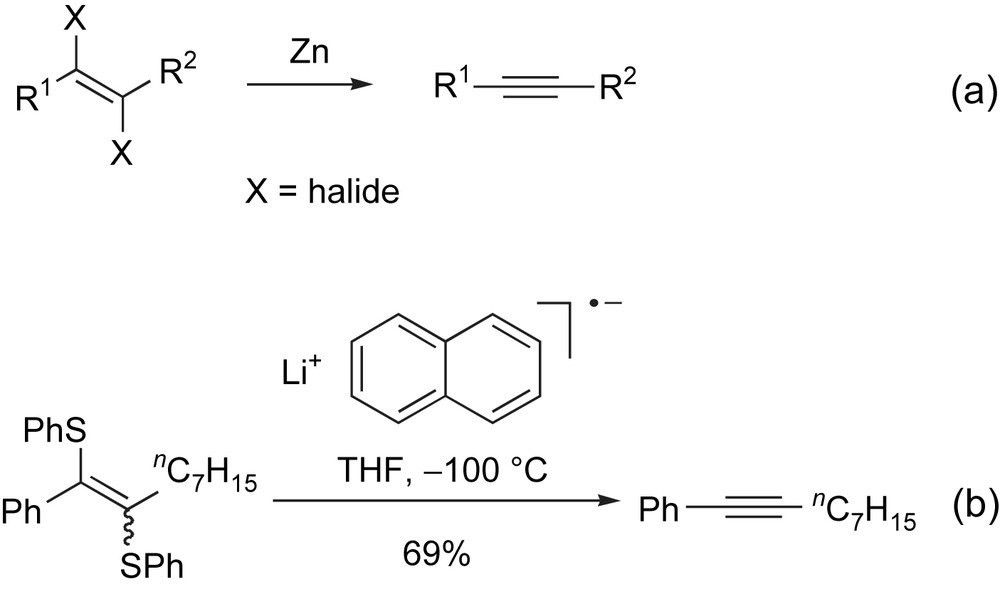
Vinyl anions possessing a vicinal heteroatom undergo elimination to provide alkynes.
Most of the elimination strategies described so far likely proceed through an anti-elimination pathway; however, syn-elimination processes have also been used to access alkynes [20]. For example, β-silylvinylsulfoxide 9 underwent a syn-specific elimination on thermolysis to provide alkyne 10 (Scheme 4a) [20b]. The silyl group in the alkyne precursor was important in this case for isolating the alkyne product: a similar reaction with the non-silylated vinylsulfoxide 11 provided the vinylsulfoxide regioisomer 12, resulting from addition of the phenyl sulfenic acid elimination by-product to the intermediate alkyne (Scheme 4b).
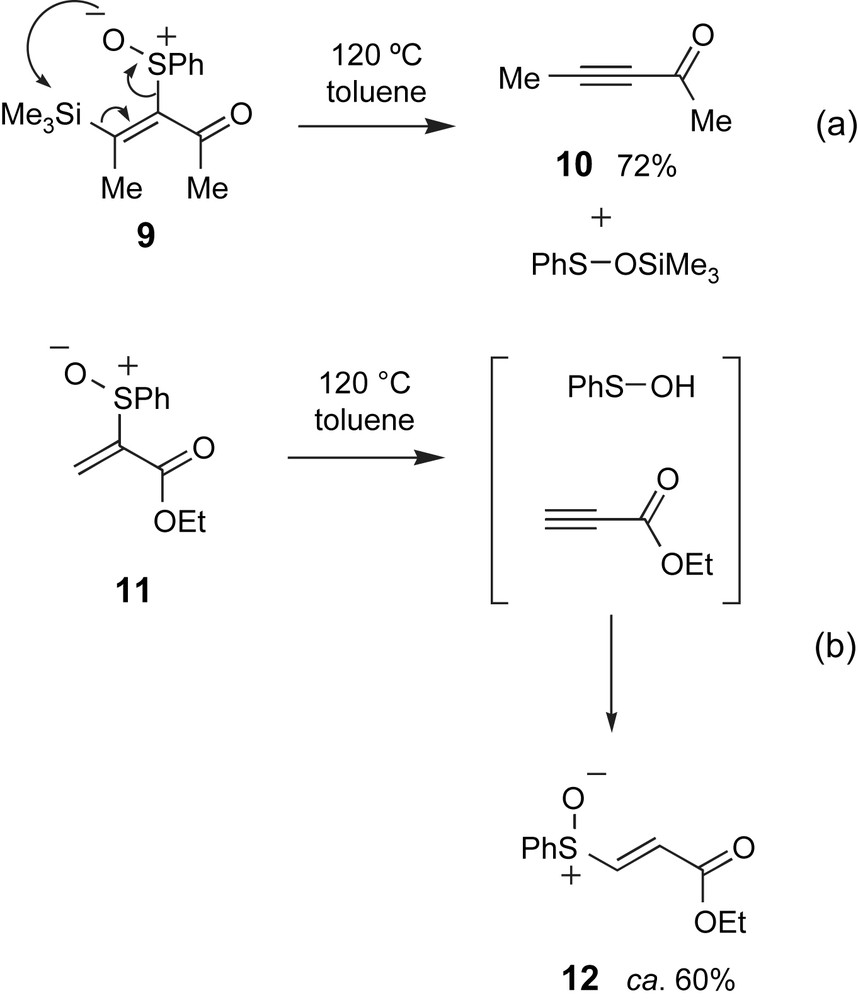
Syn-elimination strategies have been used to prepare alkynes.
The syn-elimination of vinyl sulfoxides summarised in Scheme 4 nicely illustrates the potential improvements that can be achieved in elimination reactions by replacing the most commonly employed hydrogen electrofuge for a silyl group, and conveniently serves as an introduction to our own work where we have focused on developing a mild and chemoselective elimination route to alkynes, which is amenable to oligoyne assembly.
2 Elimination strategies for oligoyne assembly
Many of the available methods of alkyne synthesis, including most elimination approaches, are not conducive to long-chain oligoyne assembly; the reaction conditions are often too harsh and lead to decomposition of the oligoyne product. Although elimination strategies have only occasionally been used to prepare oligoynes [21], we were attracted to the potential simplicity of employing such an approach. To this end, we considered the possibility of employing a β-halovinylsilane motif within a masked oligoyne (Scheme 5). Treatment with a fluoride source would effect dehalosilylation and release the oligoyne chemoselectively, and importantly, under mild conditions. The ability to incorporate a range of silyl groups and halo substituents into the masked alkyne was also attractive since this would allow us to modulate a number of parameters including the solubility and stability of the oligoyne precursor, and the conditions under which unmasking of the oligoyne could be effected. The silyl substituents might also provide a handle for developing potential encapsulation strategies for the conjugated framework (see Section 2.5).
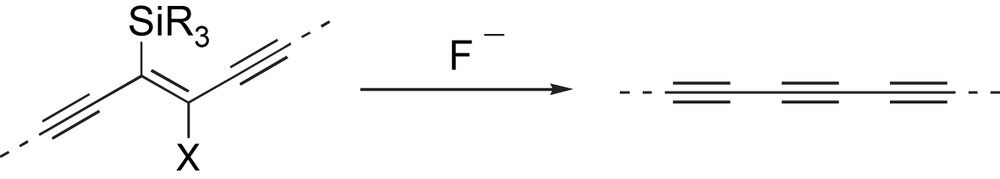
A β-halovinylsilane would provide a novel masked alkyne for oligoyne assembly.
2.1 β-Halovinylsilanes as masked alkyne precursors
Whilst there are many examples where silanes possessing a halide leaving group in the β-position have been used for alkene synthesis [22], when we first set out on this project, only a small number of β-halovinylsilanes had been reported, and even fewer had been used purposefully for alkyne synthesis [23]. Moreover, all the available methods for synthesising β-halovinylsilanes were unsuitable for the tetrasubstituted systems that we would need for our purposes (see below). For example, in an early report, Cunico and Dexheimer prepared (E)- and (Z)-β-chlorovinyltrimethylsilane from the corresponding β-chlorovinyltrichlorosilanes, which were obtained from the pyrolysis of 1,2-dichloroethene and trichlorosilane [24]. They went on to show that both (E)- and (Z)-β-chlorovinyltrimethylsilane undergo elimination to provide acetylene on exposure to a fluoride source (Scheme 6a,b) [23a]. Some years later, Danheiser et al. identified the terminal β-chlorovinylsilane 13 as a side-product in the TiCl4-mediated addition of allenyltrimethylsilane into a range of aldehydes, and again demonstrated that this side-product could be converted to the desired homopropargylic alcohol 14 on treatment with fluoride (Scheme 6c) [25]. A similar strategy has also been used by Marshall and Maxson [26].
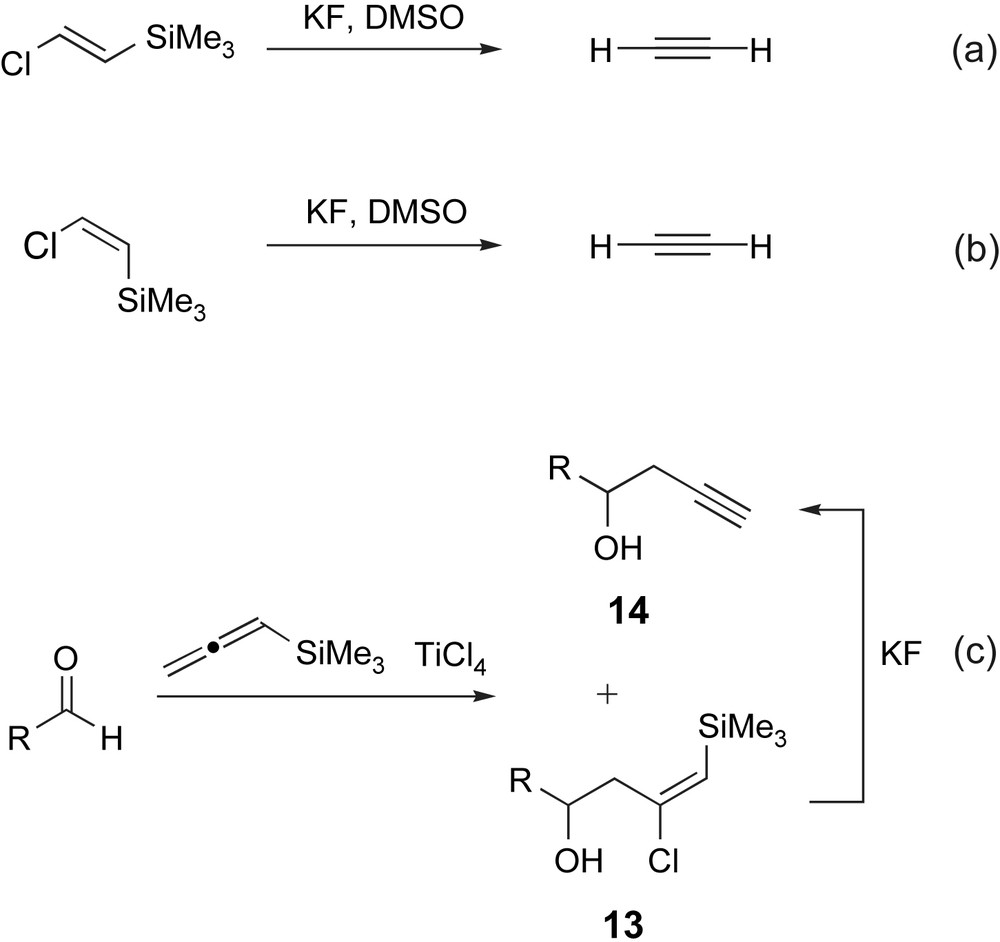
Literature precedent revealed that simple β-halovinylsilanes could be converted into the corresponding alkynes.
2.2 A masked triyne target provides a general route to a masked hexayne
Thus with limited, although encouraging, precedent, we set about designing a β-halovinylsilane building block, which could be used for oligoyne assembly. We were attracted to the possibility of incorporating this masked alkyne into an enediyne framework 15, since this would provide an interesting building block in its own right for assembling a novel class of oligo(triacetylene)s containing heteroatom substitution off the conjugated scaffold (Scheme 7) [27]. We envisaged oligomerisation of enediyne 15 could be effected under Eglinton–Glaser–Hay conditions [28]. However, owing to the unsymmetrical nature of the starting monomer, each oligomer would likely be formed as a mixture of constitutional isomers owing to the possibility of head-to-tail (e.g. 16), head-to-head (e.g. 17), and tail-to-tail coupling (e.g. 18); the dimer, for example, would be formed as a mixture of three constitutional isomers 16, 17 and 18 (Scheme 7a). Whilst this information would be lost on unmasking the oligoyne, we proposed a modification to our building block, which would circumvent this issue and also confer some additional attractive features. Protection of one of the alkyne termini in 15 with a trimethylsilyl group would provide a new masked triyne 19, which on oxidative homocoupling, would afford the corresponding masked hexayne dimer 20 (Scheme 7b). Providing the trimethylsilyl end-capping groups in 20 could be removed without effecting premature dehalosilylation, the masked hexayne 20 could now be used as a new monomer unit for oligomerisation studies. Since this masked hexayne is centrosymmetric, each oligomer in the series would be formed as a single constitutional isomer, which would greatly facilitate characterisation. Moreover, by using a relatively large repeat unit, we hoped the large difference in size between the oligomers in the series (the monomer, dimer and trimer in the series would be masked hexaynes, dodecaynes and octadecaynes, respectively) would facilitate their separation by size-exclusion chromatography.

Centrosymmetric masked hexayne 20 provided an attractive monomer for oligomerisation studies.
Our first target therefore became masked triyne 19. In analysing this molecule, we set ourselves a number of design criteria which any synthetic strategy would have to satisfy. First, we wanted to prepare the β-halovinylsilane motif as a single stereoisomer to avoid obtaining mixtures of double bond isomers in our oligomer series. At the outset, we were not too concerned whether this was the (E)- or the (Z)-stereoisomer, as Cunico and Dexheimer had demonstrated that both could be used as masked alkynes [23a]; however, we expected the (E)-stereoisomer would undergo elimination under milder conditions and therefore was our preferred target. Second, we wanted a method that would allow us to incorporate a wide range of silyl groups into the β-halovinylsilane. This would not only be important for modulating the stability of the masked alkyne motif, and the conditions under which elimination would be effected, but as will be discussed later (see Section 2.5), we were keen to investigate the possibility of using the substituents appended off the silicon atom in some form of encapsulation strategy, which we believe is likely to be important for stabilising particularly long-chain oligoynes.
Masked triyne 19 represented an interesting synthetic problem. The most challenging task would involve the stereoselective synthesis of the internal tetrasubstituted olefin. Efficient methods for accessing heavily substituted olefins in a completely stereoselective fashion are scarce [29], and at the outset of this project, a suitable route to a fully substituted olefin containing vicinal silyl and halo substituents had not been developed. A search of the literature uncovered a potentially attractive solution to this problem. In 1987, Zweifel and Leong reported a completely regio- and stereoselective metallometallation of 1-trimethylsilyl-substituted 1,3-butadiyne 21 using a stannylcopper reagent that was prepared from equimolar quantities of Me3SnLi and CuBr·SMe2 complex (Scheme 8) [30]. On their limited set of substrates, the group went on to demonstrate that treatment of the bis-vinylstannane 22 product with MeLi effected selective tin–lithium exchange of the tin residue proximal to the remaining alkyne, to generate a reactive vinyllithium intermediate 23, which could be quenched with a small selection of electrophiles to provide vinylstannane 24 (Scheme 8).
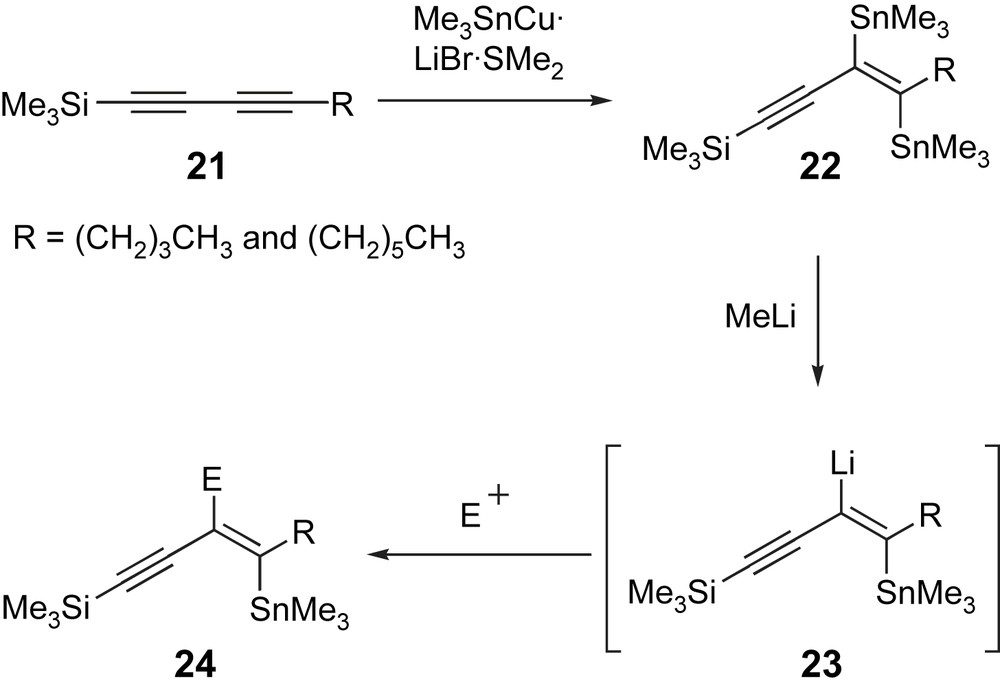
A 1,2-bis-vinylstannane provides a potential precursor to a β-halovinylsilane.
We postulated that the two vinylstannane moieties in eneyne 22 could be manipulated sequentially into the vinylsilane and vinylhalide units of our desired β-halovinylsilane. For our purposes, we would need to employ silyl electrophiles to install the vinylsilane motif, after which, we expected the remaining vinylstannane would be used to introduce the vinylhalide through a halodestannylation process [31]. Were the halo substituent to be introduced first, converting the remaining vinylstannane into a vinylsilane, presumably via a tin–lithium exchange process, would be beset with problems associated with β-halo elimination (see Scheme 3), which would simply return the starting diyne precursor 21.
Although Zweifel and Leong had shown that a small selection of (reactive) electrophiles could be used to quench the vinyllithium intermediate 23 [29,32]. We were not confident that this intermolecular trapping strategy would satisfy our needs. Fortunately, work we were carrying out on a different project using silyl tethers to deliver nucleophiles in an intramolecular fashion showed us a potential way forwards [33]. Our retrosynthesis of masked triyne 19 is summarised in Scheme 9. We had already identified a butadiyne containing a hydroxymethyl substituent, as a suitable precursor to our masked triyne. The internal alkyne would serve as the β-halovinylsilane precursor whilst the alcohol functionality would be elaborated into the terminal alkyne; anticipating the masked triyne end-product might exhibit poor stability, this would be done in a final step. Early on, we had shown that it would be necessary to protect the alcohol in our starting butadiyne in order to effect an efficient bis-stannylation reaction. We therefore postulated that if we protected the alcohol as a silyl ether, then this protecting group could serve a second role acting as a tethered silyl electrophile for introducing the vinylsilane through a 1,4-retro-Brook rearrangement [34] using Zweifel's tin–lithium exchange chemistry (Scheme 9). We expected this approach would have profound benefits for preparing systems containing bulky silyl groups, which would otherwise be difficult to incorporate were we forced to rely on an intermolecular reaction for introducing these motifs. Further elaboration into our masked triyne target would then be straightforward (Scheme 9).
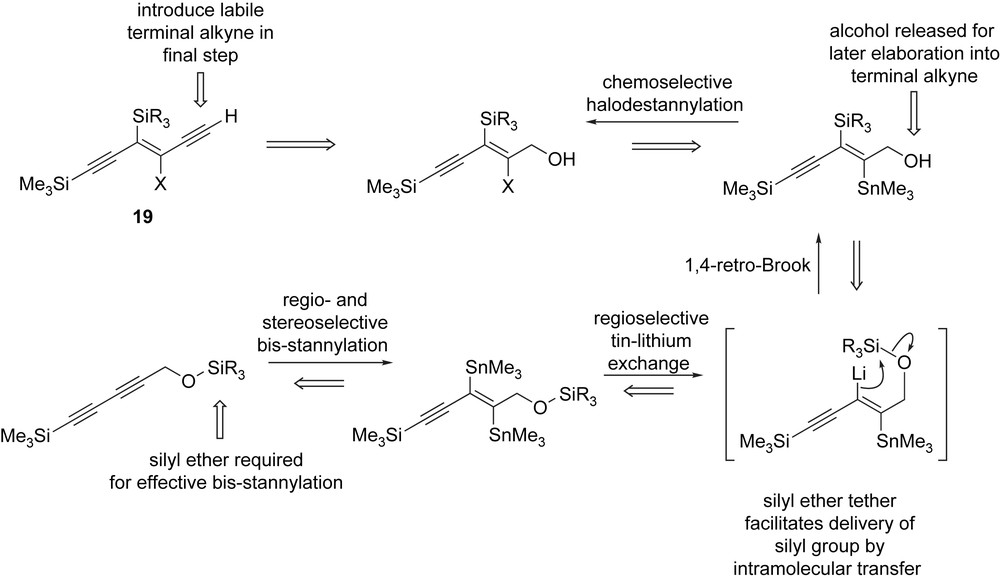
Retrosynthesis of masked triyne 19.
Since one of our principal research goals was to access long-chain oligoynes, our first target was masked triyne 19a, which contains a trimethylsilyl-substituted alkyne that would allow further elaboration (see Section 2.4). We have reported the synthesis of masked triyne 19a, which is summarised again in Scheme 10 [35].
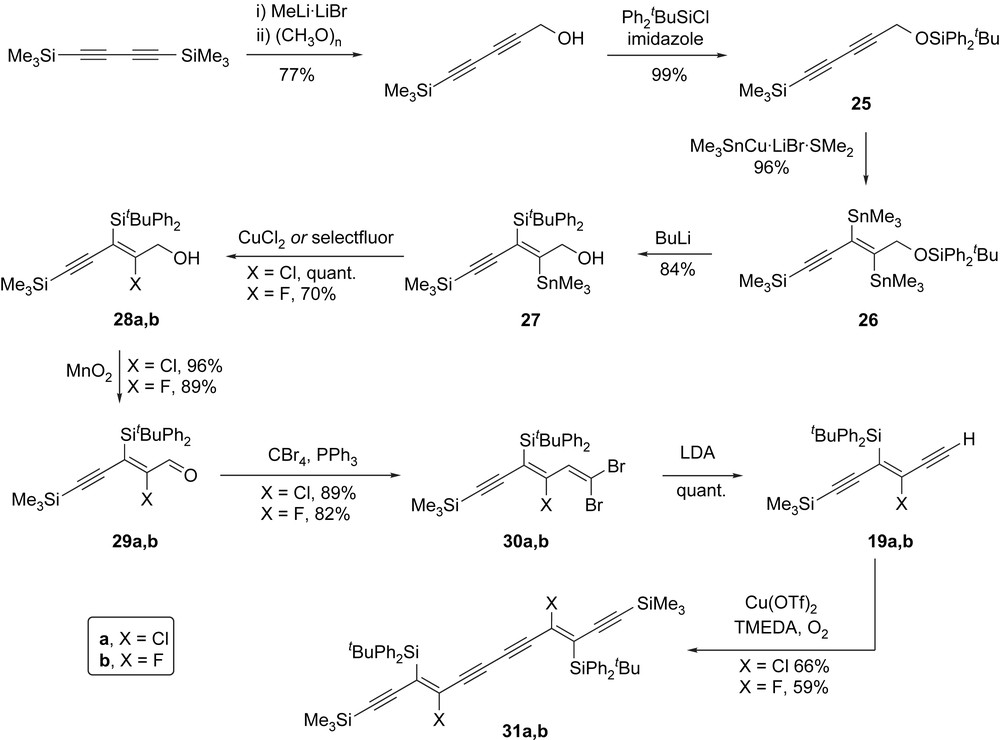
Synthesis of masked triyne 19 and centrosymmetric masked hexayne 31.
The key bis-stannylation of 1,3-butadiyne 25 proceeds with complete regiocontrol and in excellent yield, providing bis-vinylstannane 26 as a single (E)-stereoisomer. The reaction also works well using the (less toxic) tributylstannylcopper reagent although subsequent processing of the two tributylstannyl functionalities is far less efficient [36]. We do not fully understand the origins of the regioselectivity in this key reaction. One could argue that the bulky trimethylsilyl substituent controls the regiochemistry for steric reasons; however, since replacing this silyl end-cap with an aryl substituent also leads to the same regiochemical outcome, we tentatively propose that the trimethylsilyl or aryl substituents exert a mesomeric directing effect, which leads to the observed regioselectivity. That bulky silyl groups, which generally disfavour coordination of metals to the Lewis basic oxygen centre, also give the same regiochemical outcome, suggests a coordinative directing effect from the silyl ether is probably not important. Treatment of bis-stannane 26 with BuLi effects the planned 1,4-retro-Brook rearrangement and transfers the silyl group contained within the silyl ether to an internal position. Although we show the formation of the TBDPS derivative 27 in Scheme 10, this reaction works well for a range of silyl groups, including systems containing bulky ligands, which would have undoubtedly been difficult to install using an intermolecular trapping of the intermediate vinyllithium species with a silyl electrophile. Converting the remaining vinylstannane in 27 to a vinylhalide is straightforward. Most of our work to-date has focused on using β-chlorovinylsilanes as masked alkynes as we initially argued these would be sufficiently stable to avoid premature dechlorosilylation, and yet would still undergo the desired elimination under mild conditions when required. The vinylchloride in 28a is readily obtained by treatment with CuCl2. This reaction proceeds with retention of configuration, which has been confirmed by X-ray crystallography [35]. More recently we have also explored the introduction of other halogen substituents and shown that vinylstannane 27 can be converted to the corresponding fluoride, bromide and iodide [31]. The bromo and iodo derivatives proved to be too labile to be useful candidates for further investigation; however, the β-fluorovinylsilane 28b, which is obtained by treating 27 with selectfluor®, has led to a particularly useful development, about which more will be said in Section 2.3. The 1,4-retro-Brook rearrangement releases an alcohol functionality in 27, which is then elaborated to a terminal alkyne in three straightforward steps: allylic oxidation of 28 affords an intermediate aldehyde 29, which is then converted to 1,1-dibromoolefin 30 [37]. Finally, exposure of dibromide 30 to an excess of LDA effects a Fritsch–Buttenberg–Wiechell-like reaction to provide a lithiated alkyne, which can be manipulated in a number of ways. Protolytic work-up affords our masked triyne target 19; however, other electrophiles including TMSCl and acetone have also been used to provide different end-caps. Whilst we have not investigated its application in further synthesis, dehydrobromination in dibromoolefin 30a using NaHMDS provides a bromo-alkyne that could potentially be used in Cadiot–Chodkiewicz-style couplings.
Masked triyne 19 containing a free terminal alkyne is not particularly stable and for this reason, we invariably use this directly in an oxidative dimerisation to afford our second target, namely centrosymmetric masked hexayne 31. This reaction proved to be an unexpectedly troublesome step and required careful optimisation. The source of copper catalyst seems to be the most important variable for obtaining optimal yields of the dimer, with copper salts containing non-nucleophilic counteranions, in particular Cu(OTf)2, providing the best results. In summary, our route outlined in Scheme 10 is robust and flexible and allows the introduction of a range of silyl and halo substituents as well as end-capping groups to suit our needs.
2.3 Unmasking the oligoyne
Whilst silyl end-capped masked hexaynes are needed for oligomerisation studies, aryl end-capped masked hexaynes were more useful for investigating unmasking conditions since these end-caps would ensure that relatively stable hexayne products are produced on unmasking. Aryl-substituted masked hexaynes are readily prepared from the corresponding aryl-substituted butadiyne according to Scheme 10. For hexaynes containing two β-chlorovinylsilane units (e.g. 32a), treatment with two equivalents of TBAF (one equivalent per β-chlorovinylsilane) provides the free hexayne 33 in excellent yield (Scheme 11). The reaction is rapid and proceeds at room temperature or below. As described in Section 2.2, we have recently shown that masked hexaynes 32b containing two β-fluorovinylsilanes can also be prepared. This modification to the masked hexayne framework is particularly appealing because unmasking is now fluoride-catalysed rather than fluoride-mediated [38]. For example, phenyl end-capped hexayne 33 can be prepared from 32b in excellent yield using just 10 mol% TBAF (Scheme 11) [31].
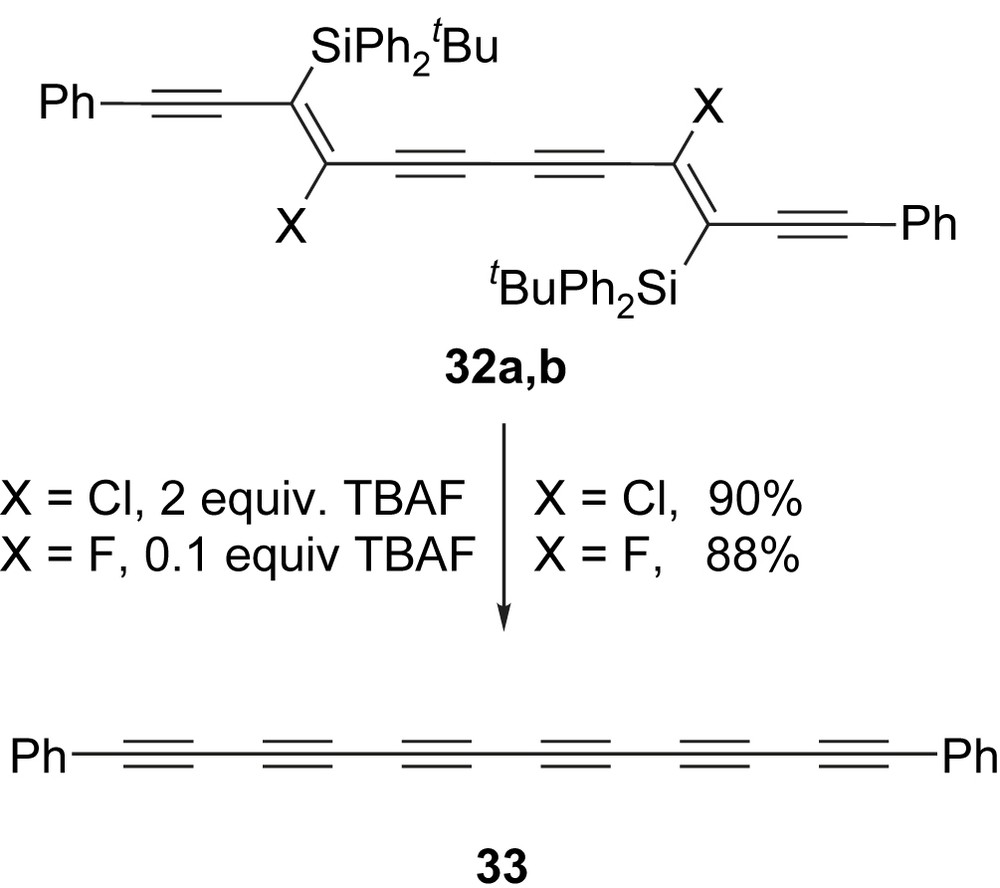
Oligoyne products are released upon treatment with fluoride.
2.4 Application to the synthesis of an aryl end-capped dodecayne
One of our research goals is to prepare long-chain oligoynes, which provide clipped analogues of a fourth allotrope of carbon known as carbyne (a poly(alkyne)). To this end, we recently dimerised masked hexayne 31a to afford the corresponding masked dodecayne 34 (Scheme 12). Exposure of 34 to TBAF provided the corresponding dodecayne 35, which represents the longest aryl end-capped oligoyne reported to date [39]. Dodecayne 35 proved to be highly unstable, even under the very dilute reaction concentrations which were used to effect unmasking; however, this oligoyne was characterised by UV–vis spectroscopy and MALDI-TOF mass spectrometry.

Using a β-chlorovinylsilane to synthesise the longest aryl end-capped oligoyne reported to date.
2.5 An encapsulation strategy for stabilising longer chain oligoynes
With some notable exceptions [40], oligoynes (triynes and longer) with hydrogen substituents at the termini exhibit very poor stability in solution as well as in the solid state, where topochemical polymerisation degradation pathways can be a particular problem. One method for stabilising the conjugated scaffold in oligoynes is to incorporate sterically bulky end-caps on the termini [41]; however, as the length of the oligoyne increases, and the end-caps constitute an ever-decreasing component of the system, this method of stabilisation becomes increasingly unsatisfactory. The instability of dodecayne 35 does not bode well for the trimer in this series, which is an octadecayne and represents our next target. For such long-chain systems, a method for providing some form of insulation along the entirety – as opposed to just the termini – of the conjugated carbon chain would be desirable, and indeed some progress has been made in recent years along these lines [42]. We postulated that the silyl substituents appended at regular intervals along the conjugated framework in the masked oligoyne precursor could provide a handle for constructing a macrocycle that encapsulates the conjugated chain. A crystal structure of the phenyl end-capped masked hexayne 32a suggested a potential strategy for achieving this (Fig. 1) [43].
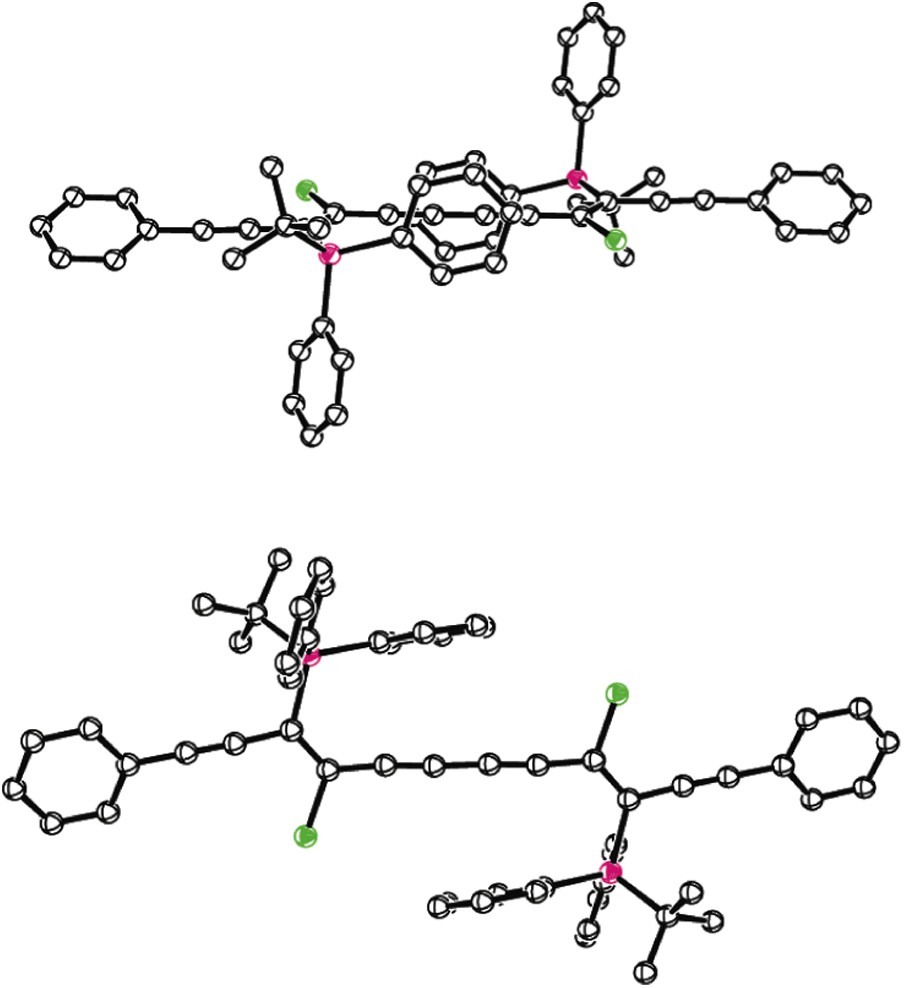
Crystal structure of masked hexayne 32a.
Of particular interest in the structure of 32a is the position of the two TBDPS substituents, which occupy opposite sides of the conjugated framework. Even more interesting is the orientation of the phenyl groups within each silyl substituent, which sandwich the conjugated framework. The average distance between the eclipsing carbons in the phenyl rings that make up the sandwich is 6.8 Å, which is less than the sum of the van der Waals radii of the carbon atoms involved (C(sp) = 1.78 Å, C(sp2) = 1.77 Å). We tentatively propose that this conformation, at least in the solid state, is a consequence of the electron-withdrawing nature of the two chloro substituents, which renders the internal region of the π-conjugated framework electron-deficient. The polarisable nature of the π-cloud within the phenyl substituents then compensates for this through π-stacking. Assuming the crystal structure of 32a represents a low-energy conformation for this type of masked hexayne, we have used this structural information to advance a novel encapsulation strategy, which would ultimately lead to a rotaxanated oligoyne. Our idea is outlined in cartoon form in Scheme 13.
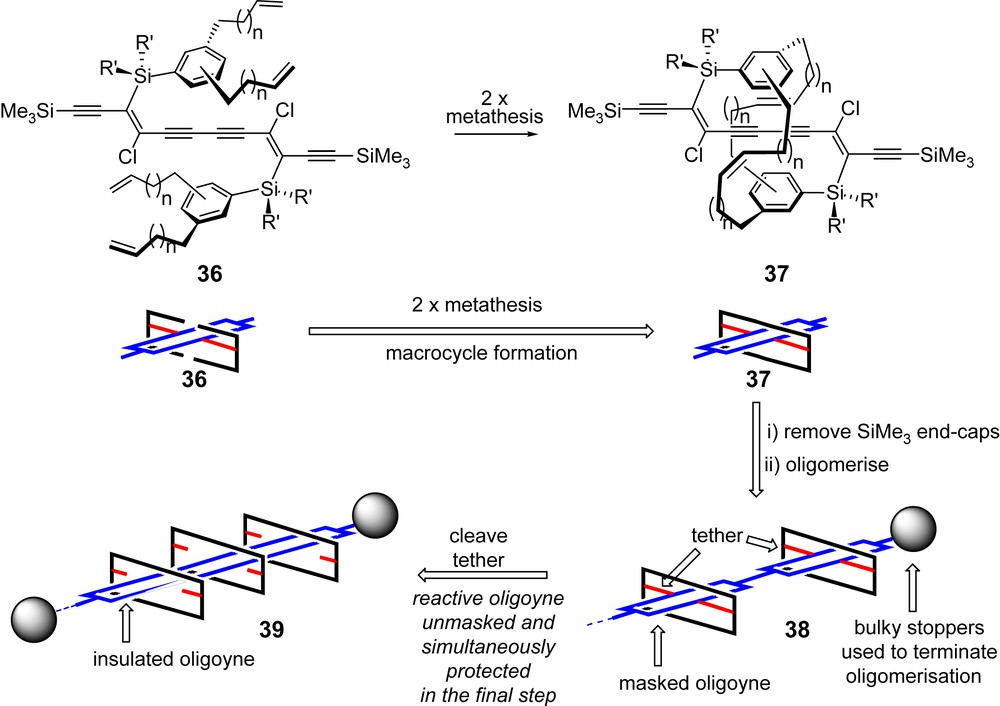
A novel oligoyne encapsulation strategy.
A phenyl group in the vinylsilane substituents would be functionalised with two alkenyl side-chains. The resulting masked hexayne 36 would then be assembled as outlined in Scheme 10. We then hypothesised that reaction of 36 in the presence of Grubbs' catalyst should effect a double metathesis operation to generate a macrocycle [44]. If the reactive conformation of 36 is similar to that found in the crystal structure of masked hexayne 32a (Fig. 1), then the macrocycle might encapsulate the conjugated enediyne framework to provide masked hexayne 37 (Scheme 13). Removal of the trimethylsilyl end-caps in 37 and oxidative coupling would generate a series of oligomers. Terminating the oligomerisation with bulky end-capping groups would provide the masked oligoyne 38. Finally, treatment with fluoride would unmask the oligoyne and simultaneously release the insulating macrocycles (one per six alkynes). Providing the end-caps are sufficiently large, the resulting macrocycles will remain around the oliogyne to afford rotaxane product 39. This insulation approach is an attractive one in that the unstable oligoyne is only generated in the final step when it is already protected by encapsulation.
In a first attempt, we have prepared masked hexayne 40 in which butenyl substituents were incorporated into the 3- and 5-positions of the aromatic group in the vinylsilane [43]. We were pleased to observe that exposing 40 to Grubbs' catalyst effected a double metathesis operation. Unfortunately in this case, a crystal structure of the product 41 revealed that the macrocycle was crowning rather than encapsulating the enediyne framework (Scheme 14) [43]. Whilst this outcome was disappointing, it is encouraging to observe that (i) the unsaturated enediyne framework is stable under the metathesis reaction conditions and (ii) that a macrocycle was formed. Efforts are now underway to modify the structure of the macrocycle precursor to encourage it to adopt a conformation that is more conducive to threading the conjugated framework.

A first attempt at molecular encapsulation of the masked hexayne framework proved unsuccessful.
3 Conclusion and future directions
Whilst oligoynes have been synthetic targets for many years, the synthesis of long-chain systems still represents a state-of-the-art challenge. Since Bohlmann's [21a–d] and Jones' [21e–g] pioneering studies in the 1950s, and Walton's important contributions in the 1970s [45], the last ten years have seen great advances in modern synthetic methods that have provided significant developments in oligoyne assembly, most notably from Hirsch et al. [46], Gladysz et al. [47] and Tykwinski [48]. Our elimination route to oligoynes provides a new approach to oligoyne assembly. The next challenges for us will involve the synthesis of even longer chain systems using molecular encapsulation methods to stabilise the entire length of the labile π-conjugated framework.
Acknowledgements
This work has been supported by The University of Birmingham, the Engineering and Physical Sciences Research Council (grant ref. EP/C532260/1) and The Leverhulme Trust (grant ref. F/00 094/T). We thank our past collaborators – Dr. Simon M. E. Simpkins and Dr. Caterina S. Aricó – for their enthusiastic contributions to the early stages of this research programme.
1 Radical intermediates may also be involved here.