

1 Introduction
With economic, infrastructural and technological advancements, the world's hunger for energy is ever increasing [1]. Finite fossil-fuel supplies, nuclear waste and global warming linked to CO2 emissions necessitate the rapid development of alternative/“green” sources of energy [2,3]. Electricity generated from renewable resources such as solar, wind and tides offers great potential to meet these future energy demands; however, the output from such sources is intermittent while available electricity is required at anytime in our daily lives. These crucial energy supply issues facing mankind, together with the rapid advance and eagerness from the electric automotive industry (e.g. Electric vehicles [EVs] and Hybrid Electric Vehicles [HEVs]) have combined to make the development of radically improved rechargeable batteries a worldwide imperative [4]. Researchers have then the responsibility for providing the world with better and more efficient batteries. However, research on batteries does not have a good track record as it took more than two centuries and the arrival of the Li-ion technology to triple the energy density of the Pb-acid batteries [5]. Besides the main difficulties in mastering electrochemical interfaces, the lack of new materials has been the overriding limitation for advanced batteries. Regardless of the application sectors considered, history is constantly reminding us of the importance of materials to make new technologies possible. With no new compound, no new exotic properties could be found. The Li-ion technology is no exception, as, back in the 1970s, its development was nested in the discovery of inorganic compounds having both a crystallographic and an electronic structure able to accept both ions and electrons, later on identified as “intercalation” compounds. Thus, it is crucial to maintain research activities towards the synthesis/design of new materials and the development of new synthesis/processing techniques to achieve materials with complex properties to ignite new applications.
Synthesis, which is the essence of chemistry, has a long history, which goes well beyond the scope of this paper that deals only with inorganic compounds, for which various synthesis techniques leading to materials with different properties have been reported. One of the oldest synthesis processes that nature taught us is biomineralization which can lead, under ambient temperature-pressure conditions, to calcite particles having different shapes and morphologies. Using either, or combining temperature and pressure, has then led to the field of solid-state synthesis; it basically consists in reacting under various temperatures and pressures two solids A and B to form a third one AB via local mass transport. Such a widely popularized approach among chemists over the last few decades has led to a colossal amount of materials with exotic properties which have led to the establishment of a wide knowledge platform providing invaluable insight and guidance for the targeted or exploratory design of new materials. However, solid-state synthesis is solely viable to prepare phases that are thermally stable within the range of temperatures required to prepare them. Eager to obtain metastable phases, chemists explored the field of low temperature synthesis enlisting hydrothermal/solvothermal synthesis as well as sol-gel synthesis, pioneered among others by Livage [6,7]. All of these low temperature techniques have been called (or referred to as) “chimie douce” by their early French promoters. Interestingly, advances in solid-state chemistry synthesis have been strongly dependent on oil supply. The first oil crisis, back in the 1970, has acted as a great push for the rapid development of “chimie douce” as low temperature processes are obviously less energetically demanding than high temperature ones, usually called ceramic processes. Similarly today, the looming energy crisis associated with fossil-fuel shortage is setting new incentives in materials synthesis, whatever the field of applications, where materials cycle life cost has become the overriding figure of merit in selecting the proper material. This applies to Li-ion battery research, as notion of sustainability, renewability, eco-efficient synthesis processes must be taken into consideration in selecting electrode materials for the next generation of Li-ion cells, and more so for those targeting high volume applications.
Addressing these urgent requests, we have developed new approaches towards the hydrothermal/solvothermal synthesis of electrode materials; they consist in either using the concept of “latent bases” [8] to tune the reacting media pH, or substituting water for ionic liquids as the reacting media [9]. The LiFePO4 phase, found in nature as triphylite, which is presently the main positive electrode contender for EVs applications, will be used to convey the benefits of the aforementioned approaches [10]. Although LiFePO4 is a natural mineral, mining is not an option considering its purity, and more so, its relative rarity. Thus, synthetic triphylite powders have to be made aiming at grain sizes as close as possible to those used in battery construction (e.g., nano) via eco-efficient synthesis processes. For reasons of clarity, the two different synthesis paths that we have recently developed and enlisted separately, the use of “latent bases” or of ionic liquids will be treated separately, emphasizing the whys and wherefores of each method. Afterwards the electrochemical performances of the obtained LiFePO4 powders vs. Li/Li+ will be presented and future synthesis trends towards returning to biomineralization discussed.
2 Use of latent bases to prepare Li 3d-metal phosphates and silicates
Besides ceramic methods, many metal-based phosphates or silicates can easily be obtained at mild temperatures via hydrothermal/solvothermal processes which simply enlist precipitation reactions. Basically, a solvothermal reaction consists in reacting in liquid media the corresponding metal/non metal-based soluble salt precursors together with the proper added bases, and increasing the temperature to promote the precipitation and growth of the desired phase via Ostwald ripening. It simply differs from a hydrothermal reaction, which requires harsher experimental conditions, namely high pressure as an extra variable, to either control the precipitation kinetics or trigger the precipitation of metastable phases.
Hydrothermal processes performed in water media and relying on the following reaction [H3PO4 + FeSO4 + 3LiOH ⇒ LiFePO4 + Li2SO4 + 3H2O] have been successfully used to synthesize lithium iron phosphate (LFP) [11]. Using the same precursors, Delacourt et al. [12] cleverly showed the feasibility of solvothermal preparation of monodisperse nanosized LiFePO4 powders by adding DMSO, as a high boiling solvent, to water in order to increase the boiling point of the reacting (water/DMSO) media to about 120 °C, the temperature at which LiFePO4 could precipitate. Although such approaches, namely the last one, provide high quality monodispersed powders, they present inherent limitations linked to the fact that:
- • they involve the local precipitation of Fe(OH)2 by LiOH with the constant risk of air oxidation leading to Fe(III) impurities in the final product;
- • they require the use of costly LiOH in three-fold excess, hence falling short of the economy of atoms.
To eschew these drawbacks, while still preserving the advantages of a precipitation process, we have pursued an approach, that we discovered had been pioneered by Makitjevij et al. [13–15] back in the late 1990s, for the precipitation of alkaline earth carbonates; this approach enlists the use of a basic medium created by the temperature-driven decomposition of urea into NH3 and CO2 upon hydrolysis at 90 °C. Mindful of this early work, we implemented this concept of “latent bases”, also called “forced hydrolysis”, to the hydrothermal preparation of various phosphates (LiFePO4, LiMnPO4, LiNiPO4, LiCoPO4), which were predicted from thermodynamic calculations to precipitate in aqueous media at basic pHs [8]. More specifically, the experiments were conducted as follows. Urea and LiH2PO4, in a molar ratio 1.2:1 so as to have a slight excess of NH3 to neutralize LiH2PO4 and raise the solution pH through the reaction, were dissolved under magnetic stirring in water during 10 minutes. Stoichiometric amounts of FeSO4·7H2O, MnSO4·H2O, NiSO4·6H2O or CoSO4·7H2O to produce LiMPO4 with either M = Fe, Mn, Ni and Co were then added to the solution, respectively. After 10 minutes of magnetic stirring, the solution, whose pH ranged from 3 to 4, was poured into a PTFE beaker placed in an autoclave. Once the atmosphere of the autoclave had been deoxygenated by flushing with argon, the solution temperature, while being maintained under constant agitation, was increased up to 180 °C at a rate of 1 °C/minute, maintained at that temperature for 3 hours, and then left to cool down to ambient temperature. The recouped solutions having pH ranging from 9 to 10 were then filtered, and the resulting solid phase washed with de-ionized water, alcohol and acetone and dried in air at 60 °C for two hours prior to being characterized by X-ray diffraction. Regardless of the nature of M, with the exception of M = Ni, the XRD pattern indicates the presence of the single phase materials (Fig. 1) whose lattice parameters are reported in Fig. 1 (insets). For M = Ni the obtained XRD reveals the presence of a not yet identified phase which transforms into the previously reported LiNiPO4 after two hours of annealing in air at 600 °C. Finally, it is worth mentioning also that the synthesis of LiCoPO4 with similar degree of crystallinity was done at slightly higher temperatures than for LiFePO4, 240 °C instead of 180 °C while maintaining the same reaction time. The need for such a higher synthesis temperature is most likely nested in the higher complexation of Co2+ as compared to Fe+2.

XRD powder patterns for various LiMPO4 phases (M = Fe, Mn, Ni, Co) prepared using urea as the “latent base”. The lattice parameters and space-groups are listed within the frame of the X-ray powder pattern for each sample. They were determined by Profile matching using the program FullProf® [23] (Windows version, March 2007). For reasons of fluorescence, the XRD patterns of Fe and Co-based phosphates were taken using a CoKα radiation as compared to those of Mn and Ni-based phosphates which were taken using a CuKα radiation. An excellent fitting can be observed for Co, Mn and Fe-based phosphates as opposed to the LiNiPO4 which show some remaining impurities. The ionothermal preparation of pure LiNiPO4 is feasible as will be reported in a forthcoming paper.
Besides urea, numerous other molecules (guanidinium or isocyanate salts, hexamethylenetetramine) capable of producing in situ, via a solvolysis reaction or via a temperature-controlled decarboxylation, Brønsted's or Lewis's bases (Fig. 2, left) necessary to the formation of LiMPO4, were successfully used to prepare the aforementioned phosphates. Additionally, it should be recognized that in most latent bases the protons can be substituted for organic groups to lead to amines, which base strength and surface tension can be controlled, hence providing a large number of candidates.
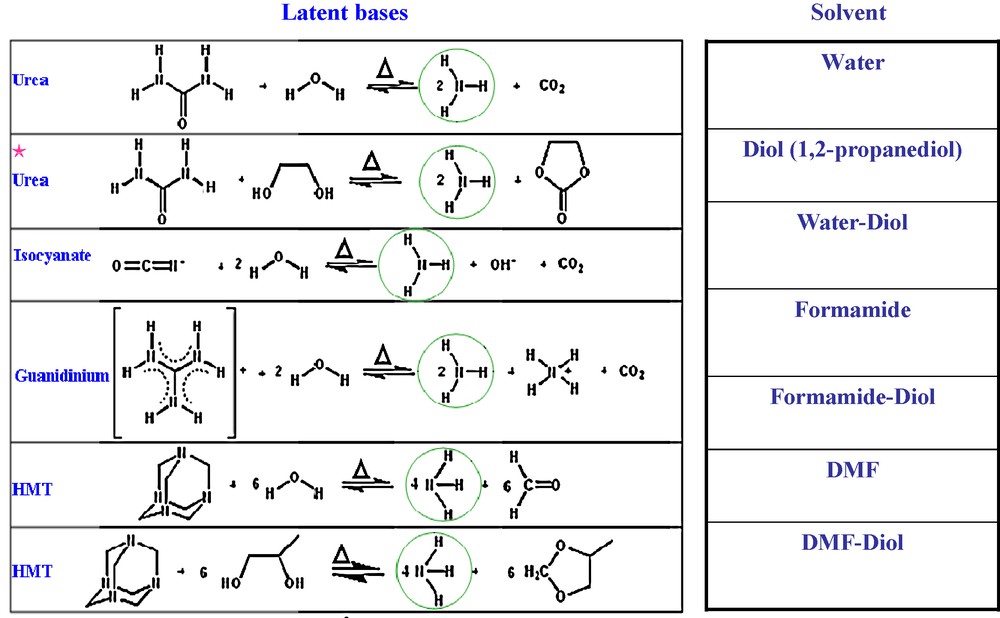
Various “latent bases”, together with the corresponding reaction by which they liberate NH3, are shown on the left; on the right the various solvents we have tried, including aqueous and non-aqueous ones. Within this figure, HMT and DMF stand for hexamethylene teramine and dimethylformamide, respectively.
Moreover, an additional attractive aspect of such a novel concept lies in the nature/choice of a solvolysis agent chemically compatible with the “latent base” to trigger the release of a base [8]. While water is by all means the most popular solvolysis agent, other polar liquids containing protic groups (OH, NH) such as diols or amides (Fig. 2, right) can be used, therefore providing myriads of opportunities to adjust the morphology/texture of the newly born oxide phases. For instance, nicely crystallized LiFePO4 were successfully produced in 1,2-propanediol using the same precursors and reacting protocol as before (24 hours at 180 °C). Moreover, we have shown that particle size and morphology can be deliberately tuned by using various water/1,2-propanediol solution mixtures (Fig. 3). Apart from these water/diols, there is also the possibility of using non-aqueous solvents to trigger the solvolysis reactions. Among the ones we have tried, formamide (F) and dimethylformamide (DMF) are worth further comments because they enable not only to conduct experiments at higher temperatures owing to their higher boiling point as compared to water but also to use a wide variety of precursors owing to their high dielectric constant.
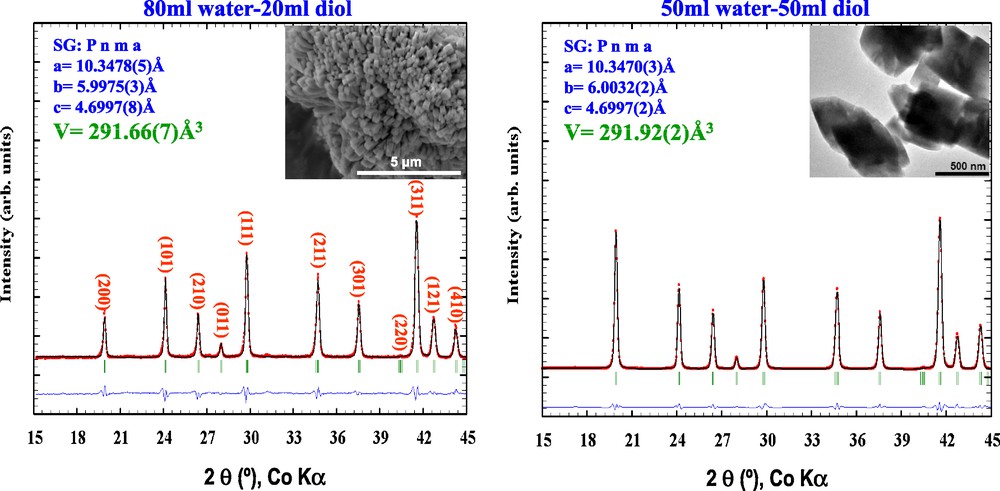
XRD diffraction patterns for LiFePO4 powders prepared using urea with different water-diol solution mixtures together with the corresponding SEM and TEM pictures for the 80–20 and 50–50 water-diol solvent mixtures, respectively as insets. A spectacular change in the (200)/(101) peak intensity with increasing the diol concentration is observed. Scanning electron microscopy images were recorded with a Philips FEG XL-30 operating at 20 kV while the TEM pictures were performed using a FEI Tecnai F20 S-Twin electron microscope operating at 200 kV. Samples for electron microscopy analysis were prepared by depositing a dispersion of nanoparticles on copper grids coated with lacey-carbon film.
Somewhat unexpected, but worth mentioning, is our success in synthesising LiFePO4 by solely reacting FeCl2 and LiH2PO4 in a formamide media in total absence of either diol or urea. The Fe precursor was changed as the solubility of FeSO4.7H2O is very limited in DMF. This implies that formamide molecules could themselves act as “latent base”. Obviously such a reaction no longer occurs via a solvolysis reaction but rather through a temperature-driven decomposition reaction. Indeed, we experimentally checked via complementary DSC–mass spectrometry measurements that, upon heating formamide, there is a release of NH3, which starts at 180 °C and becomes appreciable near 200 °C. Thus, the use of formamide offers another dimension to our concept of ‘latent base’ though the release of NH3 (+ CO) is more difficult to control.
By carefully choosing the nature of bases and solvents we reveal the countless opportunities provided by this new approach of controlling the size/shape growth of the oxides formed, although a complete study remains to be done to determine the template role played by these various additives in orienting nucleation-growth processes. Besides acting on the particle size and shape, we have also shown that a side advantage of using DMF resides in the growth of a polymer surface layer, observed by TEM and whose composition has not yet been determined, but which can lead to a carbon coating at higher temperatures, providing the electronic conductivity these materials lack when used as electrode materials. Therefore, such reactions still suffer from the mandatory need to be conducted in autoclaves. Bypassing such a limitation requires the use of non-volatile reacting media, hence the use of ionic liquids.
3 Ionothermal synthesis of LiFePO4
Like water, ionic liquids resulting from compatible cationic/anionic pairs have excellent solvent properties, but in addition they possess high thermal stability and negligible volatility so that the use of an autoclave is not mandatory. Ionic liquids have been intensively used in the field of organic synthesis, but mainly neglected within the field of inorganic synthesis. In contrast, molten salt synthesis methods are common practice for inorganic chemists. This is somewhat surprising as ionic liquids are simply molten salts that are liquids at room temperature. Besides, while the number of molten salts is somewhat limited to a handful of examples, the number of ionic liquid formulations obtained by either changing the nature of the cations (pyridinium vs. imidazolium), the anions (from fluorinated [TFSI–] to non-fluorinated [Cl−]), or by using cationic substitution (alkyl-chains vs. OH or CN termination) is practically infinite. To our knowledge, Morris [16,17] was among the first, back in 2003, to break this artificial paradigm by showing the possibility of synthesizing a wide variety of metal organic framework compounds (MOFs) in ionic liquid media. Therefore, the final MOF frequently contained the ionic liquid cation as part of the framework, whose removal was detrimental to the stability of the parent host.
Regardless of such difficulty, we decided to explore the feasibility of using an ionic liquid rather than water to conduct low temperature inorganic synthesis, and the results went beyond our expectations. The approach was first implemented on the synthesis of LiFePO4 and generalized to a wide variety of compounds. As common to any type of synthesis, a key issue is the choice of precursors based on thermodynamics, acidobasicity and solubility considerations together with tabulated intrinsic chemical-physical characteristics. Relying on the colossal amount of ground work already existing for ionic liquids [18,19], we selected for our first experiment [9] (EMI-TFSI) 1-ethyl-3-methylimadozolium bis-(TriFluoromethaneSulfonyl Imide) (Solvionic® 115 ppm water content) as reacting medium and Fe oxalate (FeC2O4.2H2O) and LiH2PO4 as Fe- and Li-based precursors owing to their anticipated slight and partial solubility in EMI-TFSI, respectively. More specifically, stoichiometric amounts of FeC2O4.2H2O (99%, Aldrich) and LiH2PO4 (99%, Aldrich) powders to prepare 0.8 g LiFePO4 together with 5 ml of EMI-TFSI were placed in a 50 ml flask. After 10 minutes of stirring, the flask temperature was increased up to 250 °C at a rate of 5 °C/minute. The solution was maintained at that temperature for 24 hours then left to cool down to ambient temperature. At the end of synthesis, powder and ionic liquid were separated by centrifugation. To recycle the IL, the supernatant was subsequently washed with concentrated HCl solutions to remove any trace of powder, which could have remained after centrifugation, water to eliminate HCl traces, and then diluted with dichloromethane (CH2Cl2) to separate it from water. Afterwards, dichloromethane was removed by distillation leaving behind the regenerated ionic liquid having low water content (< 200 ppm) that can be reused for a new synthesis, which is a plus from an economical point of view. In parallel, the recovered greenish powder was then subsequently washed with acetone, distilled water, and acetone again prior to being oven-dried at 60 °C. The resulting powders were single-phased as deduced by XRD measurements with lattice parameters (a = 10.333(3) Å, b = 6.007(1) Å, c = 4.691(1) Å; V = 291.1(1) Å3) in good agreement with the literature [10]. At this juncture we should recall that temperatures as high as 450–500 °C are needed to prepare LiFePO4 by solid-state reactions, thus indicating the benefits of ionic liquids in lowering the synthesis temperature by at least 200 °C as compared to classical ceramic approaches.
Besides, owing to the humongous variances between ionic liquids coming from their rich chemistry, we experienced this process to be quite versatile and not specific to a single type of ionic liquid. A survey of various ionic liquids differentiated either by the nature of the cations (imidazolium vs. pyridinium,) or substituants (alkyl chain length or end-groups,) or anionic groups (TFSI– vs. Cl−) was conducted (Table 1). Among the numerous ionic liquids we have tried, 1-butyl-1-methylpyrolidium bis(trifluoromethanesulfonyl)imide, containing a pyrrolidinium rather than an azolium cation, and 1-ethyl-3-methylimadozolium chlorine, containing a Cl− rather than a TFSI− anion were the only ones that could not enable the formation of LiFePO4. Therefore, we showed that by changing the nature of the Fe precursor (FeCl2 instead of the oxalate) and of the Li precursor (Li3PO4 instead of LiH2PO4) we could successfully obtain LiFePO4 at 200 °C. Finally, we demonstrated that their synthesis can be made with heating times as short as one hour using microwave heating.
List of the Ionic liquids tried to prepare LiFePO4 powders with the success of the ionothermal reaction identified by Yes or No.
| Formula | Name | LiFePO4 obtained | Reaction T (°C) |
| 1-ethyl-3-methylimidazolium Bis(trifluoromethanesulfonyl)imide (EMI-TFSI) | Yes | 250 | |
| 1-hexyl-3-methylimidazolium Bis(trifluoromethanesulfonyl)imide (C6) | Yes | 250 | |
| 1-octadecyl-3-methylimidazolium Bis(trifluoromethanesulfonyl)imide (C18) | Yes | 250 | |
| 1-butyl-3-methylimidazolium trifluoromethanesulfonate | Yes | 250 | |
| 1-(3-cyanopropyl)-3-methylimidazolium Bis(trifluoromethanesulfonyl)imide (CN) | Yes | 250 | |
| 1-(3-hydroxypropyl)-methylimidazolium Bis(trifluoromethanesulfonyl)imide (OH) | Yes | 200 | |
| 1-propyl-2,3-dimethylimidazolium Bis(trifluoromethanesulfonyl)imide | Yes | 250 | |
| 1-propylpyridinium- Bis(trifluoromethanesulfonyl)imide | Yes | 250 | |
| 1-butyl-1-methylpyrrolidinium Bis(trifluoromethanesulfonyl)imide | No | 250 | |
| 1-Butyl-3-methylimidazolium chloride | No | 250 |
Although we could prepare LiFePO4, almost regardless of the ionic liquid used, the most spectacular result from this survey remains the drastic importance of the ionic liquid nature on the morphology of the LiFePO4 particles (platelets vs. needles) that we obtained, suggesting that ionic liquids can act as structure directing agent. For instance, EMI-TFSI-type ionic liquids (Table 1) differentiated by the alkyl-chain terminating groups (CN– vs. OH–) were found to direct the growth of particles perpendicular to the [101] direction (CN) as compared to the [200] direction (OH) (Fig. 4). The pronounced dependence upon the nature of the ionic liquids used to conduct the reaction and the final shape and sizes of the crystal clearly imply a template role to the ionic liquid media. Therefore, our present understanding of the nucleation/growth mechanism of such crystals is still very sketchy. HRTEM studies have indicated that the LiFePO4 growth does occur from the solution, which at first was somewhat surprising as we know from solubility measurements that both precursors LiH2PO4 and more so FeC2O4.2H2O are weakly soluble in EMI-TFSI. Moreover, DSC measurements have shown that LiFePO4 growth does not occur until the precursors begin to decompose. In light of those aforementioned remarks, we hypothesized that the growth of LiFePO4 could result, besides the weak solubility of the precursors, from the decomposition of the precursors into soluble reacting species, which rapidly react to form the appropriate phase with the ionic liquid serving as transportation media. The feasibility of growing LiFePO4 at 180 °C in EMI-TFSI but using more soluble precursors such as FeCl2 with Li3PO4, for instance, is consistent with the above hypothesis.

XRD diffraction patterns of LiFePO4 powders prepared using different ionic liquids together with as an inset their corresponding morphology as deduced by SEM. Note that the intensity of the (101) peak is greater than that of the (200) when 1-(3-cyanopropyl)-3-methylimidazolium Bis(trifluoromethanesulfonyl) imide ionic liquid (CN-based) is used implying a [100] growth direction of the particle. In contrast, when 1-octadecyl-3-methylimidazolium (trifluoromethanesulfonyl)imide ionic liquid (C18) is used, the (101)/(200) intensity ratio is reversed suggesting a particle growth perpendicular to the [200] direction.
Mindful of this solubility issue together with the template role that the ionic liquid could play, we decided to consider non-miscible water-ionic liquid mixtures within which H2O droplets or nanodomains will act as solubilizer (high ɛ, donor number DN, acceptor number AN) — e.g. nanoreactors — and an ionic liquid as template. Several synthesis attempts were realized in autoclaves lined with Teflon® and using stoichiometric amounts of FeC2O4.2H2O (99%, Aldrich) and LiH2PO4 (99%, Aldrich) added to 5 ml of a EMI–TFSI–H2O solution having the following ionic liquid to water repartition in volume in cm3 (5 – 0; 4 – 1; 3 – 2; 2.5 – 2.5, 2 – 3, 1 – 4, 0 – 5). Once stirred for 10 minutes, the solutions were placed in an autoclave whose temperature was raised to 250 °C and maintained for 24 hours prior to being cooled down. Regardless of the IL to water ratio (Fig. 5), the XRDs of the recovered powders reveal the presence of single-phased LiFePO4 with therefore some changes in the relative intensity of the (101) peak as compared to the (200), indicative of a preferential growth along the [101] direction. The maximum preferred orientation is obtained for a 50–50 EMI–TFSI–water volume ratio, and can be brought together that this [200] growth orientation is privileged in the presence of OH-based EMI-TFSI ionic liquid. As deduced by Scanning Electron Microscopy (SEM), such powders consist of micron-sized crystals (Fig. 5 middle) with well defined facets and salient edges whose shape evolves from platelets, needles, parallelepipeds to hollow structures as a function of the IL/water ratio, in a non systematic way. Enhancing the reaction time to 48 hours instead of 24 hours resulted in a particle coarsening as expected (Fig. 5 right), offering therefore the possibility of growing well-shaped crystals for extensive transport (electronic and ionic) characterization. We experienced with similar success the possibility of carrying out similar syntheses with an ionic-liquid diol-mixture, once again illustrating the richness of such a new synthesis path.

XRD patterns of LiFePO4 powders prepared using various (EMI-TFSI)-water mixtures, after a reacting time of 24 hours at 250 °C together with the corresponding SEM pictures. Changing the (EMI-TFSI)/water ratio leads to drastic morphology changes which become more pronounced after 48 hours (right SEM pictures).
Importantly, it should be noted that through this systematic study we succeeded in preparing LiFePO4 in aqueous media from stoichiometry amounts of LiH2PO4 and FeC2O4.2H20. At first, this is quite surprising since we claimed that LiFePO4 can only precipitate in basic media, hence the addition of bases in all syntheses so far reported. The question should therefore be raised: under which circumstances could iron oxalate act as a base? It is most likely the decomposition of C2O42− ⇒ CO32− + CO raises the reaction pH via the formation of carbonate ions which are in equilibrium with CO2 (CO32− + H2O ⇔ CO2 + 2OH−) at high pHs. Thus, iron-oxalate falls to a certain extent within the family of the presently defined “Latent bases”, although being an inorganic compound.
Returning to the ionic liquid reaction, obviously we cannot talk in terms of pH, but acidic and basic considerations must prevail to explain the growth mechanistic. What is most likely happening here is that iron oxalate decomposes near 200 °C according to the following reaction, FeC2O4 ⇒ CO + CO2 + FeO, giving “nascent FeO”, which acts as the base within the ionic media once formed. Regarding LiH2PO4, it decomposes into LiPO3 that seems to indicate that the presence of protons is not needed as indicated by our ability to prepare LiFePO4 from Li3PO4 and FeCl2 or FeSO4 instead of LiH2PO4.
Ionothermal synthesis is not specific to iron phosphates but can be extended to other members of the family (LiCoPO4, LiMnPO4 and LiNiPO4) by simply changing the nature of the 3d-metal based precursor, while keeping the same ionic liquid and the same experimental conditions. LiCoPO4 was, for instance, prepared by reacting LiH2PO4 and CoC2O4.2H2O in EMI-TFSI for 24 hours at 250 °C, and the advancement of the reaction was simply followed by the colour change of the solution; the latter evolves from brownish to purple when the reaction is completed as deduced from the XRD of the recovered powder. Additionally, such an approach was successfully applied to the synthesis of fluorophosphates, as we will be reporting in a forthcoming paper.
4 Conclusions
The ever growing demand for electrode materials made via eco-efficient processes has set a new challenge to solid-state chemists. Tackling this challenge we came out with two different approaches, a hydrothermal one relying on the use of “Latent bases” [20] and a solvothermal one based on the use of ionic liquids [21], capable of producing highly divided LiFePO4 powders having electrochemical properties (Fig. 6) comparing equally with the best ever reported LiFePO4 powders. Both methods necessitate only low temperatures (e.g. < 250 °C), do not require excess of reactants (e.g., economy of atoms), produce benign by-products such as, for instance, (NH4)2SO4, a fertilizer, use recoverable solvents and cheap precursors so as to be cost attractive. Nevertheless, much research effort is still needed to explore the potential of such new methods to its limits in designing tailor-made electrode materials. This applies more so to the ionothermal method, which has so far not been implemented in the field of inorganic chemistry. Despite these advances, the ultimate potential of these methods will not be reached unless we gain better insight in the reactions’ mechanisms. Questions requiring further attention include a deeper understanding of the reacting path by grasping better insight in:
- • the solvating properties of the ionic liquids (e.g. interactions between precursors and ILs);
- • the templating role of the ILs as function of their cations/anion chemistry;
- • the specificity of mixed ILs-water reacting media;
- • all aspects of the scale up of the ionothermal processes (e.g. ILs cost production, purity tolerance and efficient recovery process).

Left: room temperature voltage–composition curves collected at a C rate (1 Li in one hour) together with the capacity retention (inset). Right: rate capability (as determined by signature curves [22]) on the right for two Swagelok™-type Li/LiFePO4 cells using respectively 10 mg/cm2 of LiFePO4 powders; the latter are prepared using (1) a water(80 ml)-diol(20 ml) solution mixture containing urea as “latent base” and FeSO4.7H2O plus LiH2PO4 (red curve) and (2) ionic liquid (EMI-TFSI) as a reacting media in the presence of LiH2PO4 and FeC2O4.2H2O. Positive electrodes were prepared by mixing 85 wt% active material and 15 wt% SP® (MM, Belgium) carbon, as electronic conductor, and were cycled vs. Li using a LiPF6 (1 M) solution of ethylene carbonate (EC)/dimethyl carbonate (DMC) mixture (1/1 w/w) as electrolyte mixture. Data was collected using a VMP cycling/ data recording systems (Biologic SA, Claix, France).
Presently, the hydrothermal approach based on the use of iron-sulfate and urea as a “latent inorganic base” appears extremely attractive, cost-wise, to produce high quality LiFePO4 powders. Although less advanced, we expect the ionothermal approach to open numerous synthesis opportunities within the field of inorganic chemistry so as to make in a controlled fashion (e.g., size, morphology, and texture controlled) known phases (oxides, phosphates, silicates) as well as to stabilize new metastable ones. Additionally, the feasibility of using microwave heating to trigger the formation of these phases in ionic liquid media make the microwave-driven a high throughput synthesis method which deserves further investigation. It is therefore worth mentioning that making highly performing electrodes containing “Latent bases” or ionothermally made materials requires an additional tuning of our synthesis processes together with innovative electrode wiring which, although done in our group and to appear in forthcoming papers, are far beyond the scope of this paper.
Further into the future of such a race towards materials having the minimum footprint in nature made via eco-efficient processes, great hope is placed in the biosynthesis of electrode materials. Biomineralization processes, frequently used by nature, may be back again giving new life to materials for energy. Along that line, we are working with great success towards the development of an enzyme-mediated synthesis process for the preparation of LiFePO4 nanopowders.
Acknowledgments
The authors would like to thank all the members of the LRCS who have enabled to pursue such a type by research, they are indebted to Jacques Livage who has been our inspiration in the “chimie douce” approach.