

1 Introduction
Breast carcinoma is the leading cause of cancer mortality among women in the Western hemisphere [1]. Endometriosis is diagnosed in about 10% of women in the reproductive age [2]. It has been shown that estrogens play an important role in both diseases [3–5]. Common therapies target the systemic action of estradiol (E2) and consequently cause severe side effects. Thus, there is need for novel therapeutical approaches. 17β-Hydroxysteroid dehydrogenase type 1 (17βHSD1), which catalyses the NADPH-dependent reduction of the weakly potent estrogen estrone (E1) to the highly active E2 (Fig. 1), is often overexpressed in the diseased tissue [6–10]. Hence, a selective inhibition of this enzyme would lead to a reduction of active estrogen especially in the tissue concerned. Therefore, 17βHSD1 is considered as an appropriate new target for the therapy of estrogen dependent diseases.
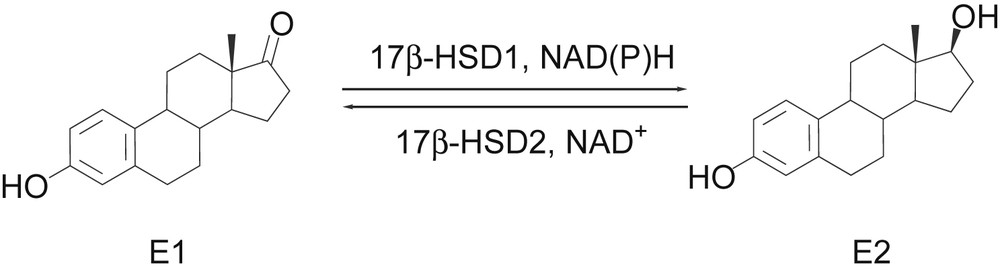
Estrogen interconversion by 17βHSDs.
Several requirements must be fulfilled for 17βHSD1-inhibitors to be applied in therapy. The major pharmacodynamic demands are high inhibitory activity at the target enzyme, selectivity towards its physiological counterpart 17βHSD2, which inactivates E2 to E1 (Fig. 1), and towards the estrogen receptors (ER) α and β, which mediate estrogenic effects. Moreover, the most important hepatic CYP-enzymes should not be inhibited to avoid interference with metabolism of other endogenous or exogenous compounds. Besides, the inhibitors must meet pharmacokinetic prerequisites such as cell permeability, metabolic stability, and a sufficient intestinal absorption. For this reason, a biological screening system was developed and applied to our compound collection of potential h17βHSD1-inhibitors [11]. Using this test sequence a number of highly active and selective inhibitors of the human target enzyme were identified to have adequate pharmacokinetic properties and hence to be suitable for further evaluations in a disease oriented animal model [12–16].
The efficacy of selected compounds was demonstrated in T47D cells. Thus, a first proof of concept for the indication of breast cancer was performed [17]. With respect on endometriosis therapy, the efficacy should be proven in an appropriate animal model like the rat. Before performing in vivo experiments it must be shown that the inhibitors of the human enzyme are active in the rat enzyme as well. For the selection of suitable compounds to investigate in the animal model, there is need for appropriate tests.
Here we report the establishment of two assays for the determination of selective inhibition of E2-production from E1 in rat tissue preparations. Using these two assays 100 potential 17βHSD1-inhibitors were evaluated for inhibitory activity of estrogen conversion.
2 Results
Ammonium sulphate precipitate of rat liver cytoplasm was used for the evaluation of the reduction of E1 to E2. Rat liver microsomes were applied for the reverse oxidative reaction. For tissue preparation fresh rat livers were homogenised and fractionally centrifuged. Ammonium sulphate precipitate was prepared and immediately tested for specific conversion of E1 into E2. Accordingly, the microsomal fraction was obtained and evaluated for activity. In both cases a specific reaction was detectable with a reasonable conversion. The amount of product formed was dependent on the dilution factor of the respective tissue preparation. The microsomal fraction showed a higher specific enzymatic activity (0.26 U/mg protein) than the ammonium sulphate precipitate (0.03 U/mg protein). Protein content was determined according to Bradford [18].
For an easy and reproducible test procedure it is desirable to preserve activity of the obtained fractions during storage. Thus, fractions were frozen in liquid nitrogen and retested for activity after they had been thawed. No activity was detected (data not shown). In a second procedure freezing buffer was supplemented with 20% of glycerol. Samples were frozen in liquid nitrogen and thawed repeatedly and after each cycle tested for activity. Under the applied conditions, enzymatic activity was maintained even after two freeze-thaw-cycles (Fig. 2).
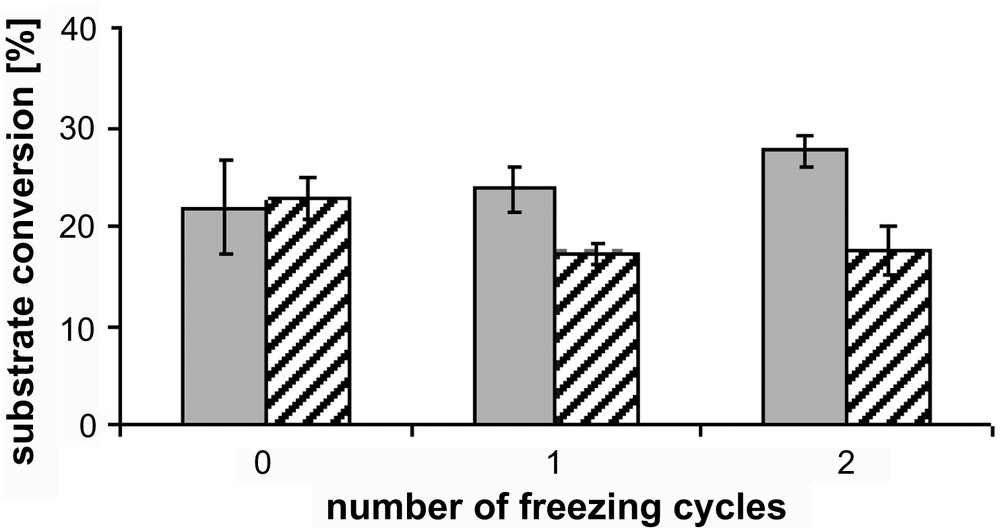
Maintenance of enzymatic activity after repeated freezing. Grey bars represent ammonium sulphate precipitate from cytosolic fraction, striped bars microsomal fraction. Conditions of E1- and E2-conversion were identical for each enzyme preparation.
Since both enzymes in principle are able to catalyse the respective back-reaction in a non cellular environment after addition of the corresponding cofactor [19], the reverse conditions were applied to each fraction. In both cases the respective back-reaction was observed running to a smaller extent indicating reasonable purity of the preparations (Fig. 3).
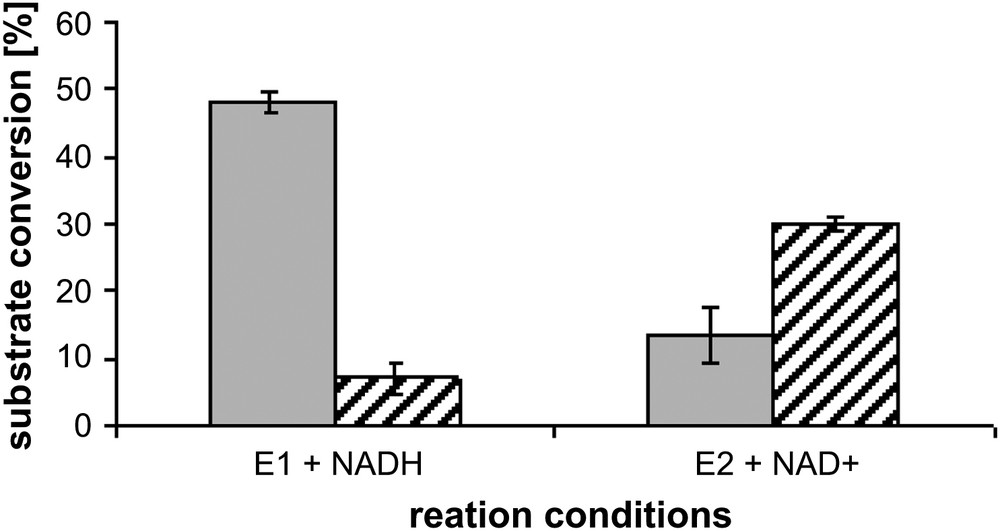
Direction of reaction under different conditions. Grey bars represent ammonium sulphate precipitate from cytosolic fraction, striped bars microsomal fraction. Conditions of E1- and E2-conversion were identical for each enzyme preparation. Concentration of E1 or E2, respectively: 500 nM, concentration of NADH: 500 μM, concentration of NAD+: 1500 μM, 37 °C.
For comparison of the results of the rat enzymes with the human enzymes, assay conditions have to be similar. Therefore, substrate concentrations (500 nM, each) were adopted from the established test procedures and respective cosubstrates were added in excess [20]. For reproducible inhibitor evaluation, the enzymatic conversion must be linear over time. Hence, time-dependent conversion was determined. In the cytosolic precipitate conversion was linear for 15 min whereas reaction ran linearly for 30 min in the microsomal fraction.
After definition of the respective reaction conditions 100 compounds were tested for inhibition of E2-formation. The compounds can be assigned to the structure classes depicted in Fig. 4, which were shown to contain selective inhibitors of h17βHSD1 [20–23]. For a comparison of human and rat data compounds with high, medium and without inhibitory activity at the human enzyme were chosen comprising a high structural diversity.

Structures of evaluated compounds.
For the identification of inhibitors of the rat enzyme, percent inhibition of the compounds at a concentration of 1 μM was determined. Although most of the screened compounds were inactive at the rat enzyme (data not shown), inhibitors could be identified both in the class of bis(hydroxyphenyl)heteroarenes and -arenes and 6-aryl-substituted 2-hydroxynaphthalenes (Table 1).
Structure of selected compounds tested for inhibition of E2-formation from E1 in rats.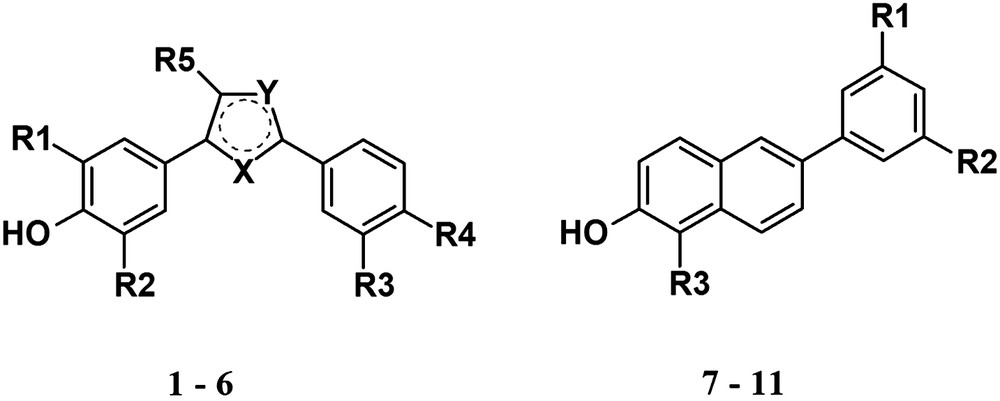
| Compound | X | Y | R1 | R2 | R3 | R4 | R5 |
| 1 | C | S | CH3 | H | OH | H | H |
| 2 | C | S | F | H | OH | H | H |
| 3 | S | C | F | H | OH | H | H |
| 4 | S | C | F | F | OH | H | H |
| 5 | S | C | F | H | OH | F | H |
| 6 | S | C | H | H | H | OH | |
| 7 | OH | H | |||||
| 8 | NO2 | H | |||||
| 9 | OH | H | |||||
| 10 | OH | H | |||||
| 11 | H | H | H |
Ten compounds turned out to be moderate or good inhibitors of the rat E2-production (% inhibition > 25%). For these compounds, IC50-values were determined. Four compounds showed IC50-values below 1 μM (Table 2).
Inhibition of rat E2- and E1-formation by selected compounds.
| Compound | h17βHSD1a | E2-formationb | E1-formationc | ||
| IC50 [μM] | Inhibition [%]d, e | IC50 [μM]d | Inhibition [%]d, e | IC50 [μM]d | |
| 1 | <0.1 | 27 | 2.60 | 51 | 0.98 |
| 2 | <0.1 | 55 | 0.85 | 83 | 0.24 |
| 3 | <0.1 | 50 | 1.08 | 68 | 0.42 |
| 4 | <0.1 | 81 | 0.27 | 95 | 0.08 |
| 5 | <0.1 | 47 | 0.96 | 69 | 0.45 |
| 6 | n.i. | 28 | 2.28 | 65 | 0.37 |
| 7 | n.i. | 49 | 1.08 | 66 | 0.56 |
| 8 | n.i. | 37 | 1.60 | 56 | 0.68 |
| 9 | <0.1 | 69 | 0.56 | 48 | 1.06 |
| 10 | <0.1 | 26 | 5.37 | <10 | 6.25 |
| 11 | >10 | <10 | n.d. | n.d. | n.d. |
a Human placenta, ammonium sulphate precipitate from cytosolic fraction, substrate [3H]-E1 + E1 [500 nM], cofactor NADH [500 μM], data given for comparison.
b Rat liver, ammonium sulphate precipitate from cytosolic fraction, substrate [3H]-E1 + E1 [500 nM], cofactor NADH [500 μM].
c Rat liver, microsomal fraction, substrate [3H]-E2 + E2 [500 nM], cofactor NAD+ [1500 μM].
d Mean values of three determinations standard deviation less than 17%.
e Inhibitor concentration 1 μM.
Interestingly, in case of the bis(hydroxyphenyl)heteroarenes only sulphur-containing heterocycles as core showed inhibition of the enzymatic reaction, the 2,5-disubstitution of the heterocycle in general leading to a higher activity than the 2,4-disubstitution. The combination of both substitution patterns, as implemented in the 2,4,5-tris(hydroxyphenyl)-thiophene (6), does not increase inhibitory potency but leads to medium activity. Moreover, it is striking that the highly active inhibitors of this series are fluorine-substituted, the most potent being compound 4.
Regarding compounds with a naphthalene system most of the identified inhibitors were 6-hydroxyphenyl-substituted 2-naphthols (7, 9, and 10). Obviously, the hydroxyl-substituent of the phenyl moiety is needed for activity in the rat enzyme (11). However, a 6-nitrophenyl-substituted 2-naphthol (8) also showed moderate inhibition of E2-production. Generally, two substitution sites in addition to the hydroxy-groups seem to be tolerated, the 5′-position of the hydroxy- or nitrophenyl-moiety and the 1-position of the naphthalene core, leading to comparable inhibitory activities (7 and 9). In 1-position of the hydroxynaphthalene core a phenyl-moiety increased inhibitory activity compared to the unsubstituted compound (11% inhibition at 1 μM vs. of 69% for 9). However, further substitution of this phenyl-ring decreased activity again (data not shown).
Comparing inhibition of human and rat E2-synthesis it is noticeable that highly active h17βHSD1-inhibitors show also moderate to good inhibition of rat E2-formation. On the other hand, there are also compounds among the ten best inhibitors in the rat which lack inhibitory activity at h17βHSD1. Remarkably, in 5′-position even a large substituent is tolerated by the rat enzyme while this compound is inactive at the human protein (Table 2, compounds 7 and 8). Nevertheless as a rule highly active h17βHSD1-inhibitors were observed to show inhibition of E2-activation in rat liver preparations.
3 Discussion and conclusion
Using our screening system we had been able to identify a number of h17βHSD1-inhibitors with suitable properties for further preclinical studies [20–23]. As a next step, their efficacy should be demonstrated in a disease oriented animal model. Currently, several animal models are available using different species and addressing the two estrogen dependent diseases, breast cancer and endometriosis.
Regarding breast cancer two models can be considered for application, both using nude mice as host for human breast cancer cells [12,15,24]. However, due to the artificial environment of the established tumour cell line the prediction of side effects of the inhibitors is limited.
Concerning endometriosis several models are potentially usable for the evaluation of inhibitors. One of them is similar to the breast cancer experiments in nude mice and therefore bears similar advantages and drawbacks. It uses human endometrial fragments, which are introduced into the peritoneal cavity of nude mice [14,25]. Furthermore, there are two monkey models available, in which an endometriosis is established by introducing autografts of endometrial tissue into the abdomen of the animals. One model is working with marmoset monkeys, non menstruating primates, the other with menstruating cynomolgus monkeys [13,26]. Both give a good insight into endometriosis and its potential treatments. However, major disadvantage of these models is the fact that they are very labour-intensive and require monkey specific 17βHSD1-inhibitors. A further described model is applicable to different rodent species such as hamster, mouse, and rat. In this experiment, endometrial autografts are implanted into the dorsal skinfold [16,27]. This is a relatively artificial model, but on the other hand has the advantage of host-derived disease tissue. Moreover, aminoacid-sequences of rat and mouse proteins are known and can possibly be used for the identification of inhibitors.
However, before performing an animal experiment in a certain species pharmacokinetic data of the active inhibitors have to be determined for the respective animal. First routine pharmacokinetic-studies in rats are promising, and we decided to screen for inhibitors of the rat E2-formation first aiming at a potential application in the mentioned endometriosis model.
For this purpose, rat liver was used as enzyme source, because 17βHSD1-activity was described for this tissue [28]. As it is the case in humans, rat 17βHSD1 is a cytosolic enzyme, whereas 17βHSD2 is membrane associated [29,30]. Consequently, tissue was homogenised and fractionated by centrifugation. In both obtained fractions (cytosol and microsomes) the expected conversions were found to run specifically. The finding that there is obviously a pronounced E2-inactivation might be related to the discovery that enzymatic activity in case of E2-oxidation is age dependent as was shown before [31].
Screening of our substance library containing of highly active 17βHSD1-inhibitors for selective inhibition of E2-formation in the rat led to ten compounds with moderate to good activity. Keeping in mind that the compounds were designed as substrate mimetics a higher number of compounds active in the rat enzyme were expected. On the other hand, those compounds with high inhibitory activity towards the human enzyme were also moderate to good inhibitors of rat E2-formation. Structure-activity-relationships are similar for both species although there is only an aminoacid identity of 68% between the two proteins [32]. Inhibitors of the rodent E1-reduction which were not active in the human enzyme (6–8) have large substituents in positions which differ from those of the highly active h17βHSD1-inhibitors. This finding indicates, that there is additional space available in the rat enzyme which is not present in the human.
In conclusion, we successfully established assays for the inhibition of E2-formation from E1 and vice versa in rats. Using these tests, we are able to determine the potency of a compound to selectively inhibit E2-formation in rats. Applying the developed assays a screening for rat 17βHSD1-inhibitors was performed and compounds with a satisfying inhibitory activity were identified.
4 Experimental section
[3H]-E1 and [3H]-E2 were purchased from Perkin Elmer, Boston. Quickszint Flow 302 scintillator fluid was bought from Zinsser Analytic, Frankfurt. Other chemicals were purchased from Sigma, Roth or Merck.
4.1 Tissue preparation and determination of inhibition
Cytosol and microsomal fractions from rat liver tissue were separated according to a procedure described previously [31,33]. Ammonium sulphate precipitate from the cytosolic fraction was used for evaluation of E2-formation. Microsomal fraction was purified and used for the determination of E2-inactivation.
For determination of enzymatic activity, the preparations were incubated with NADH and [3H]-E1 (precipitate) or NAD+ and [3H]-E2 (microsomes), respectively, at 37 °C in a phosphate buffer (pH 7.4) supplemented with 20% of glycerol and EDTA 1 mM. Reaction was stopped with HgCl2 and steroids were extracted into ether. Substrates and products were separated using an acetonitrile/water (45/55 v/v) mixture as mobile phase in a C18 rp chromatography column (Nucleodur C18, 3 μm, Macherey-Nagel, Düren) connected to a HPLC-system (Agilent 1100 Series, Agilent Technologies, Waldbronn). Detection and quantification of the steroids were performed using a radioflow detector (Berthold Technologies, Bad Wildbad).
4.2 Specific enzymatic activity
Protein amount of the preparations was determined as described by Bradford [18]. Specific activities were given in U/mg protein.
Conversion rates were evaluated and the formation of one micromole of product per minute was defined as representing 1 U enzyme activity.
Acknowledgement
We are grateful to the Deutsche Forschungsgemeinschaft for financial support (HA1313/8-1) of this work. P.K. is grateful for a scholarship of European Postgraduate School (DFG) GRK532. We thank Beate Geiger for her help in evaluation of inhibitors and Pharmacelsus CRO for providing us with rat livers.