

1 Introduction
Cytochrome P450s are a large family of heme-containing enzymes, catalyzing a variety of reactions including hydroxylations, epoxidations, N-dealkylations, O-dealkylations and S-oxidation. Although exceptions can be found, most of them act as monooxygenases, catalyzing the insertion of a single oxygen atom into their substrate. Regarding their function, they can be divided into two groups: the hepatic enzymes unspecifically convert endogenous and exogenous compounds, like drugs, into more hydrophilic products. The other group consists of enzymes with a very specific role in the biosynthesis of bioactive molecules like hormones. In this work we will focus our attention on the latter group and therein especially on the CYP enzymes involved in the human steroid biosynthesis. These enzymes show in contrast to the hepatic CYPs high substrate specificity and are therefore principally suitable as drug targets.
In steroidogenesis six CYP enzymes are involved (Scheme 1), namely CYP11A1 (cholesterol side chain cleavage enzyme, SCC, desmolase), CYP21 (steroid-21-hydroxylase), CYP17 (17α-hydroxylase-C17, 20-lyase), CYP19 (aromatase), CYP11B1 (steroid-11beta-hydroxylase) and CYP11B2 (aldosterone-synthase). As inhibition of CYP11A1 affects formation of all steroid hormones it is not appropriate as drug target. The same is true for CYP21 being involved in the biosynthesis of both gluco- and mineralocorticoids.
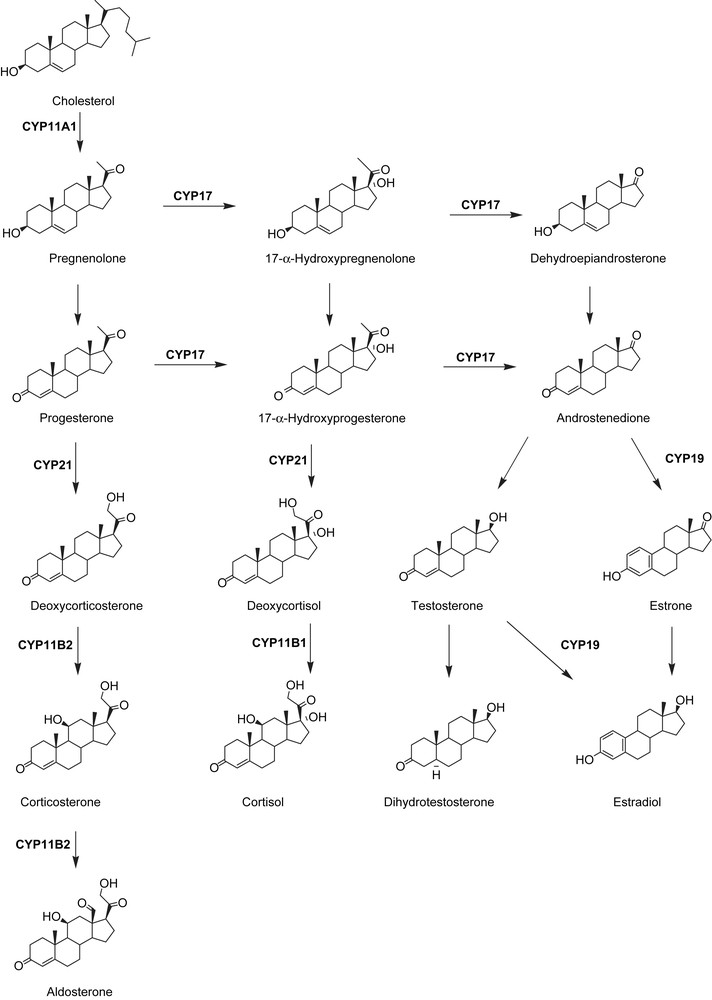
CYP enzymes in steroid biosynthesis.
So far, the most successful example of targeting CYP enzymes is presumably the introduction of aromatase (CYP19) inhibitors for the treatment of hormone dependent breast cancer (HDBC). With these drugs it was possible to overcome the drawbacks of the anti-estrogen tamoxifen, which used to be the standard treatment of HDBC for years, although having many side effects due to its affinity to the estrogen receptor. In 1977 the aromatase inhibitor formestane was proven to be an efficient therapeutic for HDBC [1]. In the following years some steroidal and non-steroidal inhibitors have been approved. Although there are several inhibitors available today it is worthwhile to engage in the development of new entities in this field for several reasons, e.g. new estrogen dependent indications like endometriosis are emerging.
A similar approach was followed for the treatment of hormone dependent prostate cancer (HDPC). After Huggins and Hodges had shown in 1941 that androgen deprivation is effective against a high percentage of prostate tumours [2,3], the surgical castration became the standard treatment. Later “medical castration” using GnRH analogues (agonists [4] and very recently also antagonists [5]) came up as an alternative. However, neither treatment is able to affect adrenal androgen production. Testicular and adrenal androgen production could be suppressed by CYP17 inhibitors. The first clinically administered, rather unselective CYP17 inhibitor was the antimycotic ketoconazole. It turned out to be highly effective against prostate tumours [6,7], but finally was not accepted as it showed severe side effects such as liver toxicity [8]. The basic problem that CYP17 inhibitors also reduce glucocorticoid levels (Scheme 1) could be solved in these clinical studies with ketoconazole. Low dose treatment regimens are capable of reducing testosterone levels but do not affect glucocorticoid concentrations [9].Very recently, the steroidal CYP17 inhibitor abiraterone has passed phase II clinical trials. It showed high activity and seems to have no dose-limiting toxicity [10]. Nevertheless, steroidal drugs are known for their potential side effects due to their affinities for other steroid hormone receptors. Therefore we investigated extensively in the development of non-steroidal CYP17 inhibitors.
The third enzyme to be regarded as a potential drug target is aldosterone-synthase (CYP11B2). As the aldosterone antagonists spironolactone and eplerenone have been proven to reduce mortality in patients suffering from congestive heart failure and myocardial fibrosis [11,12], we expect the inhibition of aldosterone formation to be a milestone for the treatment of these diseases. Although no selective inhibitor is on the market yet, a few compounds, like fadrozole or ketoconazole, are known to deprive aldosterone production [13,14]. However, these compounds unselectively interact with other CYP enzymes and show only moderate activity. Very recently, we reported on the discovery and development of highly active and selective CYP11B2 inhibitors with oral bioavailability in the rat [15].
As fourth steroidogenic CYP enzyme 11-β-steroidhydroxylase (CYP11B1) has recently been propagated as a novel target for diseases with abnormal cortisol levels like Cushinǵs syndrome [14], and first inhibitors have been described [16–18].
In the last two decades we have discovered a large number of highly active and selective CYP19 [19–22], CYP17 [23–26] and CYP11B2 [15–17,27] inhibitors, respectively. All of them contain a heterocycle with sp2-hybridized nitrogen, which is complexing the heme iron of the active site of the enzyme (Scheme 2). This heterocycle is substituting directly or via a linker a core structure consisting of at least two more rings. Furnished with appropriate substituents, highly active inhibitors (A, B and C) of the corresponding enzymes were obtained (Scheme 3).

Proposed model for CYP inhibitors: active site of the enzyme (heme and substrate binding site).
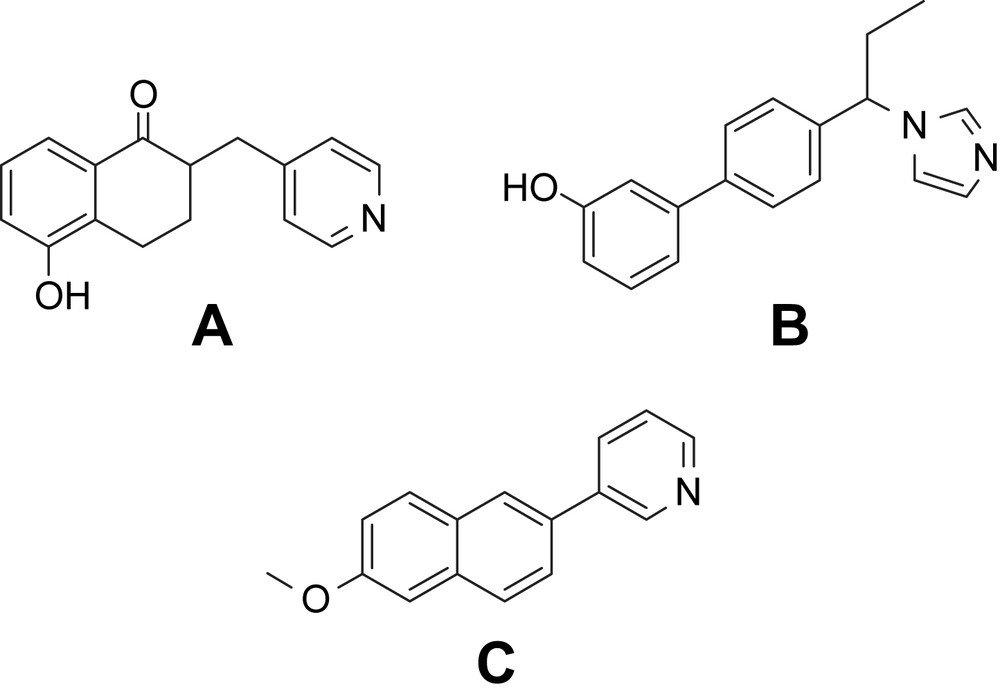
Highly active inhibitors of CYP19 (A) [21], CYP17 (B) [26], and CYP11B2 (C) [27].
In the following we describe a novel approach for the discovery of new hit compounds. Using our CYP19 (A), CYP17 (B) and CYP11B2 (C) inhibitors, we here have studied new combinations of their substructures (core: naphthalene, tetrahydronaphthalene, biphenyl; linker: no, methylene, substituted methylene; heterocycle: 3-pyridine, 4-pyridine, 1-imidazole) leading to compounds 1–7 (Table 1). The synthesis and biological evaluation regarding their activity and selectivity toward the four CYP enzymes are reported.
List of synthesized compounds 1–7. 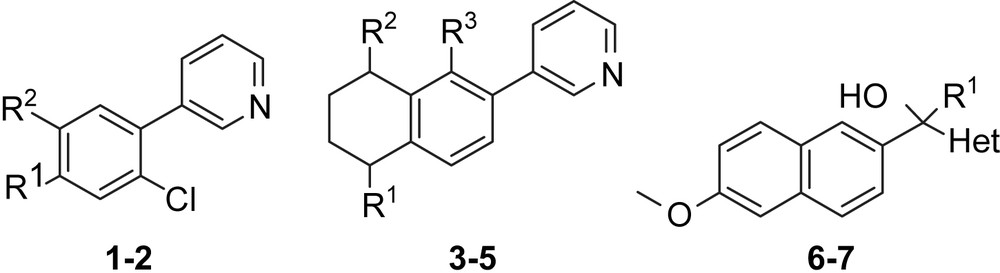
| Compound | R1 | R2 | R3 | Het |
| 1 | 2-OMe-Ph | H | ||
| 2 | H | 3-OMe-Ph | ||
| 3 | O | H | Cl | |
| 4 | H | O | H | |
| 5 | H | OH | H | |
| 6 | Et | 3-Py | ||
| 7 | H | 4-Py |
2 Chemistry
The syntheses of compounds 1–7 are shown in Schemes 4–7. In our aim to explore different core structures with improved activity and selectivity toward the most important steroidogenic CYP enzymes, a variety of structural quite different compounds were synthesized. They can be divided into biphenyls, naphthalenes and tetralones linked either directly or via a methylene bridge to heme-complexing pyridine.
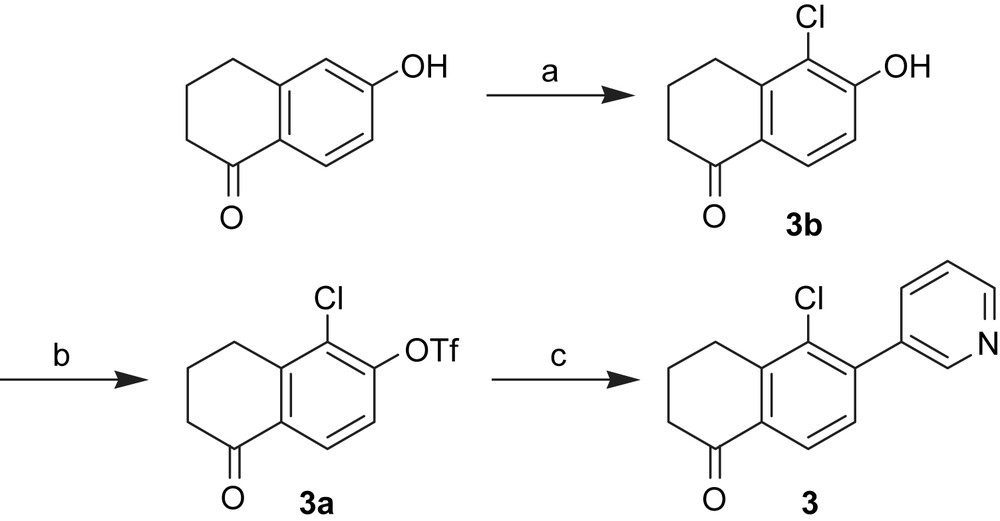
Synthesis of 6-substituted tetralone derivative. Reagents and conditions: (a) tert-BuOCl, DCM, 0 °C, 2 h; (b) Method A: Tf2O, pyridine, DCM, 0 °C – rt, 3 h; (c) Method B: 3-pyridinylboronic acid, Pd(PPh3)4, Na2CO3, toluene, H2O, reflux, 8 h.
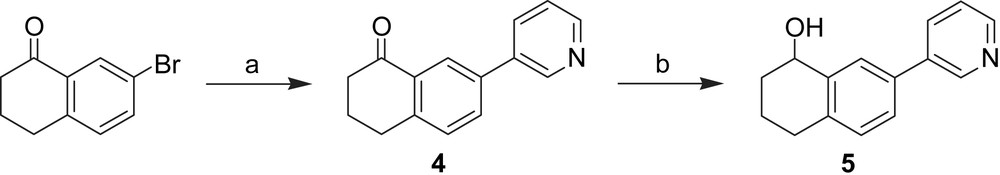
Synthesis of 7-substituted tetralone derivatives Reagents and conditions: (a) Method B: 3-pyridinylboronic acid, Pd(PPh3)4, Na2CO3, toluene, H2O, reflux, 8 h; (b) NaBH4, MeOH, THF, 0 °C – rt, 2 h.
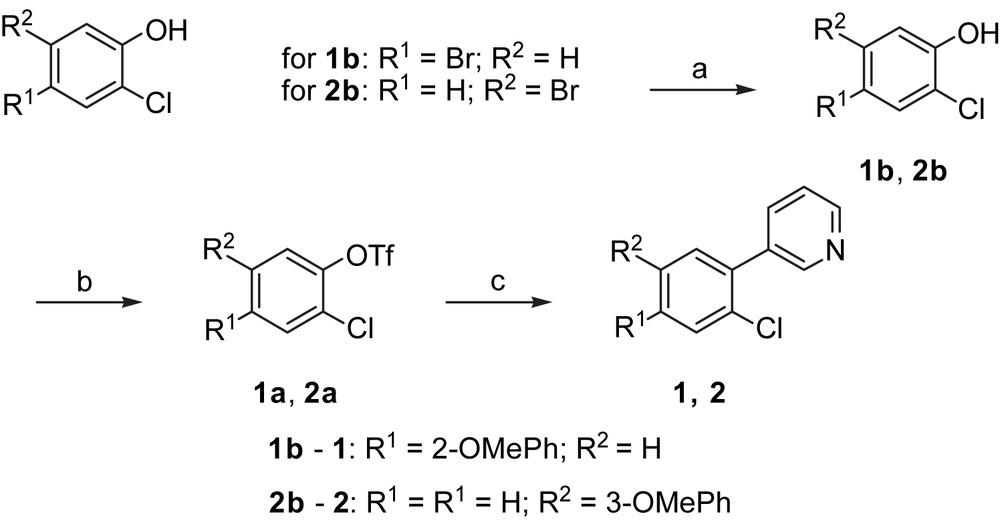
Synthesis of biphenylic derivatives. Reagents and conditions: (a) Method B: R1 B(OH)2 or R2 B(OH)2, Pd(PPh3)4, Na2CO3, toluene, H2O, reflux, 8 h; (b) Method A: pyridine, Tf2O, DCM, 0 °C – rt, 3 h; (c) Method B: 3-pyridinylboronic acid, Pd(PPh3)4, Na2CO3, toluene, H2O, reflux, 8 h.
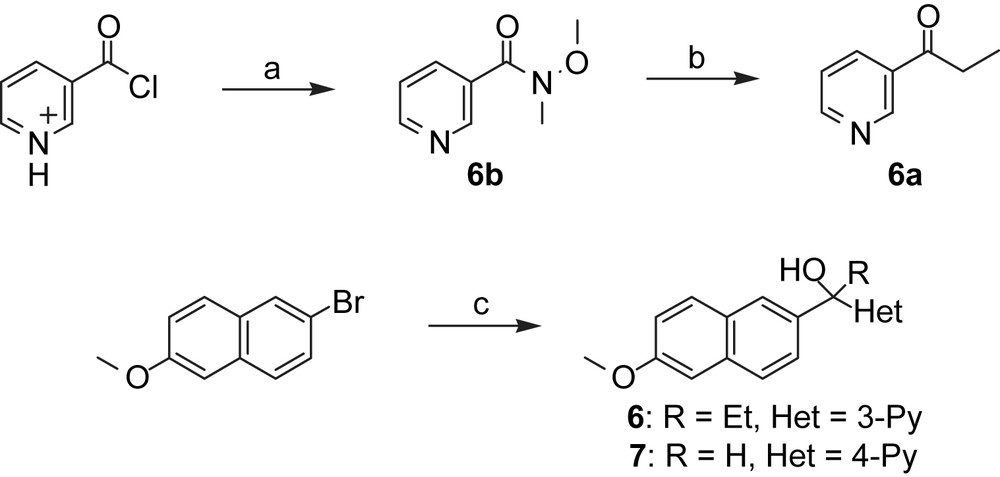
Synthesis of bridged naphthalene derivatives. Reagents and conditions: (a): NEt3, NH(OMe)2 × HCl, DCM, rt, 16 h; (b): EtMgBr, THF, rt, 8 h; (c): Method C:, tert-BuLi, THF, −78 °C, then 6a or isonicotinaldehyde, −78 °C–0 °C, 2 h.
The synthesis of biphenyls C–C bonds was carried out by palladium-catalyzed Suzuki coupling reactions using commercially available boronic acids and aryl bromides (4, 1b, 2b) or triflates (3, 1, 2), which were synthesized from the corresponding alcohol (3a, 1a, 2a).
Following the procedure of Weinstock [28] and Bravo [29], a chlorine substituent was introduced in compound 3b. Reduction of the tetralone 4 led to its corresponding alcohol 5. The introduction of a methylene bridge between the naphthalene moiety and the pyridyl group (compounds 6 and 7) was obtained by a bromine/lithium-exchange in 2-bromo-6-methoxynaphtalene and nucleophilic addition of the resulting organolithium compound to either nicotinealdehyde to afford the secondary alcohol 6 or to 6a yielding the tertiary alcohol 7. Compound 6a was prepared by conversion of nicotinoyl chloride to its Weinreb amide 6b and the subsequent introduction of an ethyl group by Grignard reaction with EtMgBr.
3 Biological results
Inhibition of human CYP17 was determined by performing our previously described assay [30]. As source of human CYP17, Escherichia coli coexpressing human CYP17 and NADPH-P450 reductase was used [31]. After homogenization, the 50.000 g sediment was incubated with progesterone (25 μM) and inhibitor. Separation of the product was performed by HPLC using UV-detection. The IC50 values determined for compounds 1–7 are shown in Table 2 with well-known reference compounds.
Inhibition of CYP19, CYP17, CYP11B2 and CYP11B1 by compounds 1–7. 
| Compound | Structure | IC50 [nM] | ||||
| R1 | Het | CYP19a,d | CYP17a,b | CYP11B2a,c | CYP11B1a,c | |
| 1 | >1000 | >5000 | >1000 | >5000 | ||
| 2 | >1000 | >1000 | >1000 | >1000 | ||
| 3 | 19 | >1000 | 10 | 495 | ||
| 4 | O | >5000 | >5000 | 114 | 1262 | |
| 5 | OH | >1000 | >50,000 | 100 | 2152 | |
| 6 | Et | 3-Py | >1000 | >50,000 | 1144 | 429 |
| 7 | H | 4-Py | >1000 | >1000 | 140 | 391 |
| KTZe | 2780 | 67 | 127 | |||
| FDZe | 1 | 10 | ||||
| ABTe | 72 | 1751 | 1608 |
a Data shown were obtained by performing the tests in duplicate. The deviations were within <±5%.
b Concentration of progesterone (substrate) was 25 μM.
c Concentration of 11-deoxycorticosterone (substrate) was 300 nM.
d Concentration of androstenedione (substrate) was 500 nM.
e KTZ: ketoconazole; FDZ: fadrozole; ABT: abiraterone.
All of the seven compounds showed only very little inhibitory activity toward CYP17 with IC50 values >1000 nM.
For the determination of the CYP11B2 inhibitory activities, V79MZh11B2 cells expressing human CYP11B2 were used as well as [14C]-labeled 11-deoxycorticosterone (300 nM) as substrate. The product formation was monitored by HPTLC using a phosphoimager [3,16,32–35]. The IC50 values determined for compounds 1–7 are shown in Table 2. While compounds 1, 2 and 6 showed only very little inhibition (IC50 values >1000 nM) of the mineralocorticoid-forming enzyme, a high inhibitory activity could be observed for compounds 3–5 and 7 (IC50 = 10–140 nM). Compound 3 turned out to be the most active one showing an IC50 of 10 nM.
Determination of CYP11B1 inhibition has been performed in analogy to the procedure for CYP11B2 described above with V79MZh11B1 cells. The IC50 values determined for compounds 1–7 are shown in Table 2. Compounds 1, 2, 4 and 5 showed only very little (IC50 >1000 nM) and compounds 3, 6 and 7 rather low (IC50 = 391–495 nM) activity toward CYP11B1.
The in vitro aromatase inhibitory activity was evaluated using the microsomal fraction of human placental tissue [36] according to a described procedure [37,38]. Labeled [1β-3H]androstenedione (500 nM), NADPH-generating system and inhibitor were incubated. The formed 3H2O was measured [39]. The IC50 values determined for compounds 1–7 are shown in Table 2. Here compound 3 turned out to be a strong aromatase inhibitor (IC50 = 19 nM). Compounds 1, 2 and 4–7 showed only very little activity (IC50 >1000 nM).
4 Discussion
Regarding the main core, the compounds can be divided into biphenylic structures (1 and 2), in tetrahydronaphthalenes (3–5) and in naphthalenes (6 and 7). In compounds 1–5 the heterocycle is bound directly to the core, while in compound 6 and 7 a hydroxy substituted methylene bridge, carrying an additional ethyl group in compound 7, is introduced.
It becomes apparent, that the biphenyls, which are derived from the highly active CYP17 inhibitor B, only show very little inhibition of all target enzymes (IC50 >1000 nM) independent from their substitution pattern. From this it can be concluded that for the biphenyls, the kind and orientation of the heterocycle are very important, as well as the methylene spacer between the core and the heterocycle.
In the class of the naphthalenes we found a very active compound (7). Derived from compound C – a strong CYP11B2 inhibitor – the modified structures 6 and 7 showed very interesting effects: By introducing a hydroxy substituted methylene spacer connecting the core with a 4-pyridine, compound 7 keeps its inhibitory activity for CYP11B2 (IC50 = 140 nM) with a moderate selectivity toward CYP11B1 (IC50 = 429 nM) and high selectivity toward CYP17 and CYP19 (IC50 >1000 nM). On the contrary, the inhibitory activity of the naphthalenes is decreased by introduction of an additional ethyl substituent at the methylene bridge and moving the 4-pyridyl nitrogen into the 3-position as shown by compound 6. This compound exhibits moderate activity toward CYP11B1 (IC50 = 491 nM) and very low activity toward the other enzymes (IC50 >1000 nM).
The tetralone 3 derived from aromatase inhibitor A, which is connected to the pyridine moiety in a different way compared to the parent compound, shows a very interesting activity profile. As expected, this compound is highly active regarding its inhibition of aromatase (IC50 = 19 nM), but, additionally, it is the most potent CYP11B2 inhibitor of this series (IC50 = 10 nM). It is very selective toward CYP17 (IC50 >1000) and shows only low inhibition of CYP11B1 (IC50 = 495 nM). Therefore this compound might be an attractive starting point for the development of dual inhibitors.
In contrast to this finding, the tetralone 4 and its respective alcohol 5 completely loose their aromatase inhibitory activity (IC50 >1000 nM), while keeping their potency toward CYP11B2 (IC50 = 114 resp. 100 nM). Furthermore only very little activity toward CYP11B1 and CYP17 could be observed. Therefore these two compounds can be regarded as highly active and selective CYP11B2 inhibitors.
5 Conclusion and outlook
From the data shown in this paper it becomes apparent that our new approach for the discovery of novel hit compounds targeting steroidogenic CYP enzymes was successful. Derived from the aromatase (CYP19) inhibitor A, we succeeded in finding a new lead structure for the development of novel inhibitors of CYP11B2, namely the tetralone derivatives 4 and 5. Furthermore a dual inhibitor reducing the activities of CYP19 and CYP11B2 (compound 3) could be identified. The optimization of these new compounds is presently under investigation.
6 Experimental section
6.1 CYP17 preparation and assay
As source of human CYP17, our E. coli system [40] coexpressing human CYP17 and NADPH-P450 reductase was used. The assay was performed as previously described [30] using unlabeled progesterone as substrate and applying HPLC with UV-detection for separation.
6.2 CYP19 preparation and assay
As source of human CYP19, microsomal preparations of human placenta were used [36,41]. The assay was performed using the 3H2O-method as described [37,38] using [1β-3H]androstenedione and unlabelled androstenedione as substrate and monitored by β-counter detection.
6.3 Inhibition of CYP11B1 and CYP11B2
V79MZh11B1 or V79MZh11B2 cells expressing CYP11B1 or CYP11B2 [33–35] were used and our assay procedure with [4-14C]-11-deoxycorticosterone was applied [16,32] using [14C]-labeled 11-deoxycorticosterone as substrate and monitored by TLC and by use of a phosphoimaging.
6.4 Chemistry section
6.4.1 General experimental
Melting points were determined on a Mettler FP1 melting point apparatus and are uncorrected. IR spectra were recorded neat on a Bruker Vector 33FT-infrared spectrometer. 1H NMR spectra were measured on a Bruker DRX-500 (500 MHz). Chemical shifts are given in parts per million (ppm), and TMS was used as an internal standard for spectra obtained in CDCl3. All coupling constants (J) are given in Hz. ESI (electrospray ionization) mass spectra were determined on a TSQ quantum (Thermo Electron Corporation) instrument. Elemental analyses were performed at the Department of Instrumental Analysis and Bioanalysis, Saarland University. Column chromatography was performed using silica gel 60 (50–200 μm), and reaction progress was monitored by TLC analysis on Alugram® SIL G/UV254 (Macherey-Nagel). Boronic acids and bromoaryls used as starting materials were obtained commercially (CombiBlocks, Chempur, Aldrich, Acros) and used without further purification.
6.4.2 Method A: synthesis of triflates
To a solution of the corresponding alcohol and pyridine in dry DCM (10 mL/mmol), trifluoromethanesulfonic anhydride was carefully added over 1 min at 0 °C. The reaction mixture was stirred at room temperature for 3 h. Afterwards, excess trifluoromethanesulfonic anhydride was neutralized with Na2CO3, and the crude was washed two times with water. The organic phase was dried over Na2SO4 and concentrated. Purification of the residue was performed by flash chromatography using SiO2.
6.4.2.1 3-Chloro-2′-methoxy-biphenyl-4-yl trifluoro-methanesulfonate (1a)
Synthesized from 1b (0.85 g, 3.6 mmol), pyridine (0.30 mL, 3.7 mmol) and trifluoromethanesulfonic anhydride (1.22 mL, 7.24 mmol) in dry DCM (10 mL) according to Method A. Yield: 85%. The obtained crude triflate was used directly for the Suzuki coupling without further purification.
6.4.2.2 4-Chloro-3′-methoxy-biphenyl-3-yl trifluoro-methanesulfonate (2a)
Synthesized from 2b (0.93 g, 4.0 mmol), pyridine (0.32 mL, 4.0 mmol) and trifluoromethanesulfonic anhydride (1.35 mL, 8.02 mmol) in dry DCM (10 mL) according to Method A. Yield: 82%. The obtained crude triflate was used directly for the Suzuki coupling without further purification.
6.4.2.3 1-Chloro-5-oxo-5,6,7,8-tetrahydronaphthalen-2-yl trifluoromethanesulfonate (3a)
Synthesized from 3b (6.50 g, 31.2 mmol), pyridine (3.76 mL, 46.7 mmol) and trifluoromethanesulfonic anhydride (10 mL, 59.0 mmol) in dry DCM (20 mL) according to Method A. Yield: 33%. 1H NMR (CDCl3, 500 MHz): δH (ppm) = 2.15 (q, J = 6.4 Hz, 2H), 2.61 (t, J = 6.5 Hz, 2H), 3.02 (t, J = 6.2 Hz, 2H), 7.27 (d, J = 8.7 Hz, 1H), 8.00 (d, J = 8.7 Hz, 1H). 13C NMR (CDCl3, 125 MHz): δC (ppm) = 21.98, 27.43, 37.89, 114.4, 117.3, 119.8, 120.7, 127.2, 127.3, 127.9, 133.4, 144.8, 148.9, 195.8.
6.4.3 Method B: Suzuki coupling
The corresponding naphthalene or benzene derivative and the boronic acid were dissolved in toluene (20 mL) and aq. Na2CO3 (2.0 M, 5.0 mL). The mixture was deoxygenated under reduced pressure and flushed with N2. After having repeated this cycle three times, Pd(PPh3)4 (5 mol%) was added, and the resulting suspension was heated under reflux for 8 h. After cooling, the phases were separated and the water phase was extracted two times with EtOAc. The combined organic extracts were dried over Na2SO4, and concentrated under reduced pressure. The purification was performed by flash chromatography using SiO2.
6.4.3.1 3-(3-Chloro-2′-methoxy-biphenyl-4-yl)-pyridine (1)
Synthesized from 1a (0.98 g, 2.7 mmol) and 2-pyridinylboronic acid (0.49 g, 4.0 mmol) according to Method B. Pale oil. Yield: 72%. IR (ATR) υ (cm−1) = 2927, 1601, 1466, 1435, 1412, 1251, 1181, 1082, 1026, 833, 807, 752, 714, 680; 1H NMR (CDCl3, 500 MHz): δH (ppm) = 3.87 (s, 3H), 7.01–7.08 (m, 2H), 7.34–7.40 (m, 4H), 7.53 (d, J = 7.8 Hz, 1H), 7.70 (s, 1H), 7.86 (d, J = 7.8 Hz, 1H), 8.64 (d, J = 3.8 Hz, 1H), 8.75 (s, 1H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 55.60, 111.3, 120.9, 124.3, 124.6, 122.9, 128.3, 128.4, 128.5, 128.6, 129.4, 130.7, 130.8, 131.3, 136.9, 140.1, 148.6, 150.1, 156.4; MS (ESI): m/z = 296.2 [M + H]+.
6.4.3.2 3-Chloro-2′-methoxy-biphenyl-4-ol (1b)
Synthesized from 5-bromo-2-chlorophenol (1.00 g, 4.82 mmol) and 3-methoxyphenylboronic acid (1.10 g, 7.23 mmol) according to Method B. Yield: 75%. 1H NMR (CDCl3, 500 MHz): δH (ppm) = 3.74 (s, 3H), 5.49 (s, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.93 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 7.19 (dd, J = 1.6 Hz, J = 7.4 Hz, 1H), 7.21–7.25 (m, 1H), 7.26 (dd, J = 2.1 Hz, J = 8.4 Hz, 1H), 7.43 (d, J = 2.1 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 55.53, 111.2, 115.7, 119.1, 120.8, 128.7, 128.9, 129.6, 129.8, 130.5, 131.9, 150.3, 156.3.
6.4.3.3 3-(4-Chloro-3′-methoxy-biphenyl-3-yl)-pyridine (2)
Synthesized from 2a (0.58 g, 2.3 mmol) and 3-pyridinylboronic acid (0.43 g, 3.5 mmol) according to Method B. Pale oil. Yield: 83%. IR (ATR) υ (cm−1) = 2924, 1583, 1465, 1410, 1319, 1288, 1090, 1043, 1011, 874, 780, 713, 696; 1H NMR (CDCl3, 500 MHz): δH (ppm) = 3.86 (s, 3H), 6.93 (dd, J = 2.3 Hz, J = 8.2 Hz, 1H), 7.11 (s, 1H), 7.17 (d, J = 7.7 Hz, 1H), 7.35–7.41 (m, 2H), 7.53–7.56 (m, 3H), 7.84–7.86 (m, 1H), 8.66 (d, J = 3.8 Hz, 1H), 8.75 (s, 1H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 55.59, 112.3, 120.9, 122.8, 124.7, 128.3, 128.6, 129.4, 130.7, 131.0, 132.3, 133.3, 136.9, 140.0, 141.2, 148.6, 150.1, 156.4; MS (ESI): m/z = 296.2 [M + H]+.
6.4.3.4 4-Chloro-3′-methoxy-biphenyl-3-ol (2b)
Synthesized from 5-bromo-2-chlorophenol (1.00 g, 4.82 mmol) and 3-methoxyphenylboronic acid (1.10 g, 7.23 mmol) according to Method B. Yield: 82%. 1H NMR (CDCl3, 500 MHz): δH (ppm) = 3.78 (s, 3H), 5.53 (s, 1H), 6.82 (dd, J = 2.4 Hz, J = 8.2 Hz, 1H), 6.99–7.02 (m, 2H), 7.05 (d, J = 7.7 Hz, 1H), 7.17 (d, J = 2.0 Hz, 1H), 7.25–7.29 (m, 2H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 55.30, 112.7, 113.2, 114.8, 119.1, 119.5, 120.2, 129.1, 129.8, 141.3, 141.7, 151.4, 159.9.
6.4.3.5 5-Chloro-6-(pyridin-3-yl)-3,4-dihydronaphthalen-1(2H)-one (3)
Synthesized from 3a and 3-pyridinylboronic acid according to Method B. Orange oil. Yield: 86%. 1H NMR (CDCl3, 500 MHz): δH (ppm) = 2.15 (m, 2H), 2.62 (t, J = 7.2 Hz, 2H), 3.04 (t, J = 6.2 Hz, 2H), 7.24 (d, J = 7.9 Hz, 1H), 7.34 (dd, J = 4.8 Hz, J = 7.9 Hz, 1H), 7.99 (m, 2H), 8.60 (d, J = 4.8 Hz, 1H), 8.61 (s, 1H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 22.18, 27.66, 38.10, 123.0, 125.7, 129.0, 132.7, 133.9, 135.2, 136.8, 142.0, 142.9, 149.1, 149.6, 197.1; MS (ESI): m/z = 258.0 [M + H]+.
6.4.3.6 7-(Pyridin-3-yl)-3,4-dihydronaphthalen-1(2H)-one (4)
Synthesized from 7-Bromo-3,4-dihydro-2H-naphthalen-1-one and 3-pyridinylboronic acid according to Method B. Pale oil. Yield: 89%. IR (ATR) υ (cm−1) = 3028, 2940, 1677, 1608, 1427, 1325, 1252, 1220, 1180, 1028, 905, 808, 720, 555; 1H NMR (CDCl3, 500 MHz): δH (ppm) = 2.15–2.20 (m, 2H), 2.70 (dd, J = 6.0 Hz, J = 7.0 Hz, 2H), 3.02 (dd, J = 5.8 Hz, J = 6.1 Hz, 2H), 7.37–7.39 (m, 2H), 7.70 (dd, J = 2.1 Hz, J = 7.9 Hz, 1H), 7.91 (ddd, J = 1.5 Hz, J = 7.1 Hz, J = 7.9 Hz, 1H), 8.26 (d, J = 2.1 Hz, 1H), 8.60 (dd, J = 1.5 Hz, J = 4.9 Hz, 1H), 8.86 (d, J = 2.2 Hz, 1H); MS (ESI): m/z = 224.2 [M + H]+.
6.4.4 7-(Pyridin-3-yl)-1,2,3,4-tetrahydronaphthalen-1-ol (5)
A suspension of sodium borohydride (0.090 g, 2.3 mmol) in MeOH (10 mL) was added to a solution of 4 (0.26 g, 1.16 mmol) in THF (10 mL) at 0 °C and stirred at room temperature over 2 h. The reaction mixture was extracted three times with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated. No further purification was necessary. Yellow oil. Yield: 96%. IR (ATR) υ (cm−1) = 3290, 2934, 1475, 1430, 1187, 1071, 1026, 988, 800, 710; 1H NMR (CDCl3, 500 MHz): δH (ppm) = 1.74–1.94 (m, 2H), 1.96–2.07 (m, 2H), 2.70–2.86 (m, 2H), 4.82 (dd, J = 4.7 Hz, J = 5.7 Hz, 1H), 7.15(d, J = 7.9 Hz, 1H), 7.27 (ddd, J = 1.0 Hz, J = 5.0 Hz, J = 8.2 Hz, 1H), 7.32 (dd, J = 1.9 Hz, J = 7.9 Hz, 1H), 7.66 (d, J = 1.9 Hz, 1H), 7.79 (ddd, J = 1.9 Hz, J = 2.5 Hz, J = 7.9 Hz, 1H), 8.45 (dd, J = 1.0 Hz, J = 4.7 Hz, 1H), 8.68 (dd, J = 1.0 Hz, J = 2.5 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 18.96, 28.94, 32.36, 67.79, 123.5, 125.8, 127.2, 129.6, 134.2, 135.3, 136.5, 137.2, 140.0, 147.7, 147.7; MS (ESI): m/z = 226.2 [M + H]+.
6.4.5 5-Chloro-6-hydroxy-3,4-dihydronaphthalen-1(2H)-one (3b)
The synthesis of 3b and the required tert-butyl hypochlorite was carried out according to Weinstock et al. [28] and Bravo et al. [29], respectively. Yield: 79%. 1H NMR (CDCl3, 500 MHz): δH (ppm) = 2.03–2.12 (m, 2H), 2.55 (t, J = 6.4 Hz, 2H), 2.92 (t, J = 6.2 Hz, 2H), 6.23 (m, 1H), 7.97 (m, 1H).
6.4.6 N-Methoxy-N-methylnicotinamide (6b)
Nicotinoyl chloride hydrochloride (2 g, 11.2 mmol) and N-dimethoxy-methylamine HCl hydrochloride (12 g, 12.4 mmol) were dissolved in DCM (30 mL), and triethylamine (5.15 mL, 37.0 mmol) was added dropwise. After stirring at room temperature overnight, the reaction mixture was extracted with EtOAc and the solution concentrated under reduced pressure. Further purification was performed by flash chromatography using SiO2. Yield: 82%. 1H NMR (CDCl3, 500 MHz): δH (ppm) = 3.33 (s, 3H), 3.50 (s, 3H), 7.31 (ddd, J = 1.0 Hz, J = 4.9 Hz, J = 7.9 Hz, 1H), 7.97 (ddd, J = 2.0 Hz, J = 2.2 Hz, J = 7.9 Hz, 1H), 8.63 (dd, J = 1.6 Hz, J = 4.9 Hz, 1H), 8.90 (d, J = 1.6 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 33.05, 61.13, 122.8, 129.8, 136.0, 149.1, 151.2, 167.3.
6.4.7 1-(Pyridin-3-yl)propan-1-one (6a)
6b (1.3 g, 7.8 mmol) was dissolved in THF (20 mL), and ethylmagnesium bromide in THF (1.0 M, 10.1 mL, 10.1 mmol) was added dropwise. After stirring at room temperature for 8 h, water was added and the mixture was stirred for additional 15 min. The layers were separated and the organic phase extracted three times with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The purification was performed by flash chromatography using SiO2. Yield: 62%. 1H NMR (CDCl3, 500 MHz): δH (ppm) = 1.21 (t, J = 7.3 Hz, 3H), 2.99 (q, J = 7.3 Hz, 2H), 7.39 (ddd, J = 1.0 Hz, J = 4.8 Hz, J = 7.9 Hz, 1H), 8.21 (ddd, J = 1.9 Hz, J = 2.2 Hz, J = 7.9 Hz, 1H), 8.73 (dd, J = 1.0 Hz, J = 4.7 Hz, 1H), 9.14 (dd, J = 1.0 Hz, J = 2.2 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 7.78, 32.07, 123.6, 132.0, 135.2, 149.4, 153.2, 199.4.
6.4.8 Method C: addition of the heterocycle
2-Bromo-6-methoxynaphthalene (6 mmol) was dissolved in THF (20 mL) at −78 °C and tert-BuLi in hexanes (1.5 M, 7.3 mL, 11 mmol) was added dropwise. This solution was added to a solution of ketone or aldehyde (5 mmol) in THF (20 mL) and the mixture was left stirring at 0 °C for 2 h. Afterwards, water and EtOAc were added, the phases separated, and the aqueous phase was extracted three times with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The purification was performed by flash chromatography using SiO2.
6.4.8.1 1-(6-Methoxynaphthalen-2-yl)-1-(pyridin-3-yl)propan-1-ol (6)
Synthesized from 6a according to Method C. Yellowish solid. Mp: 136.5 °C. Yield: 43%. IR (ATR) υ (cm−1) = 2969, 1603, 1503, 1264, 1171, 1030, 985, 883, 804, 718, 560; 1H NMR (CDCl3, 500 MHz): δH (ppm) = 1.21 (t, J = 7.3 Hz, 3H), 2.99 (d, J = 7.3 Hz, 2H), 3.90 (s, 3H), 6.97–7.03 (m, 2H), 7.16–7.19 (m, 3H) 7.40–7.51 (m, 2H), 8.21 (ddd, J = 1.9 Hz, J = 2.2 Hz, J = 7.9 Hz, 1H), 8.73 (dd, J = 1.0 Hz, J = 4.7 Hz, 1H), 9.14 (dd, J = 1.0 Hz, J = 2.2 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 7.96, 34.17, 55.30, 105.5, 119.0, 123.1, 124.2, 125.2, 127.1, 128.4, 129.7, 133.6, 134.4, 141.0, 142.5, 147.5, 157.9; MS (ESI): m/z = 294.2 [M + H]+.
6.4.8.2 (6-Methoxynaphthalen-2-yl)(pyridin-4-yl)methanol (7)
Synthesized from isonicotinaldehyde according to Method C. Yellowish solid. Mp: 167.8 °C. Yield: 59%. IR (ATR) υ (cm−1) = 2926, 1603, 1507, 1270, 1169, 1056, 845, 772, 676; 1H NMR (CDCl3, 500 MHz): δH (ppm) = 3.69 (s, 3H), 5.65 (s, 1H), 6.90–6.93 (m, 2H), 7.16–7.19 (m, 3H), 7.47–7.51 (m, 2H), 7.55 (m, 2H), 8.28–8.30 (m, 2H); 13C NMR (CDCl3, 125 MHz): δC (ppm) = 54.74, 73.82, 105.2, 118.5, 121.1, 124.0, 125.0, 126.7, 128.0, 128.9, 133.5, 138.3, 148.7, 153.5, 157.2; MS (ESI): m/z = 266.3 [M + H]+.
Acknowledgement
UEH is grateful to the European Postgraduate School 532 (DFG) for a scholarship. We thank Profs Hermans (U. Maastricht) and Bernhardt (Saarland U.) for providing us with V79MZh11B1 and B2 cells, respectively, and Dr. Müller-Vieira for the CYP17 tests.