

1 Introduction
Advances in transition metal catalyzed asymmetric processes have been traditionally guided by the concept that stereochemically rigid enantiopure ligands are required to achieve high enantioselectivities [1]. Chiral diphosphines proved to be one of the most successful and widely used ligands for this purpose in particular the highly versatile BINAP which belongs to the atropos1 class of diphosphine ligands since its axial chiral conformation can be resolved [1c]. A conceptually new approach has emerged where achiral and meso ligands can be used to convey asymmetry in enantioselective catalysis [2]. Therefore, conformationally flexible tropos1 diphosphine ligands are intentionally used to either magnify the stereochemical induction of a chiral ligand or act as the only source of asymmetry for enantioselective transformation. In the latter case, coordination of the ligand to transition metal slows interconversion of its conformations and as a consequence a metastable enantiopure assembly can be resolved by a chiral controller and used for catalysis with or without it (Scheme 1). Enantiopure complexes with tropos diphosphines such as BIPHEP [3], NUPHOS [4], cyclo-NUPHOS or DPPF [5] demonstrate good to excellent enantioselectivities and can efficiently replace complexes bearing classical enantiopure ligands.
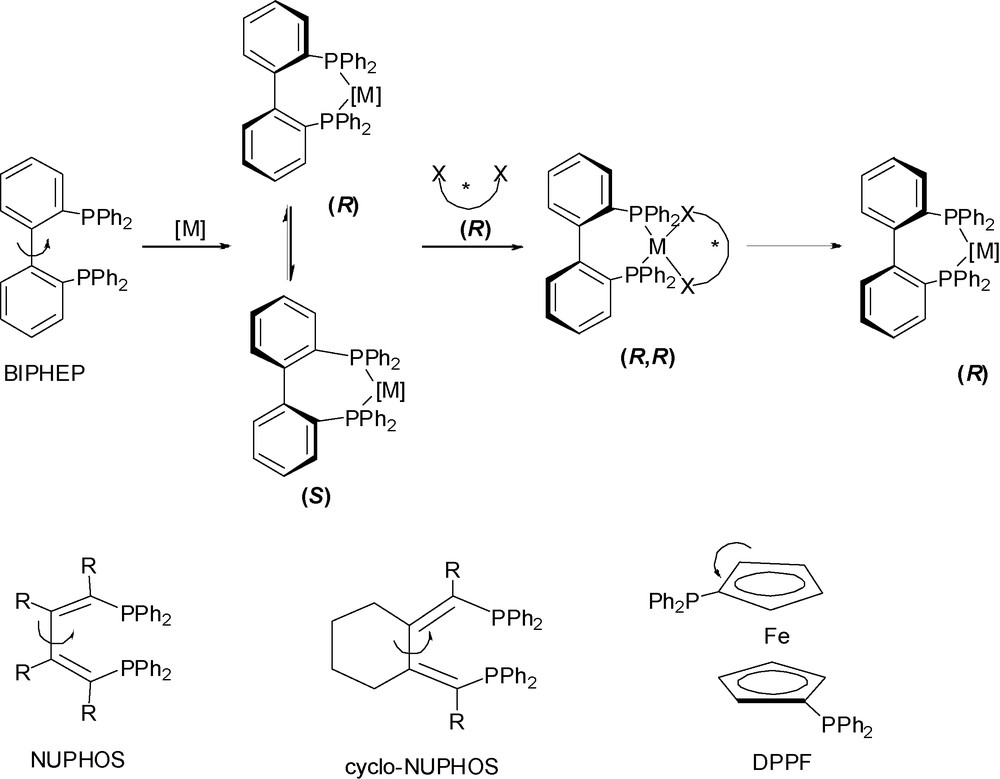
BIPHEP, NUPHOS, cyclo-NUPHOS, DPPF.
The advantage of this approach to asymmetric catalysis over the traditional one is that catalysts can be optimised by the synthesis of achiral or meso ligands instead of the synthesis of enantiopure ones which requires elaborate and expensive methods (e.g., asymmetric synthesis or resolution). Consequently, the design of new catalysts, based on dynamic chirality control (e.g., N-chirality control [6] or dual chirality control of axial and N-chirality [7]) by chiral activator through complexation is actually an important field.
In a related approach, we have examined the dual chirality control of axial and central phosphorus chiralities of 2,2’-biphosphole ligands [8]. Some years ago, we reported the first application of the chiral stereochemically dynamic 2,2’-biphophole ligand (BIPHOS) to asymmetric allylic substitution involving crystallization-induced spontaneous resolution and kinetic stabilization by coordination to Pd centre2 [9]. In a more convenient procedure, we have discovered that a dual chirality control can be achieved by introducing a chiral linker between the two phosphorus atoms that favours a single enantiomeric form upon coordination to a metal [10]. In this article, we report our approach to stereodynamic diphosphines and diphosphinites derived from 2’2-biphospholes for asymmetric catalysis.
2 Synthesis of stereodynamic diphosphines and diphosphinites
This approach requires, in the first step, the synthesis of new 2,2’-biphosphole ligands with restricted configurations. The axial and central chiralities of the 2,2’-biphosphole framework could be controlled and limited by introducing a chiral linker between the two phosphorus atoms. Thus, we have investigated the introduction of enantiomerically pure carbon and alkoxy linkers in order to obtain, respectively, diphosphines and diphosphinites derived from 2,2’-biphosphole.
2.1 Diphosphines derived from 2,2’-biphosphole
Chiral stereochemically dynamic diphosphines [11] derived from 2,2’-biphosphole are prepared in two steps starting from the tetraphosphole (Scheme 2). In the first step, cleavage of the two phosphorusphosphorus bonds using sodium naphtalene lead to the 2,2’-biphospholyl anion [13]. Then, the asymmetric alkylation in high dilution conditions using various enantiomerically pure diol ditosylates or dimesylates affords the expected diphosphines 1 (Scheme 2) in good yields (35 to 78%) as a mixture of three diastereoisomers among the six expected. The three diastereoisomers of each diphosphines 1 are fully characterized as disulfide derivatives since these diphosphines become stereorigid as disulfide derivatives, allowing structural and stereochemistry studies by X-ray diffraction analysis.
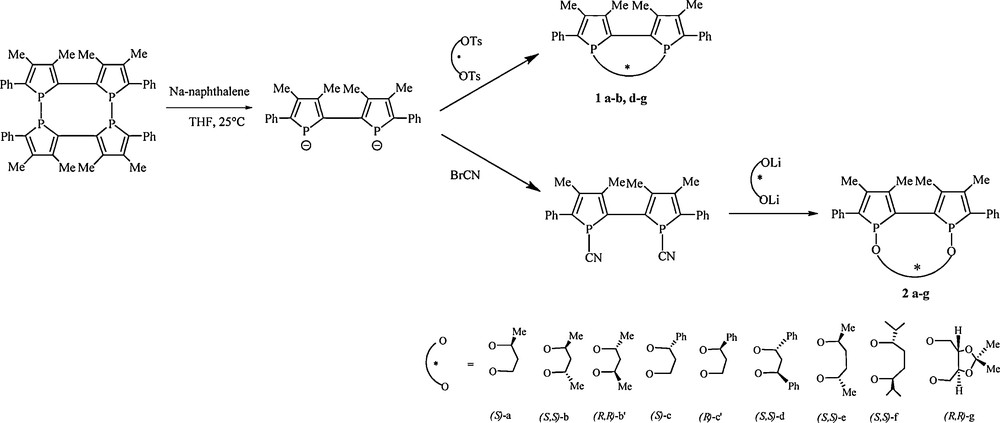
Synthesis of the diphosphines 1 and diphosphinites 2 derived from 2,2’-biphosphole.
The stereochemical analysis of the different possibilities of combining axial and central chiralities in 2,2’-biphosphole shows the occurrence of six really inequivalent diastereoisomers (Fig. 1)3.
The introduction of a short chain between the two phosphorus atoms prevents the formation of the A and B diastereoisomers and favours the C, D, E and F forms. Indeed, the three diastereoisomers (C, D and E [or F] forms) are observed in solution as an equilibrium mixture at room temperature. The isomerization process, studied by desulfurization of diphosphine sulfides using a fully stereoselective method [14] that affords, at low temperature, chiral phosphines with retention of configuration, occurs at temperature below −60 °C. Since the chiral bridge prevents the formation of A and B forms, the only possible pathway for this isomerisation process requires a phosphorus-inversion4 inducing atropo-inversion, the driving force being the pyramidal inversion barrier of the phosphorus atom as illustrated in Fig. 2.
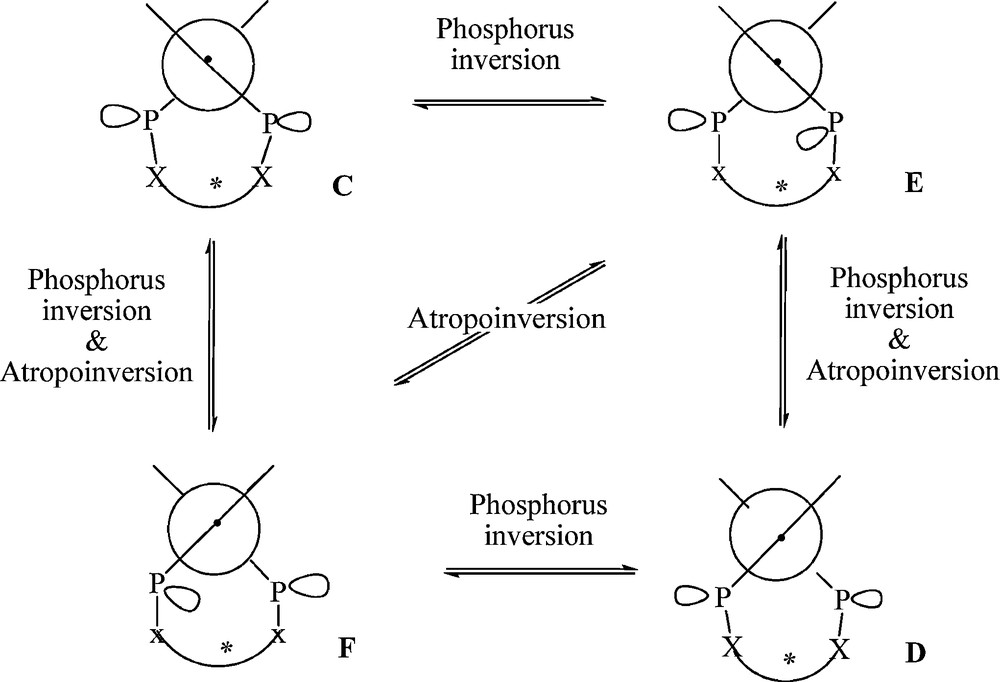
Isomerization process of diphosphines 1 or diphosphinites 2.
2.2 Diphosphinites derived from 2,2’-biphosphole
The strategy to obtain chiral stereochemically dynamic diphosphinites 25 derived from 2,2’-biphosphole is based on the introduction of an enantiomerically pure alkoxy linker between the two phosphorus atoms which involves a nucleophilic substitution reaction on 1,1’-dicyano-3,3’,4,4’-tetramethyl-5,5’-diphenyl-2,2’-biphosphole 2 [13] by using enantiomerically pure diols. The synthetic pathway involves three steps starting from tetraphosphole 1 [12] as shown in Scheme 2. The first step leads to the 2,2’-biphospholyl anion which reacts in the next step with BrCN6 to give the dicyano compound 2. Treatment of this intermediate with the lithium derivative of diols a–g in high dilution conditions affords in the last step the air-stable diphosphinite compounds 2a–g in high yields (71–86%). These compounds have been fully characterized and the molecular structures of 2a and 2b’ have been established by X-ray diffraction. Compounds 2a and 2b’ are enantiomerically pure in solid state according to X-ray diffraction analysis7 [15] (Figs. 3 and 4). The absolute configuration is R[Sp,Sp,Sc] for compound 2a and S[Rp,Rp,Rc,Rc] for compound 2b’ (axial configuration [phosphorus configuration, carbon configuration]).
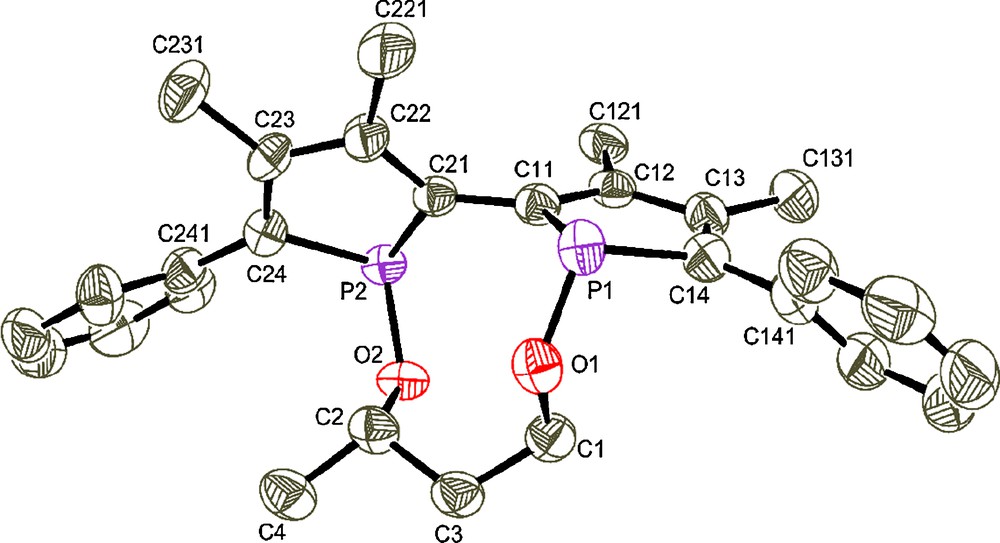
Molecular view of R[Sp,Sp,Sc]- 2a space group: P212121, Flack's parameter: 0.06(14).
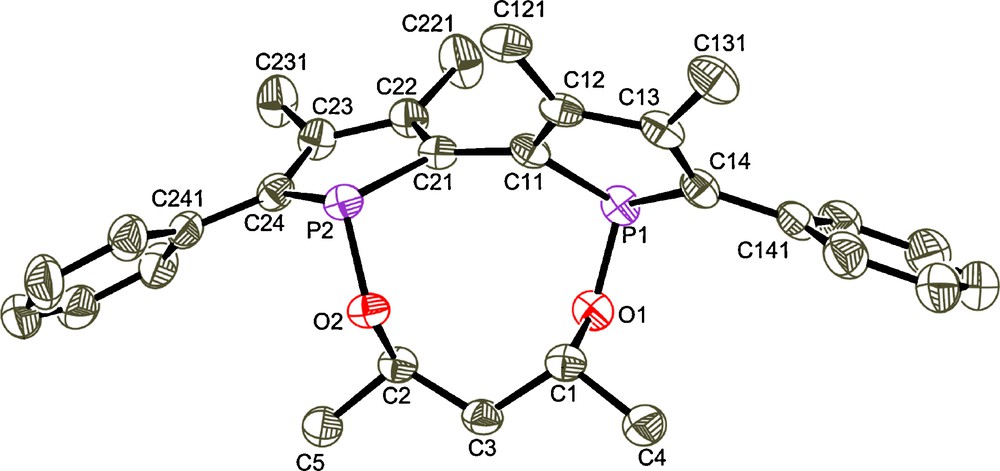
Molecular view of S[Rp,Rp,Rc,Rc]-2b’ space group: P212121, Flack's parameter:-0.03(6).
It is worth mentioning the influence of the configuration of the starting diol on the axial and central configuration of the skeleton 2,2’–biphosphole in solid state as the (S) configuration provides the R[Sp,Sp] configuration of 2a whereas the (R,R) configuration leads to the S[Rp,Rp] configuration of 2b’.
Contrary to the diphosphines 1, the diphosphinites 2 have been observed in solution by NMR as a single compound even within the temperature range −60 °C to +85 °C, corresponding probably to an equilibrium mixture of several diastereoisomers interconverting rapidly on the NMR time scale. Indeed, as the pyramidal inversion barrier of the phosphorus atom is not affected by π-donor substituents on the phosphorus [16], a similar isomerization process occurs rapidly in solution between the most four favoured diastereoisomers (C, D, E or F) of the diphosphinites 2 (Fig. 2).
These studies show that a partial chirality control of the axial and the central chiralities of 2,2’-biphosphole ligands can be achieved by introducing an enantiomerically pure linker between the two phosphorus atoms. In addition, this partial control maintains some degree of freedom to accomplish a metal dynamic resolution.
3 Dynamic resolution by coordination to a transition metal
We have examined the dynamic chirality control of the axial and central chiralities upon coordination since the use of these stereodynamic ligands in asymmetric catalysis relies on obtaining enantiopure complexes.
3.1 Palladium complexes
Complexation of the diastereoisomeric mixture of equilibrating diphosphines 1a, 1a’ and 1b with [PdCl2(CH3CN)2] in dichloromethane at room temperature produces [PdCl2(diphosphine)] 3a, 3a’ and 3b [12] respectively in excellent yield (90%) and an unidentified complex (10%)8 in each case (Scheme 3).
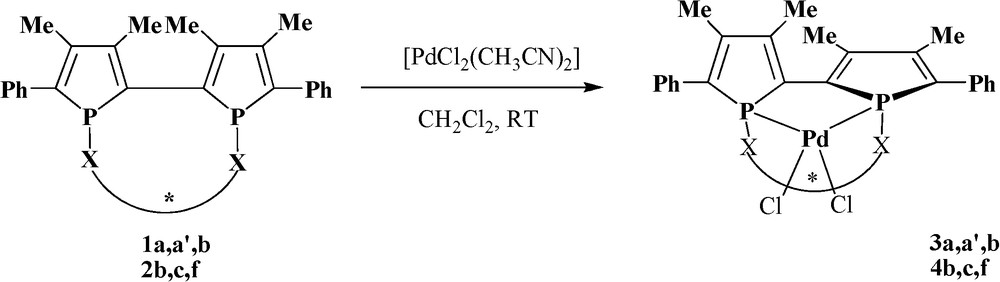
Synthesis of the palladium complexes.
Palladium complexes 3 are obtained in a pure diastereoisomeric form in solution according to 31P NMR and no dynamic configuration change of the 2,2’-biphosphole chelated to palladium can be observed within the temperature range −60 °C to +85 °C.9 According to these results, stabilization of the 2,2’-biphosphole framework chiralities has occurred by complexation as the result of the hindered inversion of the phosphorus configurations as well as axial configuration of 2,2’-biphosphole on metal center.
In addition, complexes 3a, 3a’ and 3b are also enantiomerically pure in the solid state according to X-ray diffraction analysis [15]. Complexes 3a and 3a’ [17], which differ in the configuration of atom C1 within the backbone linking the two P atoms, are enantiomers to each other. The molecular views of the these two enantiomers, S[Sp,Rp,Rc]-3a and R[Rp,Sp,Sc]-3a’ (axial configuration [phosphorus configuration, carbon configuration]) are presented in Figs. 5 and 6.

Molecular view of S[Sp,Rp,Rc]-3a space group P21, Flack's parameter 0.03(5).
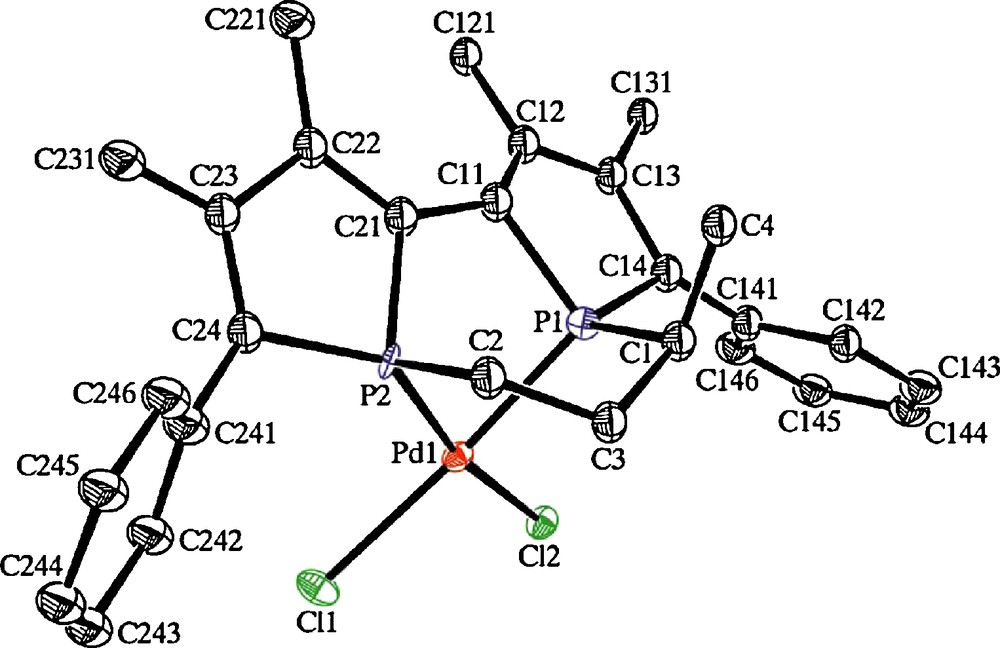
Molecular view of R[Rp,Sp,Sc]-3a’ space group P21, Flack's parameter -0.02(11).
According to these results, the chiral stereodynamic diphosphines 1 derived from 2,2’-biphosphole afford diastereo- and enantiopure complexes through a dual chirality control locked by palladium coordination. Structural studies by X-ray analysis reveal that the 2,2’-biphosphole adopts a single configuration in these palladium complexes in which the two phosphorus have opposite configurations, [Sp,Rp] or [Rp,Sp]. In addition, these analyses prove unambiguously the influence of the configuration of the carbon backbone on the axial and central configuration of the 2,2’-biphosphole skeleton in complexes 3a and 3a’, as the R configuration provides the S[Sp,Rp] configuration of 3a, whereas the S configuration leads to the R[Rp,Sp] configuration of 3a’.
In contrast, palladium-complexes of diphosphinites 2 cannot be as well structurally characterized. Treatment of diphosphinites 2b,c,f with [PdCl2(PhCN)2] leads to the complete consumption of the ligand to probably afford the corresponding [PdCl2(diphosphinite)] 4b,c,f (Scheme 3). But in each case, the Pd-complex gives a broad signal in 31P and 1H NMR even within the temperature range −70 °C to +85 °C which can be explained by an aggregation phenomena in solution.10 Unfortunately, no structural determinations by X-ray analysis can be performed.
3.2 Platinum complexes
Enantiopure platinum (II) complexes were also synthetized with chiral stereodynamic diphosphines 1 and diphosphinites 2 [18].
Diphosphines 1b,e,g react with [PtCl2(CH3CN)2] in 24 h in dichloromethane at room temperature to quantitatively afford a diastereomerically pure complex 5b,e,g (Scheme 4) as evidenced by 31P NMR spectra. Complexes 5b,e,g, are also enantiomerically pure in solid state as confirmed by X-ray analysis performed on two of these three complexes [18]. The molecular view of complex 4b’ is presented in Fig. 7. The configuration of the ligand 1b’ in the platinum complex, described as S[Rp,Sp,Sc,Sc], reveals again the opposite configuration for the two phosphorus atoms.
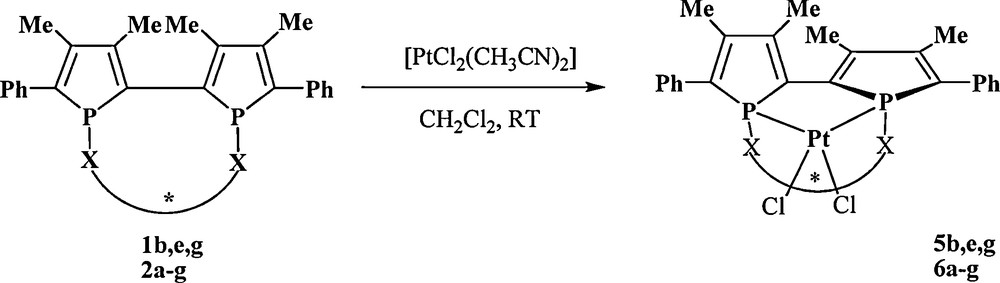
Synthesis of the platinum complexes.
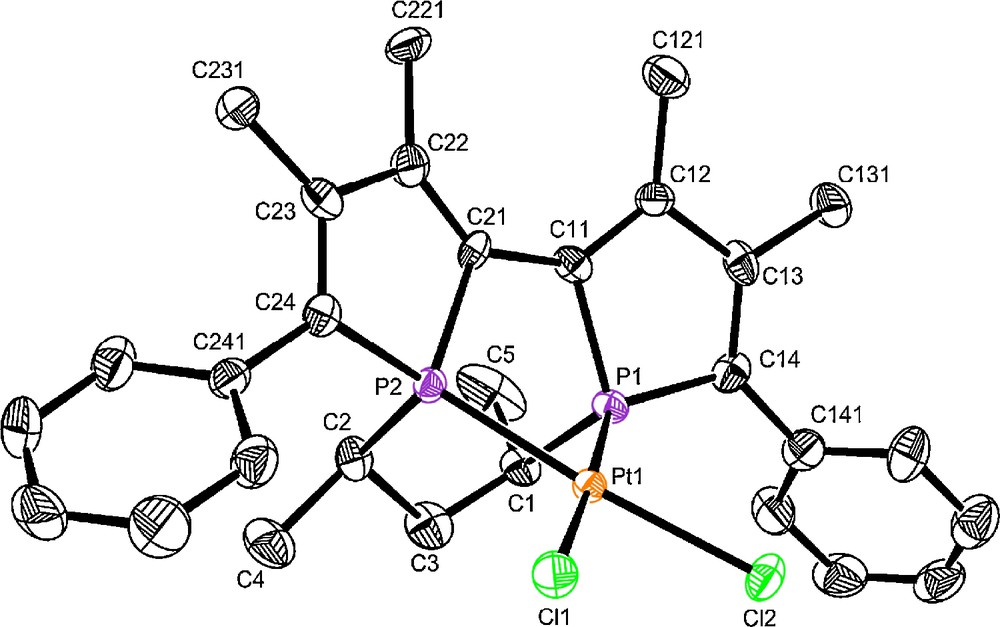
Molecular view of S[Rp,Sp,Sc,Sc]-3b’ Space group : P21, Flack's parameter : 0.04(2).
Diphosphinites 2a–g are more reactive towards [PtCl2(CH3CN)2] than diphosphines as the complete consumption of the ligand is obtained in 3 h at room temperature in dichloromethane (Scheme 4). Contrary to diphosphine-complexes 5, these platinum complexes give broad signals in 31P and 1H NMR, except for complexes 6d and 6f, even within the temperature range −70 °C to +85 °C. The well-resolved 31P NMR spectra of complexes 6d and 6f clearly indicate the formation of a pure diastereoisomer as in the case of diphosphine complexes 5. In addition, X-ray structural determinations of 6f [18] reveal the formation of an enantiopure complex (Fig. 8).
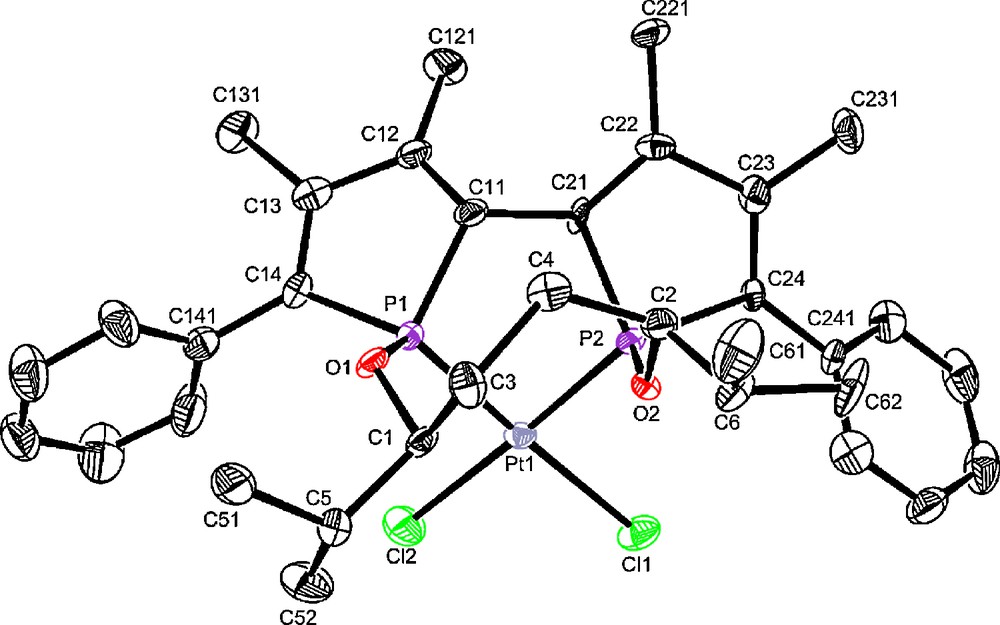
Molecular view of complex S[Sp,Rp,Sc,Sc]-4f Space group : P21, Flack's parameter : 0.013(8).
As already observed in others complexes, the two phosphorus atoms have opposite configuration [Sp,Rp] in complex 4f.
3.3 Rhodium complexes
Dynamic chirality control of diphosphines 1 and diphosphinites 2 can be also obtained by coordination to the rhodium centre. For example, reaction of diphosphine 1b and diphosphinites 2a,d with [Rh(COD)2]BF4 or [Rh(COD)2]CF3SO3 (Scheme 5) in dichloromethane quantitatively leads, in each case, to the formation of a diastereomerically pure complex, 7b and 8a,d, respectively, as evidenced by 31P{1H} NMR. Unfortunately, these rhodium complexes give a red hygroscopic and water sensitive powder after evaporating the solvent and all attempts to purify this complex failed. No single crystals could be obtained.
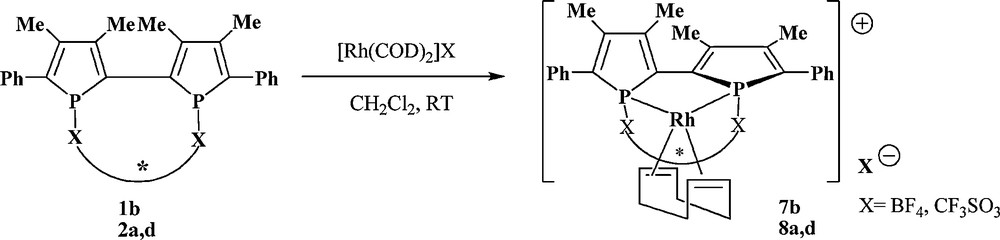
Synthesis of the rhodium complexes.
However, as complexing to a metal centre locks both the central phosphorus configuration and the axial configurations of 2,2’-biphosphole, the rhodium complexes 7 and 8 are obtained as a diastereo- and enantiopure complex.
4 Enantioselective catalytic reactions
To demonstrate the potential of our stereodynamic ligands for enantioselective catalysis, we studied Pd-catalyzed allylic substitution, Pt-catalyzed hydroformylation and Rh-catalyzed hydrogenation reactions.
4.1 Asymmetric allylic substitution
We screened various palladium catalysts in the allylic substitution of 1,3-diphenylprop-2-enyle acetate, which is widely used as a model substrate, by the dimethylmalonate anion (Scheme 6). The reaction is carried out in dichloromethane at room temperature in the presence of a catalyst generated in situ from 1 mol% of the corresponding ligand, 0.5 mol% of π-allyl-palladium chloride dimer [Pd(η3-C3H5)Cl]2.

Allylic substitution of 1,3-diphenylprop-2-enyle acetate by the dimethylmalonate anion.
All of the diphosphines 1 and diphosphinites 2 [11,15] ligands give palladium catalytic systems that are active in allylic substitution producing the corresponding allylic substitution product with enantiomeric excess in the range of 4 to 66%. The best stereoselectivity is achieved with the palladium catalyst derived from diphosphine 1d.
The stereoselectivities are, however, moderate but these results demonstrate the direct use of the stereodynamic diphosphines and diphosphinites to convey enantioselectivity in metal-catalyzed reaction through an in situ dual chirality control.
4.2 Asymmetric hydroformylation
The enantiopure platinum complexes of diphosphines and diphosphinites are examined in the asymmetric hydroformylation of styrene [18], which is an attractive catalytic approach to the synthesis of the 2-arylpropionic acid type of anti-inflammatories (Scheme 7).
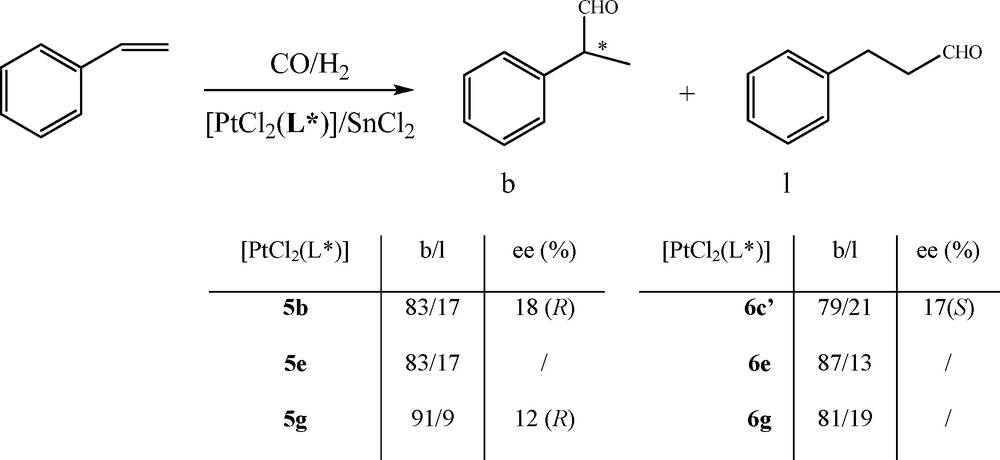
Hydroformylation of styrene.
In this case, experiments are performed using the preformed complexes [PtCl2(L*)] 5 and 6 with anhydrous SnCl2 as co-catalyst at room temperature in dichloromethane under 100 atm of CO:H2 (1:1). Excellent chemoselectivities are observed with both diphosphine and diphosphinite complexes. In addition, high regioselectivities are recorded in each case giving predominantly the branched aldehydes (76–91%). However, low activities and enantioselectivities are obtained with both diphosphine and diphosphinite complexes at room temperature (up to 18%).
4.3 Asymmetric hydrogenation
Finally, we have evaluated the catalytic performance of the diphosphines 1 and diphosphinites 2 in the asymmetric hydrogenation of dimethyl itaconate [17] (Scheme 8).
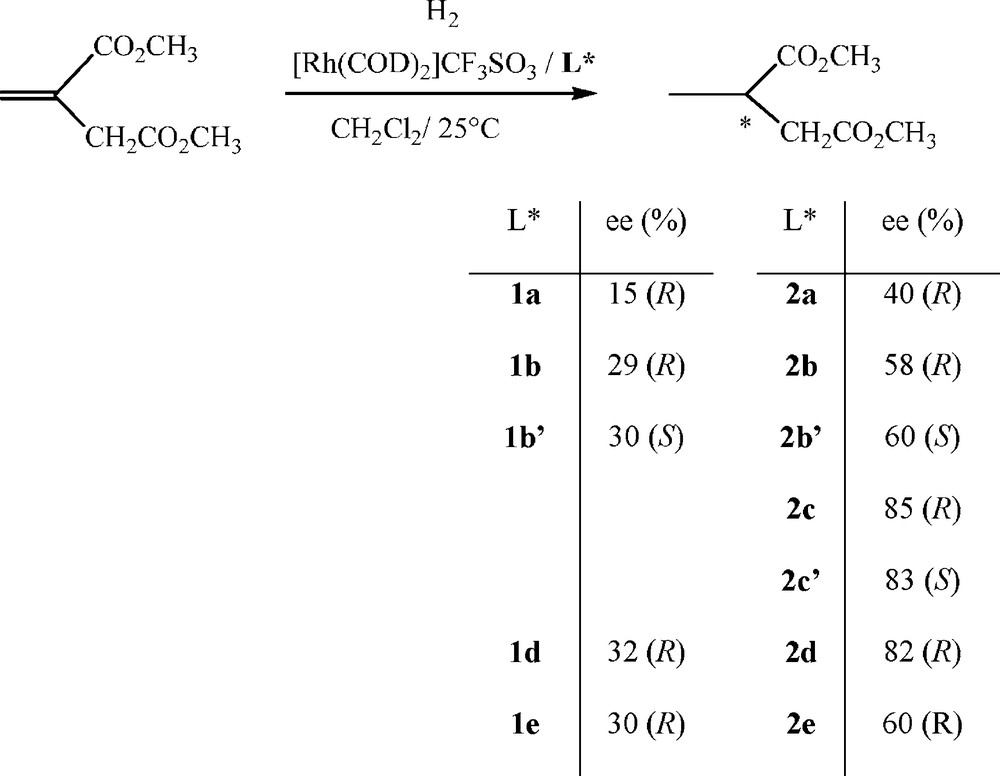
Asymmetric hydrogenation of dimethyl itaconate.
We used catalysts prepared in situ11 by adding the ligand 1 or 2 to [Rh(COD)2]CF3SO3.12 All of the ligands give rhodium catalytic systems that are active in hydrogenation of dimethyl itaconate producing enantiomeric excess in the range of 31 to 85%. Diphosphines 1 give catalytic systems that are more active as complete conversion is achieved in 3 hours at room temperature under an hydrogen pressure of 20 bar whereas diphosphinites 2 require an hydrogen pressure of 40 bar during 6 hours to reach a complete conversion. The best result in terms of enantioselectivities is obtained with diphosphinites 2c and 2d. The enantiomeric excess values are strongly influenced by the nature of the backbone whereas the sense of the enantioselection is determined by the configuration of the stereocentres as observed in asymmetric allylic substitution. The (R) enantiomer of the methylsuccinate is obtained with ligands 1–2 (a, b, c, d, e) derived from the (S) or (S,S) diols whereas the (S) enantiomer is obtained with the ligand 1–2 (b’, 2c’) derived from the (R) or (R,R) diols. The enantioselectivity observed in the hydrogenation of dimethyl itaconate is likely to be governed by the electronic properties rather than the steric factors of the ligand.
5 Conclusion
Stereodynamic chiral diphosphines and diphosphinites derived from 2,2’-biphosphole have been synthetised by a versatile methodology. By the introduction of a relatively simple backbone between the two phosphorus, the axial and P-chiralities can be controlled and locked upon coordination to a metal centre to afford diastereo- and enantiopure palladium, platinum or rhodium complexes.
These stereodynamic ligands have been evaluated in asymmetric Pd-catalyzed allylic substitution, Pt-catalyzed hydroformylation and Rh-catalyzed hydrogenation.
Notable features are the direct use of these stereodynamic diphosphinites to convey enantioselectivity in Rh-catalyzed hydrogenation through an in situ dual chirality control.
The modular construction of these stereodynamic ligands by combination of the 2,2’-biphosphole framework and chiral linker offers immense scope for structural variations and catalysts tuning. The application of the stereodynamic chiral diphosphines and diphosphinites derived from 2,2’-biphosphole in a broad range of rhodium group metal-catalyzed asymmetric transformations are currently in progress.
Acknowledgments
We are grateful to the Centre national de la recherche scientifique, the ministère de l’Éducation nationale et de la recherche and the université Paul-Sabatier.
1 The word atropos consists of “a” meaning “not” and “tropos” meaning “turn” in Greek.
2 These diphosphanes are chirally flexible ligands because of the configurational instability of the axial chirality generated by the 2,2’-biphosphole framework and of the central chiralities at the phosphorus atoms.
3 The different possibilities of combining axial and central chiralities in 2,2’-biphosphole lead to 8 diastereosiomers. Among these eight diastereosimers, six are really inequivalents because the four diastereoisomers with opposite configurations of the two phosphorus atoms are equivalent two by two. For more details on the stereochemical analysis [8].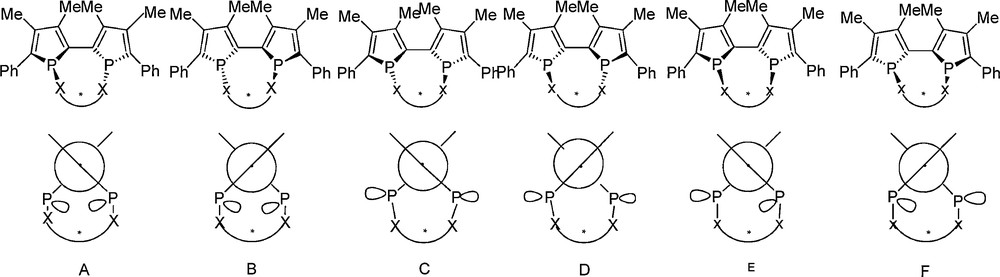
4 In phospholes, the inversion barrier for the pyramidal phosphorus is reduced, relative to that in phosphines, as a result of the increase in aromatic character of the phosphole in the transition state. The activation barrier to phosphorus inversion in 2,2’-biphosphole is measured to be only 16.5 kcal/mole leading to phosphorus inversion at −60 °C [8,10].
5 The refinement of the Flack's parameter clearly indicates that these compounds are enantiomerically pure in solid state.
6 Cyanogen bromide is toxic by inhalation of its vapors or by the hydrogen cyanide from decomposition.
7 π-donor substituents on the phosphorus of phospholes do not affect the activation barrier according to experimental measurements and calculations.
8 These unidentified products are palladium complexes with four diastereotopic phosphorus having opposite phosphorus configurations according to 31P NMR.
9 This lack of changes shows that the energy barrier for the stereochemical interconversion is higher than ∼18 kcal/mol at 85 °C.
10 The broadness of the line can be explain by a short T2 relaxation associated to an aggregation phenomena in solution as no dynamic effect have been observed by variable temperature experiment.
11 Experiments carried out by catalytic systems formed in situ provide essentially the same enantioselectivitiy values as those obtained from preformed complexes.
12 In situ catalyst were prepared by adding 1.4 eq. of ligand to [Rh(COD)2]CF3SO3. In the absence of free ligand, demetallation was observed in each case.