

1 Introduction
In contrast to the extensive organometallic chemistry of 1-phosphabenzenes (phosphinines) summarised by Le Floch [1] and others [2,3], the exploration of the behaviour of the corresponding 1,3,5-triphosphabenzene ring systems is much less developed. We described previously several η1-complexes of P3C3But3 with square-planar Pt(II) fragments [4] and Binger et al. reported the first examples of η6-ligated compounds typified by [M(CO)3(η6-P3C3But3)] (M = Mo, W), [Mn(η5-C5H5)(η6-P3C3But3)] and [Ru(η4-COD)(η6-P3C3But3)], (obtained directly from P3C3But3), which were not, however, structurally characterised [5]. Subsequently, Jones and coworkers [6] extended the range of η6-complexes to include [Ru(η5-C5Me5)(η6-P3C3But3)][PF6] and [Rh(η4-COD)(η6-P3C3But3)][BArf4], whose structures were established by single-crystal X-ray diffraction studies.
Elsewhere [7], we have structurally characterised the remarkable triple-decker compound [{Sc(P3C2But2)}2-μ-(η6-P3C3But3)] in which the 1,3,5-triphosphabenzene ring bridges two scandium(I) centres. Furthermore, in unpublished work, we have synthesised and fully structurally characterised the first simple sandwich compounds [M(η6-P3C3But3)2] (M = Zr, Hf) containing these ring systems, (albeit in very low yield), by metal-vapour syntheses directly from the phosphaalkyne, tBuC≡P [8].
Photoelectron spectroscopic studies and DFT calculations showed that the 1,3,5-triphospharene complexes [M(CO)3(η6-P3C3But3)] (M = Cr, Mo, W) have a higher first IE compared with their corresponding carbocyclic counterparts [M(CO)3(η6-C6H3But3)]. An electronic structure analysis indicated significantly stronger bonding in the former complexes as a result of the greater metal-ligand back donation to lower lying LUMOs of the phosphorus substituted ring [9].
Complexation of phosphinines is related to the aromaticity of these compounds, which is comparable to that of benzene [10,11]. Accordingly η6-complexation of phosphinines is a viable possibility, competing with the usual η1-coordination mode via the phosphorus lone-pair electrons [1]. Computations on the complex stabilities of the parent phosphinines (mono-, di- and triphosphinines) with alkaline and alkaline earth cations predict similar stabilities for the η1- and η6-coordination [12]. With increasing bulkiness of the substituents at carbon η6-ligation becomes the preferred coordination mode. In the case of the λ5-phosphorus phosphinines, the η1-coordination at phosphorus is no longer possible, whereas the π-system is able to complex transition metals.
The extent of cyclic delocalization, which results in a significant stabilisation in these systems [13], differs however from that of the λ5-analogue and magnetic measurements indicate only a small degree of aromaticity [14] for λ5-phosphabenzene. Furthermore, the electronic nature of the substituents at phosphorus has a significant effect, with the strongly electron accepting fluorine atoms increasing the nucleus independent chemical shift (NICS) value considerably [11,14]. Likewise, amino substituents should also have a similar effect to fluorine, as exemplified in the 1λ5,3λ5,5λ5-triphosphabenzene ring, P3C3H3(NMe2)6, synthesised by Fluck and coworkers, which is planar [15] whereas the parent molecule P3C3H9 is not planar [13].
Some of the λ5-phosphorus containing ylides are known to form complexes via the carbon atom of the ylidic bond, however these complexes are formed with late transition metals, for which there is no possibility to form η6-complexes with the ring π-system [16–19]. We wish to report herein new syntheses as well as structural, computational and bonding aspects of several molybdenum tricarbonyl complexes derived from the 2,4,6-tritertiarybutyl-1,3,5-triphosphabenzene ring, P3C3But3 1, and some of its λ5-alkylated derivatives.
2 Results and discussion
As mentioned in the Introduction, [Mo(CO)3(η6-P3C3But3)] 2 in which the 1,3,5-triphosphabenzene ring acts as a 6-electron donor has been previously described [5], however its molecular structure was not determined due to difficulties in obtaining suitable single crystals from the synthetic route involving displacement of cycloheptatriene from [Mo(CO)3(η6-C7H8)].
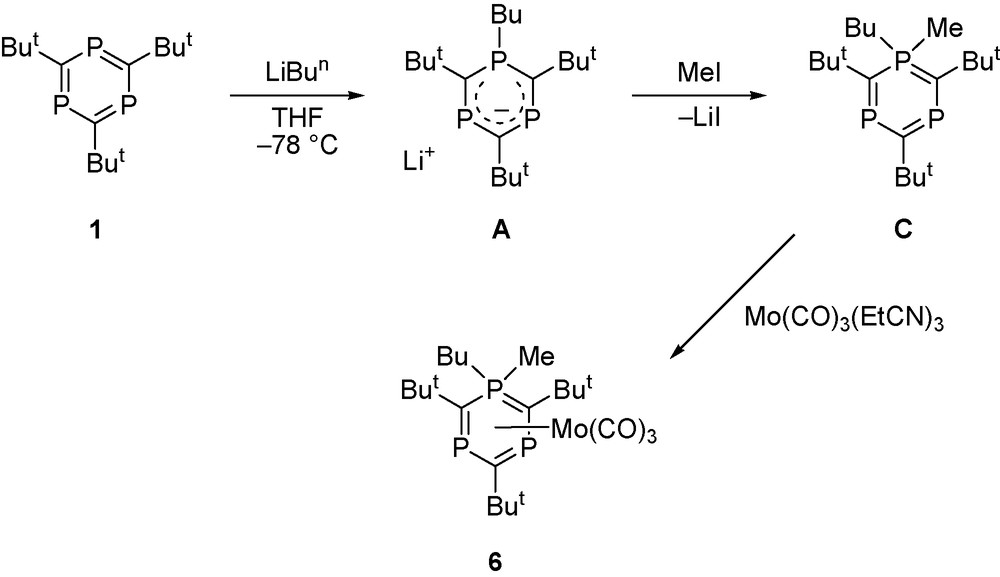
We have, however, developed an alternative synthesis by treatment of 1 with the very labile complex [Mo(CO)3(EtCN)3] at room temperature, resulting in [Mo(CO)3(η6-P3C3But3)] 2 as crystalline product which can readily be separated from the displaced EtCN (Scheme 1). A single crystal X-ray diffraction study enabled its molecular structure to be determined which is shown in Fig. 1 together with selected bond length and bond angle data (Fig. 1).
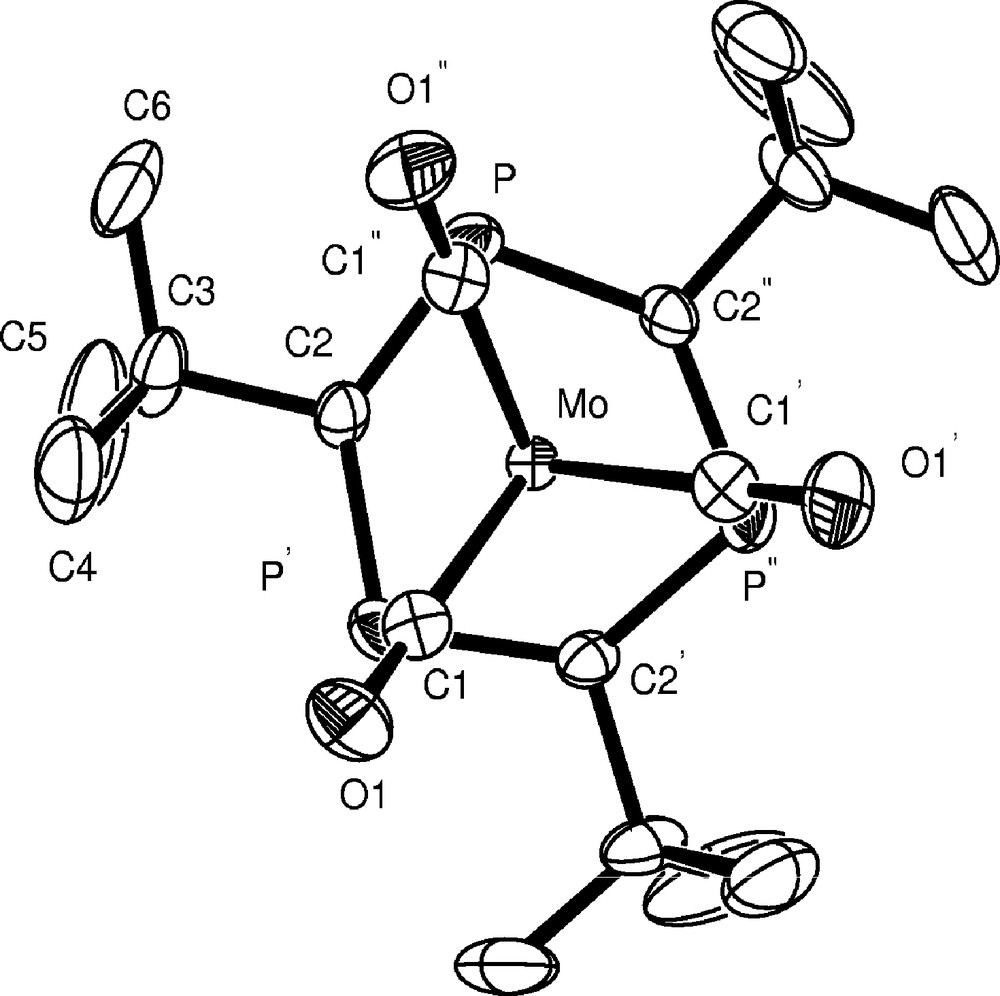
Molecular structure of [Mo(CO)3(η6-P3C3But3)] 2. Selected bond lengths (Å) and angles (°): Mo–M(1) 1.800(4), Mo–C(1) 1.994(4), Mo–C(2) 2.437(3), Mo–P 2.5772(9), P–C(2) 1.752(3), P–C(2)’ 1.754(3). M(1)–Mo–C(1) 127.4(2), P–C(2)–P” 131.8(2). M(1) is the centroid of the P3C3But3 ring.
As expected, the heterocycle in 2 is essentially planar with equal P–C bond lengths (1.753 Å av), indicative of electron delocalisation within the ring. This observation is in contrast with the structure of the analogous benzene complex [Cr(CO)3(η6-C6H6)] which exhibits alternating C–C distances [20–22]. The P–C bond length is slightly longer than that found in free P3C3But3 (1.727 Å av) [23] but similar to the value reported in [Ru(η5-C5Me5)(η6-P3C3But3)][PF6] [6]. Each Mo–CO bond eclipses a ring phosphorus atom. Interestingly, the experimentally observed bond length data in 2 compare rather well with those we obtained previously by DFT calculations [9] (e.g., Mo–P 2.577 Å (obs), 2.574 Å (calc); Mo–C (ring) 2.437 Å (obs), 2.425 Å (calc)).
In principle, the 1,3,5-triphosphabenzene ring 1 has the remarkable potential to act as a 2, 4, 6, 8, 10 or even 12-electron donor utilising appropriate combinations of its 6π-electron system and/or one or more of the three phosphorus lone pairs. To explore this potential, 2 was treated with [{PtCl2(PEt3)}2] at room temperature and the reaction monitored by 31P{1H} NMR spectroscopy (Scheme 2). Evidence was immediately observed for a bridge-splitting reaction and the formation of the new bimetallic complex trans-[Mo(CO)3(η6-P3C3But3)PtCl2(PEt3)] 3 in which the ring acts as an overall 8-electron donor towards the two metal centres. However, even in the presence of an excess of [{PtCl2(PEt3)}2] no evidence was found for any further η1-ligation of the η6-bonded triphosphabenzene ring to platinum.
As expected, the 31P {1H} NMR spectrum of 3 shows a similar pattern of lines to those we previously reported [4] for trans-[(P3C3But3)PtCl2(PEt3)] 4 but with all resonances significantly chemically shifted and having different coupling constants, (e.g., for 3 δPA = 98.3, δPB = 67.2, δPC = 16.5 ppm; 2J(PAPB) = 16.9, 2J(PAPC) = 545, 1J(PtPA) = 2392, 1J(PtPC) = 3010 Hz). For 4 δPA = 207.1, δPB = 264.5, δPC = 12.1 ppm; 2J(PAPB) = 36.4, 2J(PAPC) = 509, 1J(PtPA) = 2378, 1J(PtPC) = 2884 Hz).
We were also interested in the nature of the ligating behaviour of 1λ5,3λ3,5λ3–triphosphabenzene ring systems towards the [Mo(CO)3] fragment in the complexes 5 and 6 shown in which one of the original ring λ3-phosphorus atoms has been changed to λ5.
Thus, treatment of the 1,3,5-triphosphabenzene P3C3But3 1 with LiBun in THF at −78 °C readily formed the known lithium salt (A, Scheme 3) of the triphospha-cyclohexadienyl anion. Subsequent reaction with [Mo(CO)3(EtCN)3] afforded the presumed η5-ligated Mo(CO)3 complex B (Scheme 3), which was not isolated but readily converts to the neutral red crystalline complex [Mo(CO)3(η5-P3C3But3)(H)Bun)] 5 on passing the solution down a silica gel column (Scheme 3). Complex 5 was identified as by its characteristic 31P NMR spectrum and its molecular structure (shown in Fig. 2) was confirmed by a single crystal X-ray diffraction study.

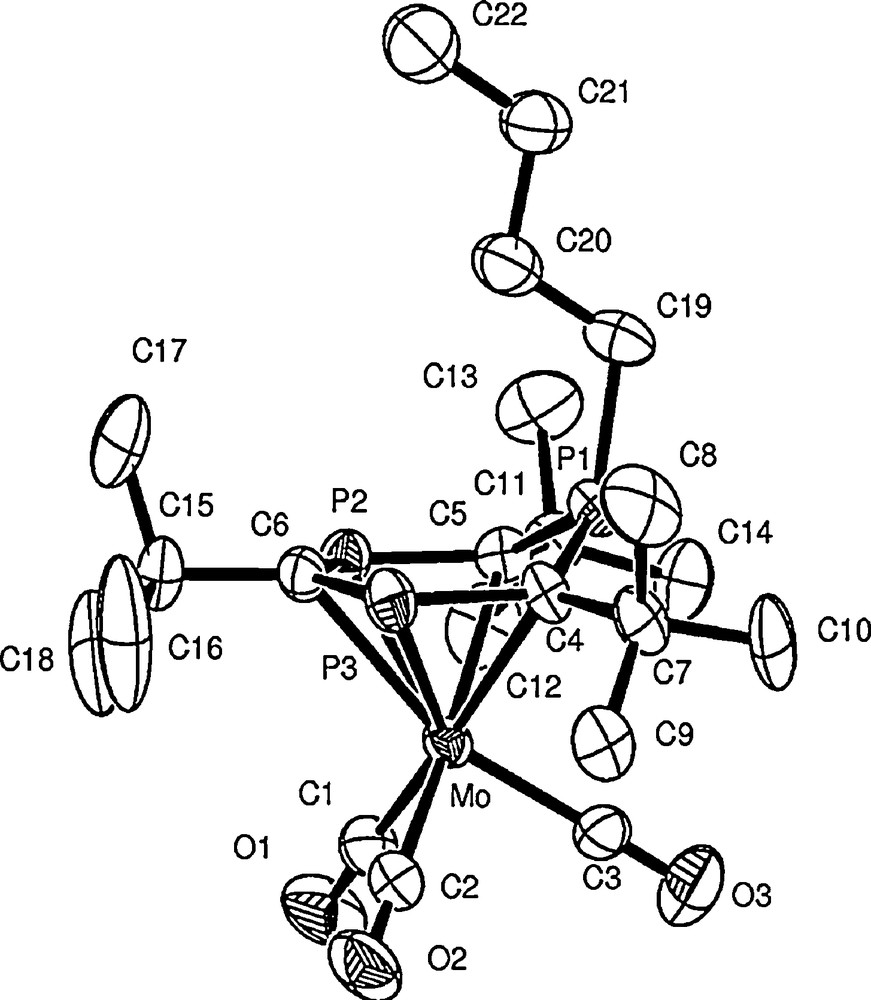
Molecular structure of [Mo(CO)3(η5-P3C3But3)(H)Bun)] 5. Selected bond lengths (Å) and angles (°): Mo–C(4) 2.418(5),Mo–C(5) 2.432(6), Mo–C(6) 2.440(6), Mo–P(2) 2.532(2), Mo–P(3) 2.545(2), Mo–P(1) 2.958(2), P(1)–C(4) 1.738(6), P(1)–C(5) 1.754(6), P(1)–C(19) 1.826(7), P(2)–C(6) 1.753(7), P(2)–C(5) 1.760(6), P(3)–C(6) 1.757(6), P(3)–C(4) 1.766(6). C(4)–P(1)–C(5) 107.1(3), C(4)–P(1)–C(19) 116.1(3), C(5)–P(1)–C(19) 117.4(3), C(6)-P(2)-C(5) 108.2(3), P(2)–C(6)–P(3) 131.4(4), P(1)–C(5)–P(2) 122.3(3), C(6)–P(3)–C(4) 107.4(3), P(1)–C(4)–P(3) 122.8(3).
The 31P{1H} NMR spectrum of 5 shows two distinct phosphorus environments, representing the two λ3- and the single λ5-phosphorus atoms of the ring, along with the expected 2JPP coupling constants (71.5 ppm, (d, 2P, 2JPP 18.3 Hz; −14.5 ppm t, 1P, 2JPP 18.3 Hz). Furthermore, the proton coupled 31P NMR spectrum confirmed that the unique P atom is directly bonded to a proton 1JPH 448 Hz.
The molecular structure of 5 can also be represented by the zwitterionic structure shown below. The P(1) atom which protrudes above the plane of the ring as expected adopts a trigonal-pyramidal geometry. The P–C bond lengths within the Mo-coordinated section of the heterocycle (C4, C5, C6, P2, P3) are very similar, indicating that there is a delocalisation of electron density over these centres. The shortest ring bond is that between P(1) and C(4) (1.738(6)Å) although the other bond involving the λ5-phosphorus atom P(1)–C(5) (1.754(6) Å) is similar. It is also noteworthy that the P–C distances in 5 are similar to those in 2.
When, however, the Li salt (A) is first methylated by treatment with MeI, the resulting non-isolated yellow compound is P3C3But3(Me)(Bun) (C) which is readily converted to [Mo(CO)3(η5-P3C3But3(Me)(Bun)] 6 by further treatment with [Mo(CO)3(EtCN)3] (Scheme 4).
The 31P{1H} NMR spectrum of 6 is similar to that observed in 5 but with an associated downfield chemical shift, especially for the λ5-phosphorus atom (37.6 ppm, 2JPP 19.1 Hz). The molecular structure of 6, which was determined by a single-crystal X-ray diffraction study, is shown in Fig. 3. A similar average P–C bond length is found for 6 as in 5 with P(1) again being located above the ring plane. The P(3)–C(5) bond length (1.743(9) Å) is slightly shorter than the other P–C distances within the ring, which are roughly equal in length.
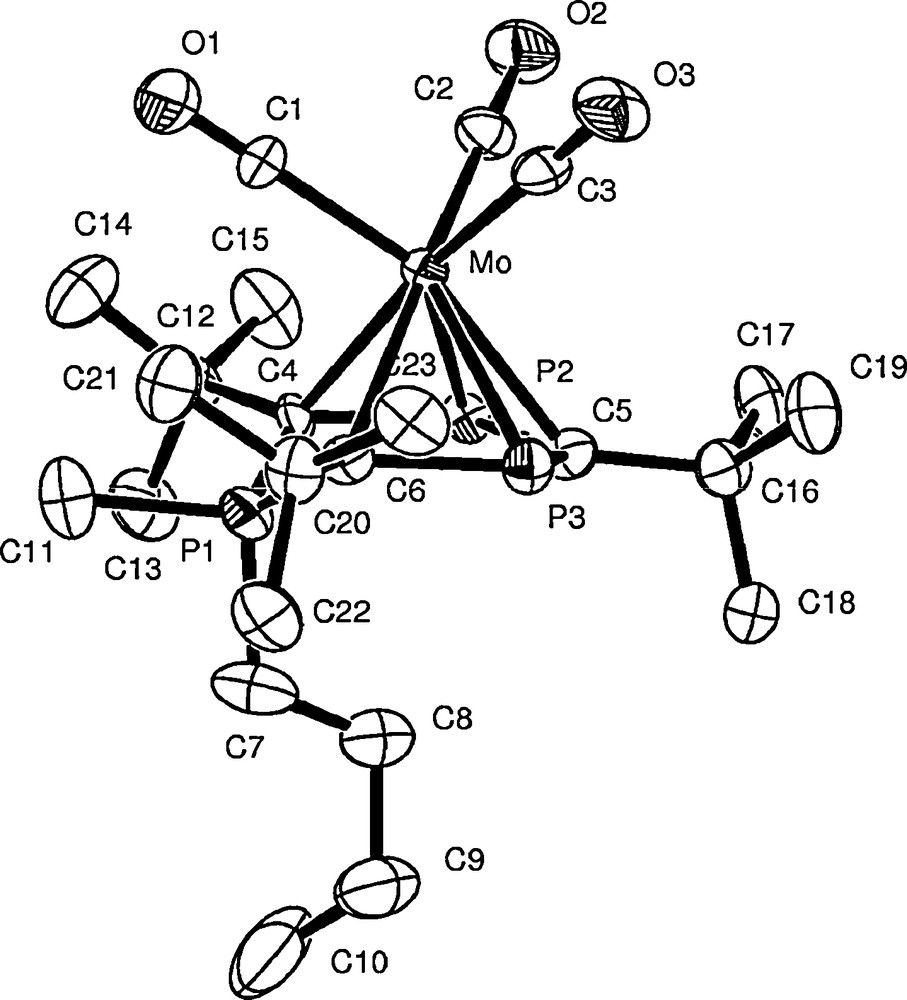
Molecular structure of [Mo(CO)3(η5-P3C3But3)(Me)(Bun)] 6. Selected bond lengths (Å) and angles (°): Mo–C(5) 2.443(9), Mo–C(6) 2.447(9), Mo–C(4) 2.474(9), Mo–P(3) 2.512(2), Mo–P(2) 2.536(2), P(1)–C(4) 1.772(9), P(1)–C(6) 1.774(9), P(1)–C(11) 1.798(11), P(1)–C(7) 1.835(11), P(2)–C(4) 1.773(9), P(2)–C(5) 1.775(10), P(3)–C(5) 1.743(9), P(3)–C(6) 1.764(9). C(4)–P(1)–C(6) 105.9(4), C(4)–P(1)–C(11) 111.9(5), C(6)–P(1)–C(11) 110.9(5), C(4)–P(1)–C(7) 113.9(5), C(6)–P(1)–C(7) 114.6(5), C(11)–P(1)–C(7) 99.8(6), P(1)–C(4)–P(2) 118.6(5), C(4)–P(2)–C(5) 108.5(4), P(3)–C(5)–P(2) 130.1(6), C(5)–P(3)–C(6) 109.2(4), P(3)–C(6)–P(1) 118.4(5).
As previously mentioned, treatment of 1 with [Mo(CO)3(EtCN)3] readily gives complex 2 which when treated with LiBun and then MeI in that order (followed by filtration through silica gel) unexpectedly results in the formation of the unusual lithium complex [Mo(CO)3(η5-P3C3But3)(Me)(Bun)LiO(THF)3] 7 in good yield (Scheme 5).
The 31P{1H} NMR spectrum of 7 reveals three distinct phosphorus environments. The λ3-phosphorus appears as a doublet of doublets at 73.9 ppm with the coupling constants showing bonding to two different λ5-phosphorus atoms (2JPP 35.4 and 27.1 Hz). The proton coupled 31P NMR spectrum clearly shows that one of these λ5-phosphorus centres is directly bonded to a proton (−3.8 ppm, 1JPH 518.6 Hz) and this was subsequently confirmed by a single crystal X-ray diffraction study which revealed other interesting and unexpected bonding features. The molecular structure of 7 is shown in Fig. 4.
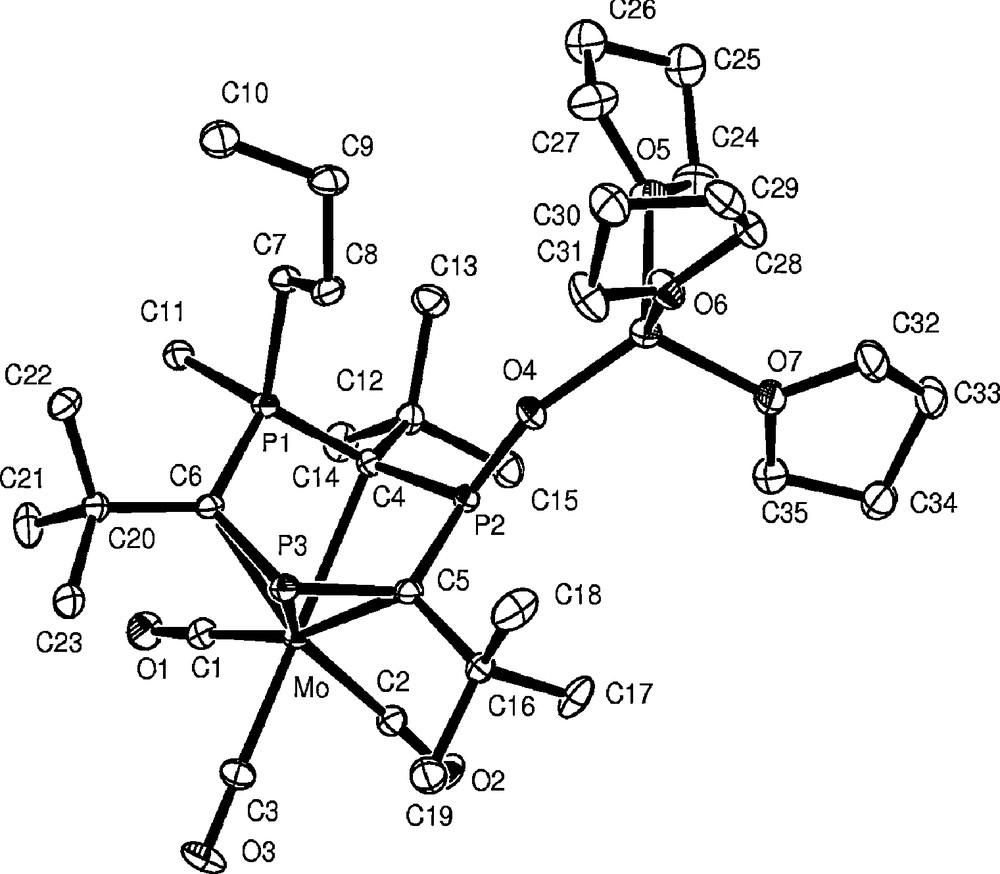
Molecular structure of [Mo(CO)3(η5-P3C3But3)(Me)Bun)LiO(THF)3] 7. Selected bond lengths (Å) and angles (°): Mo–C(6) 2.419(7), Mo–C(4) 2.433(7), Mo–C(5) 2.438(7), Mo–P(3) 2.519(2), P(1)–C(6) 1.765(7), P(1)–C(4) 1.767(7), P(1)–C(11) 1.826(7), P(1)–C(7) 1.856(7), P(2)–O(4) 1.504(5), P(2)–C(5) 1.760(8), P(2)–C(4) 1.782(7), P(3)–C(5) 1.748(8), P(3)–C(6) 1.784(7), Li–O(4) 1.841(14). C(6)–P(1)–C(4) 105.5(3), C(6)–P(1)–C(11) 111.1(3), C(4)–P(1)–C(11) 110.6(4), C(6)–P(1)–C(7) 112.7(3), C(4)–P(1)–C(7) 117.9(3), C(11)–P(1)–C(7) 99.1(3), P(1)–C(4)–P(2) 113.0(4), O(4)–P(2)–C(5) 118.8(3), O(4)–P(2)–C(4) 119.1(3), C(5)–P(2)–C(4) 103.9(3), P(3)–C(5)–P(2) 127.4(4), C(5)–P(3)–C(6) 109.6(3), P(1)–C(6)–P(3) 120.6(4), P(2)–O(4)–Li 158.6(5).
Interestingly, there are only four formal bonding contacts between the heterocycle and the metal centre as a result of the presence of two λ5-phosphorus centres which each sit out of the plane defined by the other four atoms of the ring. During the reaction, the P(1) atom has evidently undergone the desired double alkylation and now features both methyl and n-butyl groups. The presence of the P–H bond is confirmed at P(2) and there is also an unexpected Li(THF)3O− fragment incorporated into the molecular structure. The P(2)–O(4) distance falls between typical single and double bond values.
It seems likely 7 results from a reaction between the Mo-bonded ring and LiOH (which may be present in the LiBun). It is interesting that 7 is not observed in the reaction affording 6 discussed above, in which the ring was treated with LiBun and MeI before attachment of the [Mo(CO)3] fragment. It appears that when the ring is complexed to molybdenum it becomes more reactive towards LiOH. (It should be noted that an identical sample of 7 was also isolated without the use of a column chromatography in the purification step). Interestingly, we showed previously [4] that the η1-ligated triphosphabenzene ring in trans-[PtCl2(PMe3)(P3C3But3)] is very susceptible to attack by water and undergoes a remarkable trihydration reaction to afford the structurally characterised cis-[PtCl(PMe3)(P3O3C3H5But3)] containing the novel CH(But)PH(O)CH(But)PH(O)CH(But)P(O) ring system, whereas triphosphabenzene 1 itself is inert under similar conditions.
Within the heterocyclic ring of 7 the shortest bond distance P(3)–C(5) (1.748(8) Å) is located adjacent to the longest P(3)–C(6) (1.784(7) Å) and the average bond length at 1.768 Å falls almost directly between the two. A pseudo-chair type conformation is adopted by the 6-membered ring and three formal Mo–C bonding contacts which are roughly equal in length (average bond distance = 2.43 Å) can be identified. The single Mo–P bond present in the structure is within the typical range for this type of interaction and is comparable in length with those observed in structures previously discussed in this paper. The But groups associated with the carbon atoms of the ring project slightly below the C(4)–C(5)–P(3)–C(6) ring-plane and are staggered in relation to the three Mo-bonded carbonyl moieties.
3 Computational studies
Computational studies were carried out to understand the coordination abilities of the 1,3,5-triphosphabenzenes containing different numbers of λ3- and λ5-phosphorus atoms (I–IV) (Fig. 5) and their parent compounds (H atoms instead of But, noted as I’–IV’), in relation to the electronic structure and the aromaticity of these ring systems.
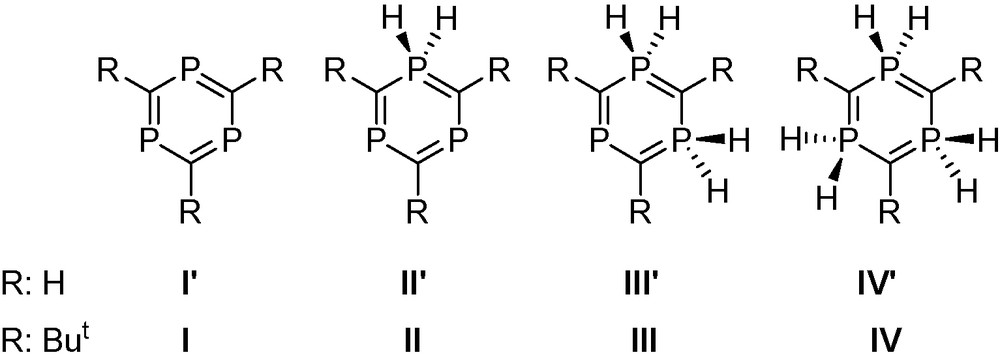
1,3,5-triphosphabenzenes containing different numbers of λ3- and λ5-phosphorus atoms (I–IV).
The computed structures of P3C3But3 (I), P2(PH2)C3But3 (II), P(PH2)2C3But3 (III) and (PH2)3C3But3 (IV) are shown in Fig. 6. At the B3LYP/aug-cc-pVTZ level (and similarly at the B3LYP/cc-pVDZ and B3LYP/6-311 + G** levels) the P–C bond lengths lie in the 1.709–1.738 Å range for I’–IV’, indicating a significant bond length equalisation for each of the investigated systems. The P–C distances of the But-substituted compounds (I–IV) are larger (1.731–1.773 Å at the B3LYP/cc-pVDZ level) presumably because of the bulky substituents. It is interesting to note that in I’ all the P–C bond lengths are the same whereas in I there is a significant difference: three bond distances are 1.743 Å and the others are 1.772 Å. These results are also similar at different levels of theory.
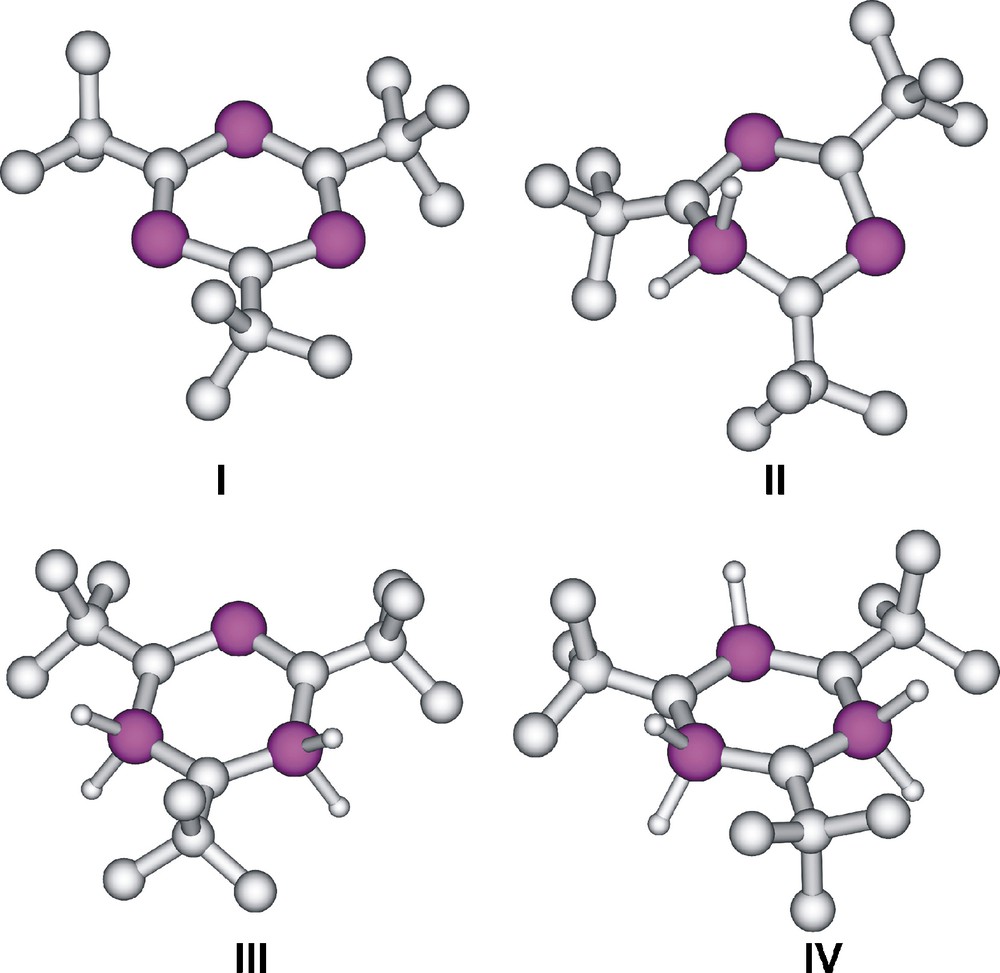
Computed structures of P3C3But3 (I), P2(PH2)C3But3 (II), P(PH2)2C3But3 (III) and (PH2)3C3But3 (IV).
In the experimentally determined X-ray structure of I [23], the bond distances vary between 1.718 and 1.729 Å; while some methyl groups appear disordered. Apparently, the rotational position of the tBu groups is responsible for the changes in the P–C distances.
Analysing the bond length and electron density [24] values, it is worth noting that the λ5-P–C bonds are slightly stronger than the λ3-P–C bonds and the covalent bond indices of the latter are much larger, supporting the ylidic description of the λ5-P–C bonds. On the basis of the natural bond orbital (NBO) analysis, the ylidic carbon and phosphorus atoms possess increasing natural population analysis (NPA) partial charges in the direction II’–IV’ (Table 1 and Fig. 7). It is interesting, however, that the charge separation between carbon and phosphorus of the formally ylidic bonds is only slightly larger than for their λ3-P–C bonded counterparts. Also noteworthy is that the λ5-P–C bonds in the rings are more polarized than in the H3P = CH2 molecule. Interestingly, the Laplacian [24] of the λ3-P–C bonds is much more positive than that of the λ5-P–C bonds. For the rings not only are the bond lengths equalized, but also the ellipticity values (ɛ), which are indicators of double bond character [24], are very similar (in general, the λ3-P–C bonds have slightly larger ellipticity than the λ5-P–C bonds). This behaviour was already noted for the isolated parent systems [25].
Bonding properties of systems I’–IV’ at the B3LYP/aug-cc-pVTZ level of theory. Bond lengths (d) in Å, WBI: Wiberg bond indices, electron density (ρ, in a.u.) and its Laplacian (▿2ρ, in a.u.) and ellipticity (ɛ) at the bond critical point, NPA partial charges of the C and P atoms [q(C) and q(P), respectively].
| Symmetry | P | d | WBI | ρ | ▿2ρ | ɛ | q(C) | q(P) | |
| HPCH2 | Cs | λ3 | 1.667 | 1.964 | 0.191 | 0.315 | 0.432 | −0.76 | 0.42 |
| H3PCH2 | Cs | λ5 | 1.678 | 1.371 | 0.198 | −0.017 | 0.467 | −1.16 | 0.85 |
| I’ | D3h | λ3 | 1.732 | 1.336 | 0.178 | 0.081 | 0.234 | −1.02 | 0.79 |
| II’ | C2v | λ3 | 1.727 | 1.287 | 0.180 | 0.051 | 0.236 | −1.17 | 0.87 |
| λ3 | 1.709 | 1.413 | 0.179 | 0.151 | 0.298 | −1.17 | 1.14 | ||
| λ5 | 1.738 | 1.077 | 0.186 | −0.126 | 0.175 | ||||
| III’ | C2v | λ3 | 1.708 | 1.367 | 0.181 | 0.126 | 0.306 | −1.25 | 0.89 |
| λ5 | 1.735 | 1.062 | 0.187 | −0.132 | 0.180 | −1.34 | 1.20 | ||
| λ5 | 1.711 | 1.116 | 0.189 | −0.031 | 0.273 | ||||
| IV’ | Cs | λ5 | 1.715 | 1.110 | 0.188 | −0.044 | 0.264 | −1.35 | 1.23 |
| λ5 | 1.711 | 1.110 | 0.189 | −0.037 | 0.263 | ||||
| λ5 | 1.719 | 1.096 | 0.188 | −0.065 | 0.254 | ||||
| IV’ planar | D3h | λ5 | 1.712 | 1.108 | 0.189 | −0.038 | 0.272 | −1.36 | 1.25 |
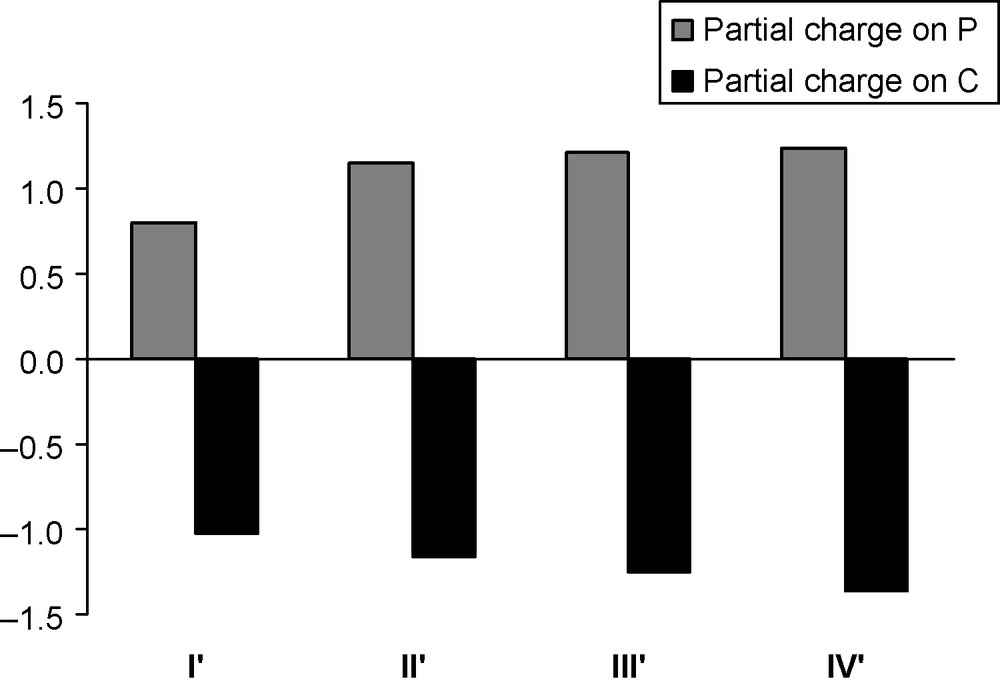
NPA partial charges on the C and P atoms at the B3LYP/aug-cc-pVTZ level (for II’, III’ and IV’ the ylidic charges are shown).
Interestingly, the computed structure of II exhibits significant non-planarity, the λ5-phosphorus atom lying out of plane by 61.3° at the B3LYP/cc-pVDZ level of theory, however, the other rings having But substituents (I, III, IV) are near planar. Since the parent 1λ3,3λ3,5λ5-triphosphabenzene (II’) was planar, it is likely that the repulsive interaction between the But- groups and the PH2 unit is responsible for the non-planarity. The presence of three PH2 units in the six membered ring of IV results in a structure very close to planarity. It is noteworthy that the parent 1λ5,3λ5,5λ5-triphosphabenzene (IV’) is non planar [13], but the planar structure is only slightly (0.5 kcal mol−l at the G3MP2//B3LYP/aug-cc-pVTZ) less stable. This behaviour could be explained by the low rotational barrier of the λ5-P–C bond, which is a result of an angle-independent effective π-type back-donating interaction [26]. Accordingly, a low energy (0.1 kcal mol−l at the G3MP2//B3LYP/aug-cc-pVTZ) C2 symmetry transition structure connects the (identical) minima of IV’.
For the parent ring systems I’–IV’ and I–IV NICS values [27–29] have also been determined (Table 2). The NICS(0) values of (I’ and I), are smaller (negative values) than for benzene, however, NICS(1) and NICS(2) [28–29], which are more characteristic for the π-system indicate significant aromaticity, while the effect of the But groups is negligible. In the presence of at least one λ5-P atom (for II’–IV’) the NICS values are significantly smaller, likewise the aromatic character of λ5-phosphabenzene was significantly reduced compared to λ3-triphosphabenzene [14]. Interestingly, II exhibits larger negative NICS values than II’, in spite of its non-planar structure. The planar 1λ5,3λ5,5λ5-triphosphabenzenes (IV’ and IV) have slightly smaller NICS values than the corresponding non-planar rings.
NICS(0), NICS(1) and NICS(2) values (in ppm) for the rings systems I’–IV’ and I–IV at the B3LYP/6-311 + G**//B3LYP/6-311 + G** level of theory.
| NICS(0) | NICS(1) | NICS(2) | |
| Benzene | −8.0 | −10.2 | −4.9 |
| I’ | −5.0 | −8.7 | −5.6 |
| II’ | + 1.1 | −1.3 | −1.1 |
| III’ | + 0.6 | + 0.1 | −0.2 |
| IV’ minimum | −2.7a | −1.2/−1.7a | −0.2/0.00a |
| IV’ planar | −3.3 | −1.6 | −0.6 |
| I | −6.8 | −9.4 | −5.9 |
| II | −8.2a,b | −9.8a,b | −4.4a,b |
| III | −1.9a | −1.4/−1.9a | −0.6/−1.0a |
| IV | −5.0a | −2.8/−2.9a | −0.9/−1.1a |
a Non-planar rings.
b NICS points were selected to minimize the close proximity of methyl groups.
The structures of the [Mo(CO)3] complexes of I’–IV’ and I–IV were also optimised at the B3LYP/cc-pVDZ(-PP) level, resulting in V’–VIII’ and V–VIII as stable complexes (Fig. 8). Additionally, the structure of benzene-molybdenum tricarbonyl was also optimised. Its minimum has C3v symmetry possessing different C–C bond lengths of 1.409 and 1.431 Å (cf. the chromium analogue [20]). A rotational transition state of C3v symmetry has also been found lying 0.2 kcal mol−1 [at the B3LYP/cc-pVDZ(-PP) level] above the minimum, in which all the C–C bond lengths are the same (1.428 Å). The extremely low barrier suggests that the rotation could take place easily. Interestingly, no structure similar to the [Mo(CO)3(C6H6)] minimum could be determined for V’. The minimum of V’ resembles the above-mentioned rotational TS having the same P–C bond lengths and the carbonyl groups lie above the CH fragments of the P3(CH)3 ring. It is worth noting that the P3C3 ring is not perfectly planar in V’ and a rotational transition state [lying 4.3 kcal mol−1 above the minimum at the B3LYP/cc-pVDZ(-PP) level] has also been found for V’. In the latter structure, the CO groups lie above the P atoms.
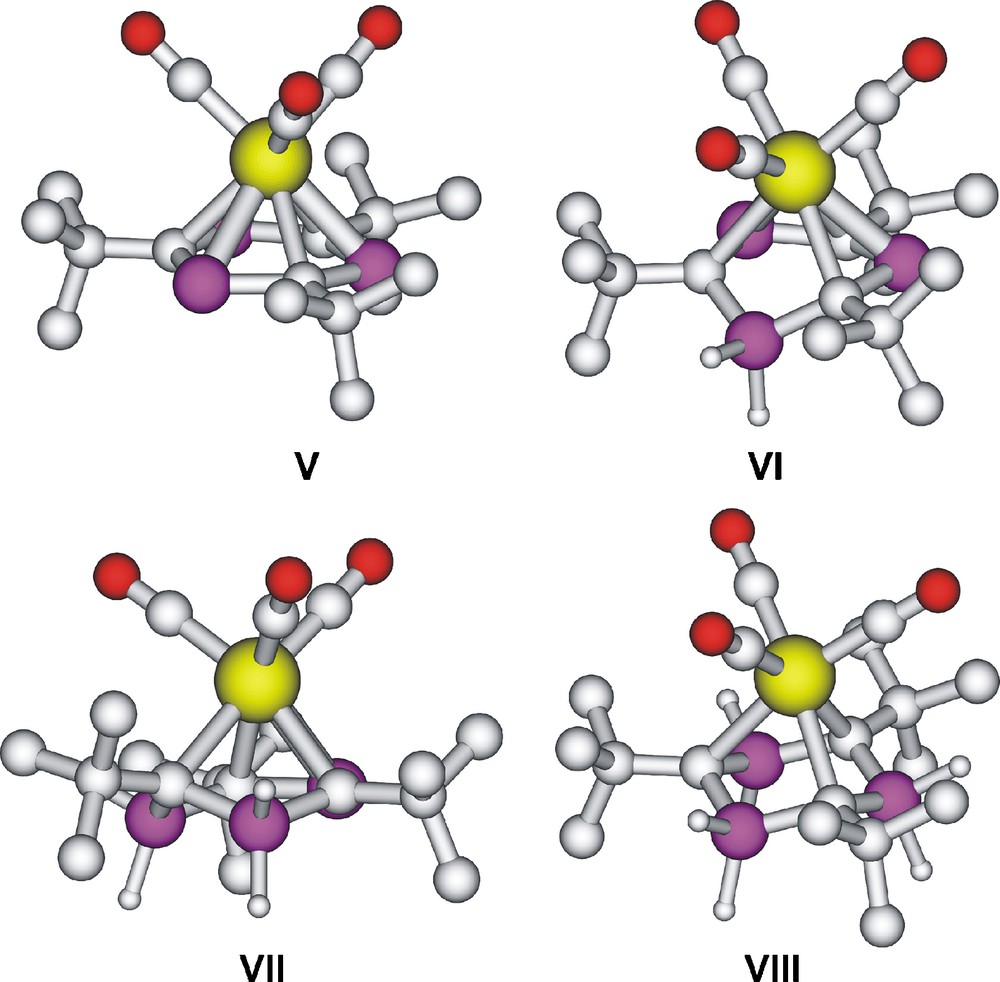
Computed structures of [Mo(CO)3P3C3But3](V), [Mo(CO)3P2(PH2)C3But3] (VI), [Mo(CO)3P(PH2)2C3But3] (VII) and [Mo(CO)3(PH2)3C3But3] (VIII).
Although V is clearly an analogue of benzene-molybdenum tricarbonyl, having P–Mo (2.67 Å) and C–Mo (2.46 Å) bond distances and an essentially planar six-membered ring; the P–C distances show some increase (to 1.779 and 1.781 Å, for I see above). While all the B3LYP/cc-pVDZ(-PP) distances are slightly longer than those determined by X-ray crystallography, the lengthening of the P–C bond upon complexation is well described by the calculations. In the case of VI–VIII, the rings are no longer planar and the λ5-phosphorus atoms do not participate in the complexation. In the case of VIII, the complexation is reduced solely to involvement of the ring carbon atoms. It is interesting to note that the λ5-P–C distances show an increased lengthening upon complexation in comparison with the λ3-analogues (for VIII the P–C distances are 1.778 Å, and for IV 1.738 Å, respectively), apparently as a result of the reduced π-back donation (negative hyperconjugation) in the ylide. This behaviour is understandable since the dative bond formation toward Mo should reduce the electron density at carbon.
Although the λ5-phosphorus atoms are excluded from the complexation, the stability of these complexes does not differ significantly for the rings with different number of pentavalent phosphorus centres (V’–VIII’) as shown by the energies (Table 3) of reactions (1)–(3). Since the Mo–C distance to the ylidic carbon is always shorter than to the other carbon atom in the complexes, the nearly constant complexation energy supports the surmise that the complexation toward the metal is in a fine balance with the π back donation toward the λ5-phosphorus within the ring.
| VI + I → V + II | (1) |
| VII + I → V + III | (2) |
| VIII + I → V + IV | (3) |
4 Experimental procedures and computations
All air and moisture-sensitive compounds were manipulated under an atmosphere of high purity argon using conventional Schlenk or glove-box techniques. Solvents were meticulously dried, distilled and freeze-thaw degassed before use. NMR spectra were recorded on a Varian Direct Drive 400 (1H 399.495 MHz, referenced to external SiMe4; 31P 161.713 MHz, referenced to external 85% H3PO4) instrument. Microanalyses were carried out by Stephen Boyer, Science Centre, London Metropolitan University, London. Single crystal X-ray structures were determined using a Nonius Kappa CCD diffractometer. All structures were refined using SHELXL-97. CCDC-775995 (2), CCDC-775995 (5), CCDC-775997 (6) and CCDC-775998 (7) contain the supplementary crystallographic data (Supplementary data) for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. P3C3But3 [30] and [Mo(CO)3(EtCN)3] [31] were synthesised by literature procedures. All other chemicals were obtained from commercial sources and used as supplied.
4.1 Synthesis of [Mo(CO)3(η6-P3C3But3)] 2
[Mo(CO)3(EtCN)3] (0.18 g, 5.2 × 10−4 mol) and P3C3But3 (0.12 g, 4 × 10−4 mol) were combined and stirred in THF (30 mL) for 24 hours. The resulting red solution was reduced to ∼2 mL and passed through a silica gel column (kiesel gel, column: 10 × 2.0 cm, eluent: n-pentane). The solvent was removed to leave a red solid which was dissolved in the minimum amount of n-pentane. Filtration and subsequent storage of the solution at −50 °C gave a crop of red crystals after 24 hours. A second batch of crystals was isolated from the filtrate at −50 °C. Yield (overall) = 150 mg, 73%. 1H NMR (C6D6, 400 MHz): 1.32 (s, 27H, But). 31P{1H} NMR (C6D6, 400 MHz): 56.2 (s).
Crystal data for 2: C18H27MoO3P3, M = 480.25, Rhombohedral, space group (No. 148), a = 16.1113(7) Å, b = 16.1113(7) Å, c = 14.6065(7) Å, α = 90°, β = 90°, γ = 120°, V = 3283.5(3) Å3, T = 173(2) K, Z = 6, Dc = 1.46 Mg m−3, μ = 0.83 mm−1, λ = 0.71073 Å, crystal size 0.08 × 0.08 × 0.04 mm3, 3987 measured reflections, 1408 independent reflections, 1206 reflections with I > 2σ(I), Final indices R1 = 0.035, wR2 = 0.079 for I > 2σ(I), R1 = 0.045, wR2 = 0.083 for all data. Data collection: KappaCCD, Program package WinGX, Abs correction MULTISCAN Refinement using SHELXL-97, Drawing using ORTEP-3 for Windows. A tiny fragment, cut from a larger crystal, was used for the data collection. The molecule lies on a crystallographic 3-fold axis. CCDC-775995.
4.2 Synthesis of trans-[Mo(CO)3(η6-P3C3But3)PtCl2(PEt3)] 3
[Mo(CO)3(η6-P3C3But3)] (0.1 g, 2.1 × 10−4 mol) and [{PtCl2(PEt3)}2] (0.08 g, 1.0 × 10−4 mol) were combined and stirred in DCM (20 mL). After 30 mins, the solvent was removed from a small portion of the reaction mixture and the 31P{1H} NMR spectrum was recorded. The 31P{1H} NMR spectrum of the reaction mixture remained unchanged over a 24 hour period. 31P{1H} NMR (CD2Cl2, 400 MHz): δPA 98.3, δPB = 67.2, δPC = 16.5 ppm; 2J(PAPB) = 16.9, 2J(PAPC) = 545, 1J(PtPA) = 2392, 1J(PtPC) = 3010 Hz).
4.3 Synthesis of [Mo(CO)3(η5-P3C3But3)(H)(Bun)] 5
P3C3But3 (0.053 g, 1.8 × 10−4 mol) was dissolved in THF (20 mL) and LiBun (0.08 mL, 2.5 m) was added dropwise at −78 °C. After allowing the resulting orange solution to reach room temperature, [Mo(CO)3(EtCN)3] (0.061 g, 1.8 × 10−4 mol) dissolved in THF (20 mL) was added. A red solution formed immediately. After stirring for 1 hour, the solution was reduced to 3 mL in volume and the crude product was purified by column chromatography on silica gel (kiesel gel, column: 10 × 2.0 cm, eluent: n-pentane). The n-pentane was removed in vacuo to leave a red solid as the desired product. Addition of hexane (1 mL) and THF (1 mL) followed by storage at −30 °C for 12 hours gave red crystals. Yield = 59 mg, 62%. Anal. Calcd for C22H37O3P3Mo: C, 49.06; H, 6.93. Found: C, 49.04; H, 7.00. 1H NMR (C6D6, 400 MHz): 1.41 (s, 9H, But), 1.13 (s, 18H, But), 0.94 (m, 6H, CH2), 0.65 (t, 3H, CH3, 7.10 Hz). 31P{1H} NMR (C6D6, 400 MHz): 71.45 (d, 2P, 2JPP 18.3 Hz), −14.50 (t, 1P, 2JPP 18.3 Hz). 31P NMR (C6D6, 400 MHz): 71.45 (d, 2P, 2JPP 18.3 Hz), −14.50 (dm, 1P, 1JPH 448.0 Hz).
Crystal data for 5: C22H37MoO3P3, M = 538.37, Orthorhombic, space group Pna21 (No. 33), a = 16.8772(6) Å, b = 9.9883(4) Å, c = 15.5881(7) Å, α = 90°, β = 90°, γ = 90°, V = 2627.76(18) Å3, T = 173(2) K, Z = 4, Dc = 1.36 Mg m−3, μ = 0.70 mm−1, λ = 0.71073 Å, crystal size 0.25 × 0.20 × 0.05 mm3, 11,790 measured reflections, 4948 independent reflections, 4102 reflections with I > 2σ(I), Final indices R1 = 0.047, wR2 = 0.112 for I > 2σ(I), R1 = 0.065, wR2 = 0.122 for all data. Data collection: KappaCCD, Program package WinGX, Abs correction MULTISCAN Refinement using SHELXL-97, Drawing using ORTEP-3 for Windows. CCDC-775995.
4.4 Synthesis of [Mo(CO)3(η5-P3C3But3)(Me)(Bun)] 6
P3C3But3 (0.059 g, 2 × 10−4 mol) was dissolved in THF (20 mL) and LiBun (0.08 mL, 2.5 m) was added dropwise at 78 °C. The resulting orange solution was allowed to reach room temperature. Addition of MeI (0.01 mL, 2 × 10−4 mol) resulted in the immediate formation of a yellow solution which was stirred for 1 hour. After this time, [Mo(CO)3(EtCN)3] (0.09 g, 2.6 × 10−4 mol) in THF (20 mL) was added to the mixture which was then stirred for 2 days. Filtration, followed by removal of THF in vacuo, resulted in a red/brown solid. Extraction with an n-pentane (30 mL)/toluene (30 mL) mixture gave a red solution which was reduced to ∼2 mL in volume before THF (1 mL) was added. Storage at −30 °C for 24 hours resulted in red crystals. A second crop of crystals was obtained from the filtrate. Yield (overall) = 74 mg, 69%. Anal. Calcd for C23H39O3P3Mo: C, 49.99; H, 7.12. Found: C, 50.11; H, 7.15. 1H NMR (C6D6, 400 MHz): 1.45 (d, 3H, Me, 11.63 Hz), 1.41 (s, 9H, But), 1.11 (s, 18H, But), 0.92 (m, 6H, CH2), 0.66 (t, 3H, CH3, 7.10 Hz). 31P{1H} NMR (C6D6, 400 MHz): 89.81 (d, 2P, 2JPP 19.09 Hz), 37.59 (t, 1P, 2JPP 19.09 Hz).
Crystal data for 6: C23H39MoO3P3, M = 552.39, Monoclinic, space group P21/c (No. 14), a = 9.9655(3) Å, b = 17.0398(7) Å, c = 15.3054(6) Å, α = 90°, β = 90.658(3)°, γ = 90°, V = 2598.84(17) Å3, T = 173(2) K, Z = 4, Dc = 1.41 Mg m−3, μ = 0.71 mm−1, λ = 0.71073 Å, crystal size 0.25 × 0.10 × 0.05 mm3, 19789 measured reflections, 5074 independent reflections, 3812 reflections with I > 2σ(I), Final indices R1 = 0.083, wR2 = 0.192 for I > 2σ(I), R1 = 0.112, wR2 = 0.203 for all data. Data collection: KappaCCD, Program package WinGX, Abs correction MULTISCAN Refinement using SHELXL-97, Drawing using ORTEP-3 for Windows. CCDC-775997.
4.5 Synthesis of the [Mo(CO)3(η5-P3C3But3)(Me)(Bun)LiO(THF)3] 7
[Mo(CO)3(η6-P3C3But3)] (0.18 g, 3.7 × 10−4 mol) was dissolved in THF (30 mL) and LiBun (0.15 mL, 2.5 m) was added dropwise at −78 °C. After allowing the solution to reach room temperature, the red solution was stirred for 45 mins. Addition of MeI (0.03 mL, 4.8 × 10−4 mol) was followed by a further 3 hours of stirring. The solution was reduced to ∼3 mL in volume and passed through a silica gel column (kiesel gel, column: 8 × 3.0 cm, eluent: THF). Removal of the solvent in vacuo left a red oil which was treated with hexane (2 mL) followed by THF (2 mL). Storage at −30 °C for 12 hours resulted in orange crystals. Yield = 198 mg, 67%. Anal. Calcd for C35H64O7P3LiMo: C, 53.01; H, 8.14. Found: C, 53.12; H, 8.20. 31P{1H} NMR (THF/C6D6, 400 MHz): 73.92 (dd, 1P, 2JPP 35.35 & 27.07 Hz), 25.54 (dd, 1P, 2JPP 27.07 & 2 Hz), −3.82 (dd, 1P, 2JPP 35.35 & 2 Hz). 31P NMR (THF/C6D6, 400 MHz): 73.92 (dd, 1P, br), 25.54 (1P, br), −3.82 (dd, 1P, 1JPH 518.6 Hz & 2JPP 35.35 Hz).
Crystal data for 7: C35H64LiMoO7P3, M = 792.65, Monoclinic, space group P21/n (No. 14), a = 12.9685(3) Å, b = 18.1937(6) Å, c = 18.1936(5) Å, α = 90°, β = 107.432(2)°, γ = 90°, V = 4095.5(2) Å3, T = 173(2) K, Z = 4, Dc = 1.29 Mg m−3, μ = 0.48 mm−1, λ = 0.71070 Å, crystal size 0.25 × 0.20 × 0.10 mm3, 39,068 measured reflections, 8031 independent reflections, 6038 reflections with I > 2σ(I), Final indices R1 = 0.088, wR2 = 0.187 for I > 2σ(I), R1 = 0.120, wR2 = 0.201 for all data. Data collection: KappaCCD, Program package WinGX, Abs correction MULTISCAN Refinement using SHELXL-97, Drawing using ORTEP-3 for Windows®. The crystals were all small and intergrown. One of the larger crystals, although visually not perfect, was used for data collection in order to get sufficient diffraction intensity. Disorder in the THF groups was treated by including alternative partial occupancy sites for C25, C26 and C35 with isotropic displacement parameters and bond length restraints (SADI). The difference map shows a residual peak of ca 1.4 e, near to H2, which is assumed to be an artefact. CCDC-775998.
5 Computations
Computations were carried out using the Gaussian03 suite of programs [32]. The structures of the parent molecules were optimized at the B3LYP/aug-cc-pVTZ level, and for the other compounds the B3LYP/cc-pVDZ level was used. For molybdenum, cc-pVDZ level was used with relativistic pseudopotentials (abbreviated as cc-pVDZ-PP) [33] from the EMSL Basis Set Exchange site. At the stationary points, second derivatives were calculated to ensure that real minima were obtained. To proof the reliability of the B3LYP calculation, all the compounds have been tested with the BP86 functional, and no systematic difference have been observed between the two functionals. For calculation of NPA charges, the NBO 5.0 program [34] was used. Similarly to previous practice, the NICS values were calculated at the B3LYP/6-311+G**//B3LYP/6-311 + G** level. For visualization of the molecules, the MOLDEN program [35] was used.
6 Note added in proof
While the present work was in the proof phase a related paper on iron complexed triphosphabenzene has appeared [36].
Acknowledgements
We thank the EPSRC for the award of a postdoctoral fellowship for CWT.