

Abbreviations
SSNMR
solid-state NMR
Crhcatabolite repression histidine-containing phosphocarrier protein
1 Introduction
Solid-state NMR (SSNMR) is a spectroscopic method to study insoluble proteins at atomic level. Structural studies of a large number of proteins, such as membrane proteins (which represent about 30% of the human proteome), fibrillar proteins (involved in many neurodegenerative diseases) and molecular nanomachines remain challenging due to the inherent insolubility and the large size of these proteins, leading to a poor representation of structures from these proteins in the Protein Data Bank (PDB) [1]. Indeed, it is often extremely difficult to grow single crystals for X-ray diffraction studies from these proteins, and their size makes studies by solution NMR spectroscopy difficult. Solid-state NMR is not limited by size or by the absence of long-range order, and is thus a promising alternative for the three-dimensional (3D) structure determination of fibrils and membrane proteins at atomic resolution.
In the last years, advances in sample preparation of solid proteins, methodological and instrumental developments have allowed structural studies of selectively and uniformly labeled proteins, leading to the sequential assignment of several microcrystalline proteins [2–8], as well as the fungal HET-s prion domain [9], the membrane protein DsbB [10] and a seven-helical membrane protein [11]. Sequential assignments open the way, in analogy to solution NMR, for 3D structure determination, as they form the basis for the measurement of distance restraints between spins. We discuss here the latest developments in SSNMR experiments used for 3D structure determination and the structural data that can be extracted from these experiments. We concentrate on the use of uniformly labeled protein samples and broad-band recoupling experiments, as these two approaches yield the most information for a minimum of samples and experiments. Selective recoupling, as well as selective labeling approaches, are however highly useful and certainly complementary to the here-described experiments. In addition to describing the experiments, we demonstrate the use of the automatic structure calculation protocol ARIA to handle the highly ambiguous NMR data.
2 SSNMR experiments for distance restraint measurements in uniformly labeled protein samples
Structural studies of proteins by solid-state NMR are usually made at high-resolution conditions under magic-angle spinning (MAS) [12]. MAS averages the chemical shift anisotropy and weak dipolar couplings, thus allowing, in combination with heteronuclear decoupling, the observation of narrow lines for each 13C and 15N spin site (typically 0.3–0.6 ppm for carbon-13 line-width in microcrystalline proteins). By averaging the dipolar couplings between heteronuclear spins, MAS suppresses the distance information encoded in the dipolar couplings, and requires its reintroduction through dipolar recoupling experiments (for a review see ref [13]). In uniformly 13C/15N labeled protein samples, homonuclear (13C13C, 15N15N) and heteronuclear (13C15N) distance measurements provide valuable structural information to calculate the 3D structure of proteins, especially long-range contacts, which correspond to through-space contacts between spins separated by at least five residues. Distance measurements in uniformly labeled protein samples are complicated [14] by a phenomenon referred to as dipolar truncation [15–17], which leads to poor transfer over weak couplings in the presence of strong ones. As a result, the spectra are dominated by correlations between covalently bonded atoms, as well as the relayed two- and three-bond transfer, and the observation of contacts between non-consecutive residues is difficult. During the past decade, different SSNMR experiments, which we will discuss in the following, have been proposed which are less sensible to dipolar truncation.
2.1 Proton-driven spin diffusion
Firstly, proton-driven spin diffusion (PDSD) [18] relies on a coherent millisecond to second time scale spin-diffusion process, which promotes 13C13C polarization transfer. Adding a simultaneous 1H1H irradiation (using a continuous-wave radio-frequency field matching the MAS frequency), as proposed in the Dipolar Assisted Rotational Recoupling (DARR) [19,20] and Radio-frequency Assisted Diffusion (RAD) [21] experiments, assists the spin-diffusion process and allows the observation of long-range contacts in a few hundreds of milliseconds mixing time [22,23]. In these experiments, the intra-residual and sequential correlations coming from one-bond and relayed one-bond transfers crowd the PDSD and DARR spectra. Nevertheless, it has been recently shown theoretically [24] and experimentally [25] that spin-diffusion based experiments are less sensitive to dipolar truncation than first order recoupling experiment [26]. Thus, the structure of Ubiquitin was resolved using DARR experiments recorded on the uniformly labeled protein at long mixing times (100, 250 and 400 ms) [25], and a large set of distance restraints was extracted from a 100 ms mixing time DARR experiment recorded on uniformly labeled ZnMMP-12 [27]. To illustrate the possibility to obtain long-range restraints on an uniformly labeled protein using DARR, we performed this experiment on a fully 13C/15N labeled Crh sample and assigned inter-residue 13C13C contacts [28]. Fig. 1a shows an extract of a 200 ms mixing time DARR experiment recorded on Crh with inter-residue correlation peaks assigned, based on the X-ray structure [29]. A total number of 196 isolated carbon–carbon contacts (including 88 long-range restraints) can be assigned in one DARR experiment, and these distance restraints were sufficient to resolve the structure of the Crh monomer without angular restraints at a good accuracy, as shown in the Fig. 1b.
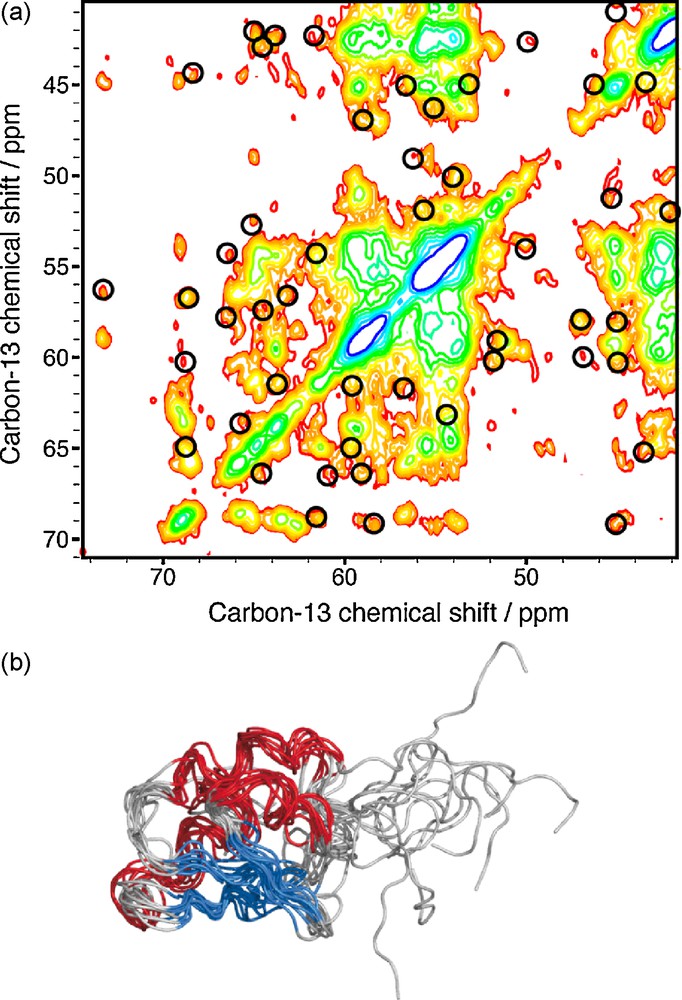
a: extract from the 13C13C DARR spectrum recorded on uniformly 13C/15N labeled Crh with a mixing period of 200ms. Circles indicate signals which can be assigned unambiguously, based on the X-ray structure; b: 10 lowest energetic structures of the Crh protein calculated with 13C13C distance restraints extracted from the DARR spectrum with a mixing period of 200 ms.
2.2 Third spin assisted recoupling
A new type of recoupling scheme named Third Spin Assisted Recoupling (TSAR) [30,31] was recently proposed. Its homonuclear version, the PAR experiment (PAR for Proton Assisted Recoupling), is a second order recoupling experiment and relies on the use of the intrinsic properties of the 1H spin (large magnetogyric ratio, high abundance) as an assisting spin to accelerate the polarization transfer between rare spins (here 13C13C31 or 15N15N [32]). In TSAR, the transfer is driven by a cross-term involving the two heteronuclear couplings in the 13C[1H]13C (or 15N[1H]15N) polarization transfer. We have demonstrated, in collaboration with R.G. Griffin, that the TSAR mechanism can efficiently provide long-range structural distance restraints [31] for uniformly labeled proteins, using the Crh protein as an example. One hundred and sixty-three isolated inter-residues distance restraints could be extracted from a single 2D 13C13C PAR spectrum (including 92 long-range contacts) and assigned using the X-ray structure; these restraints were sufficient to generate a structure of the Crh monomer with a precision for backbone atoms of 2.06 Å ± 0.3 Å.
2.3 Proton detected, rare spin correlation experiments
The experiments described above rely on the detection of distances between rare spins (13C13C, 15N15N and 15N13C), because the most abundant and structurally valuable spin, the 1H spin, is difficult to resolve in fully protonated large size molecules such as proteins, due to the important line broadening. The CHHC, NHHN and NHHC experiments [33–35] allow the indirect detection of proton–proton contacts on heteronuclear spins like 13C or 15N. In contrast to the TSAR mechanism, here the polarization is transferred from the heteronucleus to the proton via a cross-polarization (CP) with a short contact time. To allow the polarization transfer between protons, a mixing time corresponding to the initial rate regime (about 50–200 μs) for polarization transfer in a dense proton network is enclosed between the two CP steps. It is well established in solution NMR that proton contacts extracted from NOESY experiments can be used to calculate to high accuracy the 3D fold of proteins [36]. In collaboration with the group of M. Baldus, we have investigated the potential use of CHHC and NHHC experiments using the uniformly 13C/15N labeled Crh protein as a model [28,37]. Figs. 2a and b show the CHHC and NHHC spectra (respectively 200 and 100 μs of mixing time) recorded on the Crh protein. The spectral crowding is highly reduced compared to the DARR spectrum presented in Fig. 1a. In order to investigate the ability of the CHHC and NHHC experiments to provide accurate 1H1H distances, a careful analysis of the build-up curves obtained on the Crh protein was realized [37]. Figs. 2c–e depict the potential use of different mixing times to establish distance classes for these experiments. The sequential contact HaHN is characteristic of the different secondary structure elements: in an α-helical conformation, this sequential distance is about 3.4 Å, compared to 2.2 Å in β-sheet conformation. The polarization build-up curves presented in Fig. 2c allow the distinction between short and long distances, and therefore the distinction between the two conformations, shown in Fig. 2d. The use of short mixing times to reduce the distance range is possible, as demonstrated here with sequential HaHN, for contacts involving carbon and nitrogen spins with only one proton covalently attached. However, for CH2 and CH3 groups, the possible multiple relayed 1H1H transfers make it difficult to establish distance classes [37]. Fig. 2e shows the build-up curves for several long-range contacts involving CH2 and CH3 groups, where the correlation between internuclear distance and mixing time is less clear.
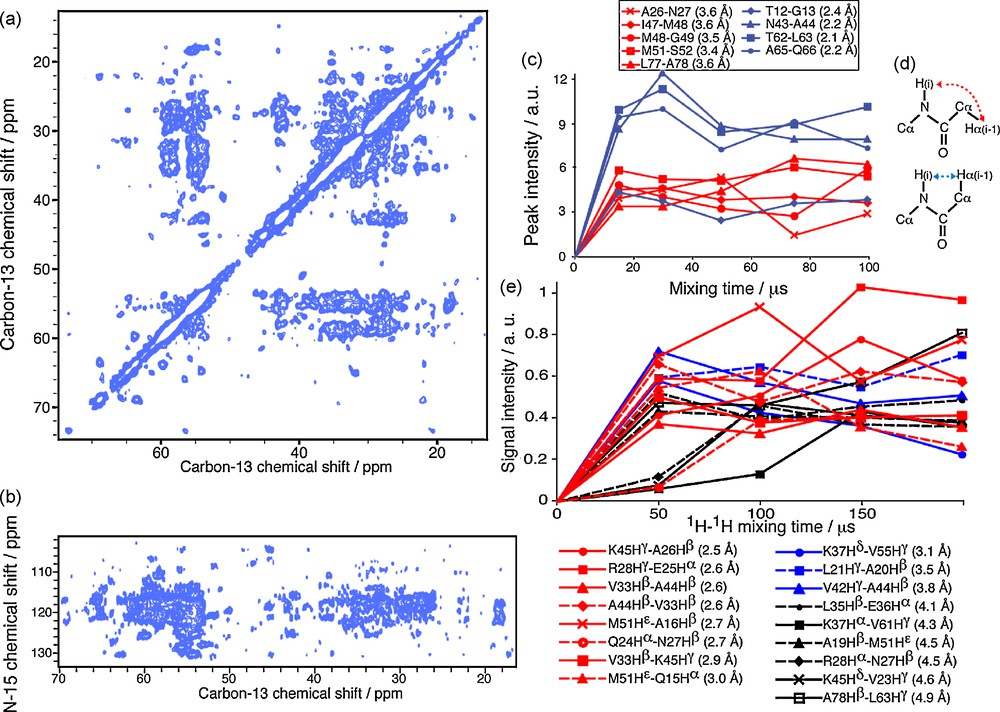
a: CHHC spectrum recorded on the uniformly 13C/15N labeled Crh protein with a mixing period of 200 μs; b: NHHC spectrum recorded on uniformly 13C/15N labeled Crh protein with a mixing period of 100 μs; c: polarization build-up curves recorded on the uniformly 13C/15N labeled Crh protein using NHHC experiments with different mixing times, for sequential HaHN spins; d: sketches of alpha-helical and beta-sheet conformations; e: polarization build-up curves for long-range CHHC contacts.
Despite the poor correlation between peak intensities and internuclear distances, the CHHC and NHHC experiments were successfully used to obtain structural restraints of several uniformly labeled proteins, including Kaliotoxin [38], Crh [28], HET-s [39], ZnMMP-12 [27] and the protofilament of β2-microglobulin [40].
One advantage of the CHHC/NHHC experiments is that, similarly to the TSAR-based methods, they are less crowded with intraresidue and sequential contacts than the DARR spectra, and they are also applicable at high-spinning frequencies. As the CHHC/NHHC pulse sequences are based on three CP steps, they however show, compared to PDSD/DARR and TSAR methods, a lower sensitivity. In addition, the three-fold rotation of the methyl group reduces the corresponding proton-carbon dipolar coupling, and the contacts involving methyl protons are weak or absent in CHHC/NHHC experiments [41,42]. To increase the signal of methyl contacts in fully labeled protein samples, we have recently introduced a complementary experiment in collaboration with the group of A. Lesage, named J-CHHC [43], which relies on the use of through-bond transfer steps as an alternative to the dipolar cross-polarization. The polarization transfer between protons and carbons is achieved by two refocused INEPT blocks [44] under proton homonuclear decoupling, and only the JCH-couplings remain active and affect the coherence transfer. As a result, the polarization transfer between methyl carbons and methyl protons is enhanced in the J-CHHC compared to classic CHHC experiment. Moreover, the J-CHHC experiment allows a gain in selectivity due to the INEPT-based transfer, as only the transfer between chemically bonded CH occurs. However, the gain in selectivity and sensitivity for the methyl group compromises the overall sensitivity, and few signals are observed for CH or CH2 groups. We have applied the J-CHHC to the uniformly labeled Crh protein [43] to determine a set of long-range restraints involving methyl groups (shown in Fig. 3a). These additional distance restraints yield a gain in precision (1.5 to 1.3 Å) and accuracy (1.8 to 1.1 Å) for the structure calculated with CHHC and J-CHHC data (Fig. 3b in green) compared to the structures calculated only with CHHC data (Fig. 3.b in black).
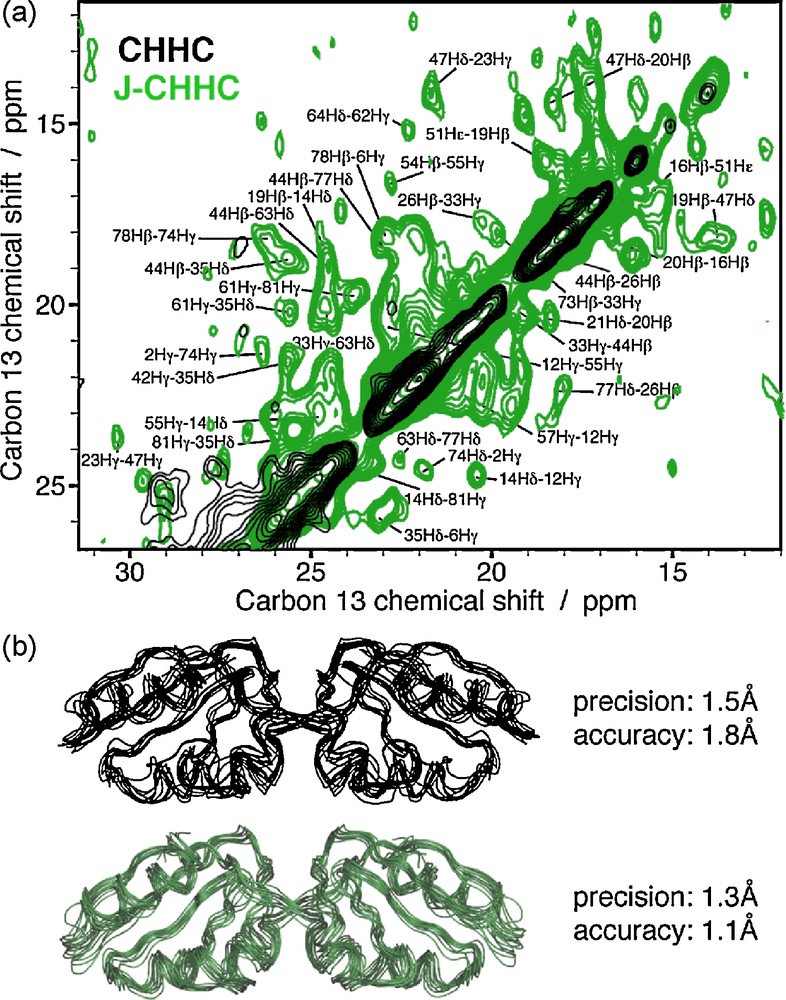
a: Comparison of the CHHC (in black) and J-CHHC (in green) spectra on uniformly labeled Crh protein; b: structure of the dimeric Crh protein calculated with CHHC data (in black) and CHHC and J-CHHC data (in green).
An analysis of the structural information potentially encoded in solid-state NMR PDSD/DARR, CHHC and PAR experiments is shown in Fig. 4a for the Crh protein. The distribution of 13C13C and 1H1H contacts as a function of the inter-nuclear distance is shown in the side panels, and the central box shows for each 13C13C contact the respective 1H1H distances of the attached protons. We can clearly see that the 13C13C distribution is dominated for short distances by the one-bond and two-bond connectivities. Below 3.5 Å, more than 90% of the possible 13C13C contacts are intra-residue, less informative to fold the protein compared to long-range contacts. For CHHC-type experiments, 25% of the 1H1H contacts below 3.5 Å are long-range (Fig. 4b), while for 13C13C distances, only a few long-range contacts can be found in this distance range. This is illustrated in the central box, in which at the distances corresponding to one- and two-bond correlations, no long-range correlations can be found for carbon-13. In contrast, numerous cross-signals can be found between proton spins. Thus, in proton correlation experiments, many valuable restraints are on roughly the same length scale as intra-residue restraints. These can then be measured in the initial rate regime. For carbon distances, the interesting long range peaks can only be measured at longer mixing times, where the initial rate for the short one and two-bond distances is already surpassed. Multi-spin effects and relay then become much more important and the measurement is accordingly less precise.
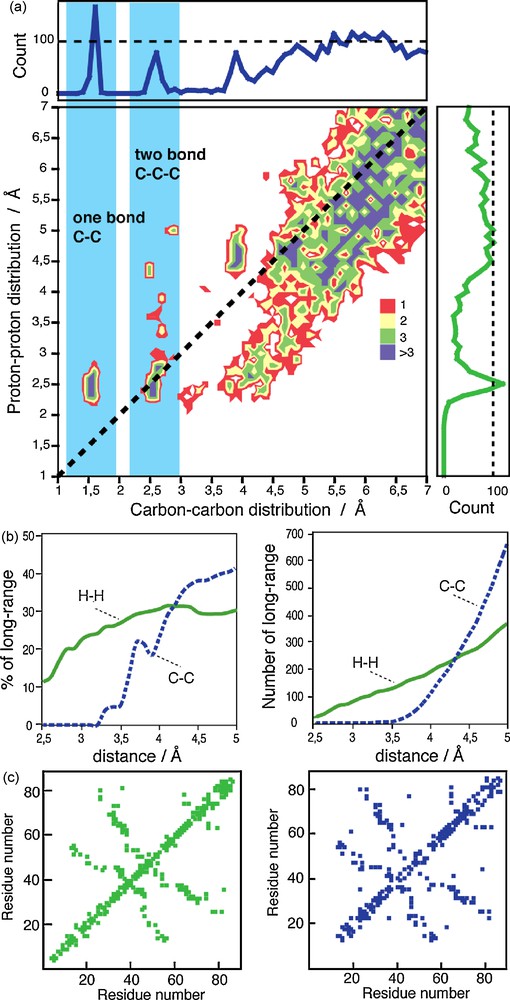
a: Distribution of the 13C13C (top box) and 1H1H (right box) distances. The central box depicts the distribution of 13C13C and 1H1H distances. For each 1H1H contact, the corresponding distance of the covalently attached carbons is plotted; b: proportion and number of long-range 13C13C and 1H1H distances as a function of the inter-nuclear distance; c: residue plot from isolated distance restraints unambiguously assigned in CHHC and DARR spectra on uniformly labeled Crh protein.
At longer distances, the proportion of long-range contacts becomes similar for 13C13C and 1H1H, as shown in Fig. 4b. Above about 4.3 Å (Fig. 4b left), the number of detectable 13C13C long-range contacts is higher than the 1H1H contacts, but the spectral crowding in 2D 13C13C experiments makes the peak analysis more difficult. Fig. 4c shows as an example the residue plots obtained from CHHC (in green) and DARR (in blue) experiments [28], where the long-range contacts allow a complete connection of the secondary structure elements.
3 Structure determination using ambiguous solid-state NMR restraints
During the 3D structure determination process, NMR relies on the assignment of cross-peaks encoding distance restraints. In solid-state NMR, the large line-width of 13C and 15N resonances often only leads to a very small amount of unambiguous distance restraints, depending on the observed line-width and the number of spins. For the uniformly labeled Crh protein, using a chemical shift tolerance window of ± 0.25 ppm, less than 12 distance restraints can be unambiguously assigned on a 2D-CHHC spectrum [28], compromising a 3D structure determination only based on unambiguous data. In uniformly labeled Ubiquitin, Manolikas et al. [25] were able to unambiguously assign 35 13C13C distances restraints using two PDSD spectra, leading to a low-resolution structure of the protein if combining these unambiguous data with TALOS restraints. Forty medium-range and 45 long-range distances could be unambiguously assigned in uniformly labeled GB1 [45]. Considering that PDSD, PAR or CHHC contain several hundreds of cross-peaks, only a small fraction of structural information could be exploitable if we just considered unambiguous data. For the Crh protein, taking a chemical shift window of 0.25 ppm, an average number of 16.3 assignments per cross peak are possible. In addition, as Crh is a dimer, each cross peak can encode an intramolecular or an intermolecular contact, which means that actually 16.3 × 2 assignments per cross-peak are possible. A method to handle this high-level of ambiguity of the data is therefore required. In solution NMR, several routines for NOE assignments have been designed, including ARIA [46–48], PASD [49], CANDID [50], AUTOSTRUCTURE [51], CYANA [52], KNOWNOE [53] or NOAH [54]. For each cross-peak, all possible assignments are encoded in an effective distance D, defined by D = [N∑(dintra−6)]−1/6. As already discussed above, the use of cross-peak volume or intensity remains difficult and thus we decided at this point not to use any distance calibration. Using these ambiguous distance restraints (ADRs) [55] as implemented in ARIA, a structure bundle is calculated, and, in an iterative manner, the less consistent assignments are discarded to define a new list of ADRs encoding less ambiguities. This iteration is repeated several times (typically 6–8) to obtain a majority of ADRs with only one possible assignment, leading to a list of unambiguous distance restraints. In collaboration with the group of M. Nilges, we have recently applied this concept of ADRs to determine the structure of the Crh protein from highly ambiguous SSNMR data [28]. We have applied the protocol to a cross-peak list obtained from CHHC and NHHC data recorded on the Crh protein. Cross-peaks were picked using the SPARKY program. Whereas many isolated signals could be picked automatically, predictions based on the X-ray structure were used to position the peaks in blobs. Only signals consistent with the crystal structure were picked to evaluate in a first time whether the program can deal with these highly ambiguous data when starting from a correct peak list. About 1000 ADRs were generated with an average value of 32.6 ambiguities. After two runs of ARIA calculations (15 iterations), a number of 643 1H1H distances restraints were assigned as unambiguous. In the second run, ARIA was used to assign both intra- and inter-monomeric restraints. Fig. 5a shows the evolution of ADRs during the ARIA calculations; the number of ambiguities decreases, and an average number of 2.5 possibilities accounts for the ambiguous ADRs at the last iteration. About 600 ADRs become unambiguous in the last iteration (including 115 long-range contacts), and these distance restraints were sufficient to obtain a bundle of high-precision (1.33 Å) and high-accuracy (2.89 Å on the dimer, 1.62 Å on the monomer) SSNMR structures (Fig. 5b). For Crh (2 × 85 residues), and also for Ubiquitin (76 residues) determined [25] using ATNOS/CANDID [50], 2D SSNMR data were used. For larger systems, as the degree of ambiguity will become a more serious obstacle, additional dimensions can be added, as already proposed in the 3D-NHHCC experiment on Ubiquitin [35] or in the 4D-CANCOCX experiment on GB1 [56]. This has been shown to reduce the number of ambiguities by a factor of 2–10, depending the protein size and the spectral resolution.
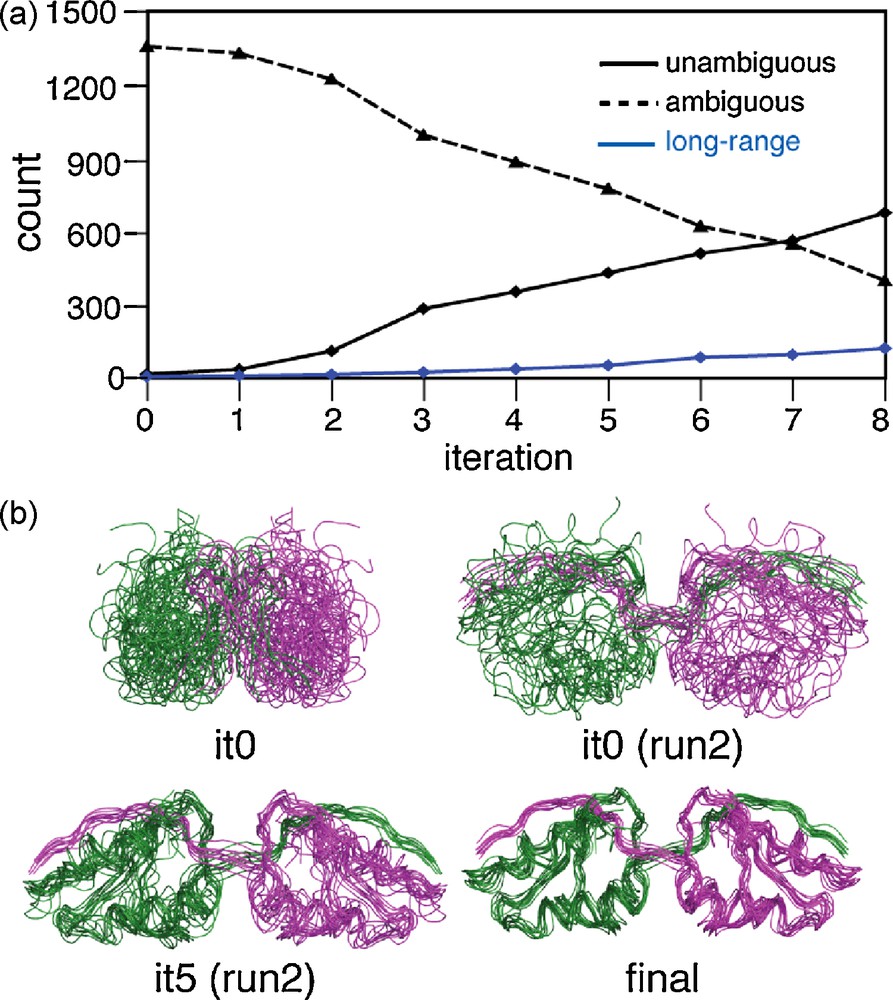
a: The number of ambiguous, unambiguous and long-range unambiguous distance restraints assigned during the ARIA calculation process; b: SSNMR structures calculated after different ARIA iterations.
4 Conclusion
Solid-state NMR spectroscopy has made important progress in the field of 3D structure determination over the last years, and currently several different proteins have been resolved at high-resolution by solid-state NMR data, including five globular proteins (SH3, GB1, Ubiquitin, Kaliotoxin and Crh) [22,25,28,38,45,57–60] and four small fibrillar proteins [39,40,61,62]. Several SSNMR experiments, particularly PDSD and CHHC, but also approaches using selective recoupling, extensive labeling schemes and deuteration, have been successfully applied to obtain high numbers of distance restraints in protein samples. The high level of ambiguities in the distance restraint assignment, as well as peak picking and peak evaluation is still a problem, but the evidence that structure calculation protocols as proposed in ARIA can handle highly ambiguous SSNMR data, at least if consistent sets of peaks are entered, presents an important step towards standard procedures. Further developments should include advanced peak-picking routines including evaluation procedures, allowing better signal identification mainly in crowded regions, as implemented for example in ATNOS/CANDID routines [50]. As the high level of ambiguity can easily introduce inconsistent distance restraints assignment, the development of structure validation tools allowing to detect incorrect assignments should help to improve the structure calculation convergence to the right fold.
The here reviewed methods all relied on broadband recoupling techniques, combined with fully labeled samples. Their combination with selective recoupling and labeling schemes (see for example ref. [63], and also the inclusion of chemical shift [64–66] and dihedral angle information [45] opens perspectives for the study of larger systems. Increase in sensitivity coming from higher fields, optimized transfers or sample preparation should allow one to progress to higher dimensional experiments [35,58,67].
Acknowledgements
The authors would like to thank their colleagues and collaborators for their substantial contributions to the work described in this article, for the generous efforts and ideas, and for the many fruitful discussions. Funding by the ANR (JC05_44957, PCV 07 PROTEIN MOTION) is acknowledged.