

1 Introduction
Phosphole, the phosphorus analog of pyrrole, possesses a low-lying LUMO due to effective σ*–π* orbital interaction between the phosphorus center and the polarizable 1,3-diene unit. In addition, the LUMO and HOMO energy levels of phosphole can be controlled by chemical functionalizations at the phosphorus center. These characteristic properties of phosphole are beneficial for the construction of unprecedented classes of optical and electronic materials [1]. In fact, phosphole-linked π-conjugated systems of general formula A have recently received increasing attention, and their fundamental properties have proven to depend strongly on the π-conjugative substituents. Most of the preceding examples of type A compounds include benzene (phenyl or phenylene) [2], pyridine (2-pyridyl) [3], and thiophene (2-thienyl, 3-thienyl, etc) [4] moieties as the α-substituents, where synergetic effects derived from each component have been exhibited in the optical and electrochemical properties of the entire π-systems. It is well known that HOMO of pyrrole is located at a higher energy level than HOMOs of benzene, thiophene, and pyridine.1 In this regard, pyrrole–phosphole–pyrrole π-systems are expected to release electrons easily as compared with known arene–phosphole–arene hybrids of type A. Such properties would be promising to construct a new class of redox active hybrid heterole π-systems. It is also anticipated that pyrrolic NH groups may act as hydrogen-bond donors toward Lewis basic functions attached to the phosphorus center. To our knowledge, however, the utilization of pyrrole as the α-substituent has gained quite a little attention in phosphole chemistry [5].2
In 2006, we established a convenient method for the synthesis of α,α′-difunctionalized phospholes via titanacycles generated from Sato-Urabe's TiII reagent [6] and the corresponding terminal-substituted 1,6-heptadiynes [7,8].3 This protocol has proven to be effective for the construction of arene-capped phosphole–vinylene [9], phosphole–acetylene [10], and phosphole–triazole [11] π-systems with characteristic properties derived from the respective components. Herein, we report a new short-step synthesis of 2,5-bis(pyrrol-2-yl)phosphole derivatives based on the titanacycle protocol and their optical, electrochemical, and coordinating properties. In this study, we aimed to elucidate the characteristic features of the pyrrole substituents by comparison with those of the other arene substituents in type A phosphole derivatives. Both experimental and theoretical results demonstrate that the attachment of the pyrrole substituents is highly promising for developing a new class of π-conjugated phosphole derivatives with multiple functions derived from these two group 15 heteroles.
2 Results and discussion
2.1 Synthesis of 2,5-bis(pyrrol-2-yl)phosphole derivatives
Scheme 1 illustrates the synthesis of 2,5-bis(pyrrol-2-yl)phosphole derivatives and 2,5-bis(5-phenylpyrrol-2-yl)phosphole derivatives. The starting materials, pyrrolyl-capped 1,6-heptadiynes 2a,b, were prepared by Sonogashira coupling of terminal-free 1,6-heptadiyne and the corresponding 1-Boc-2-iodopyrroles (Boc = t-butoxycarbonyl) 1a,b [12,13]. Reactions of 2a and 2b with (η2-propene)Ti(Oi-Pr)2, generated in situ from Ti(Oi-Pr)4 and 2 equiv of i-PrMgCl [6], followed by treatment with PhPCl2 gave N-Boc-protected 2,5-bis(pyrrol-2-yl)phosphole 3a (43%) and 2,5-bis(5-phenylpyrrol-2-yl)phosphole 3b, respectively. Compound 3b could not be separated from unreacted 2b and was used as a mixture in the following N-Boc deprotection step. The N-Boc groups of 3a,b were successfully removed by the reaction with sodium methoxide in methanol to give σ3 derivatives 4a,b. The phenyl-capped derivative 4b was isolated as an air stable solid in 28% yield based on 2b, whereas the α-free derivative 4a was difficult to completely purify due to its instability in atmospheric environment. Therefore, 4a was used for subsequent transformations without isolation. On treatment with hydrogen peroxide or m-chloroperbenzoic acid (mCPBA), each σ3-phosphorus center of 4a,b was readily oxygenated, affording the corresponding P-oxides 5a,b. When treated with elemental sulfur, 4a,b were converted to P-sulfides 6a,b.
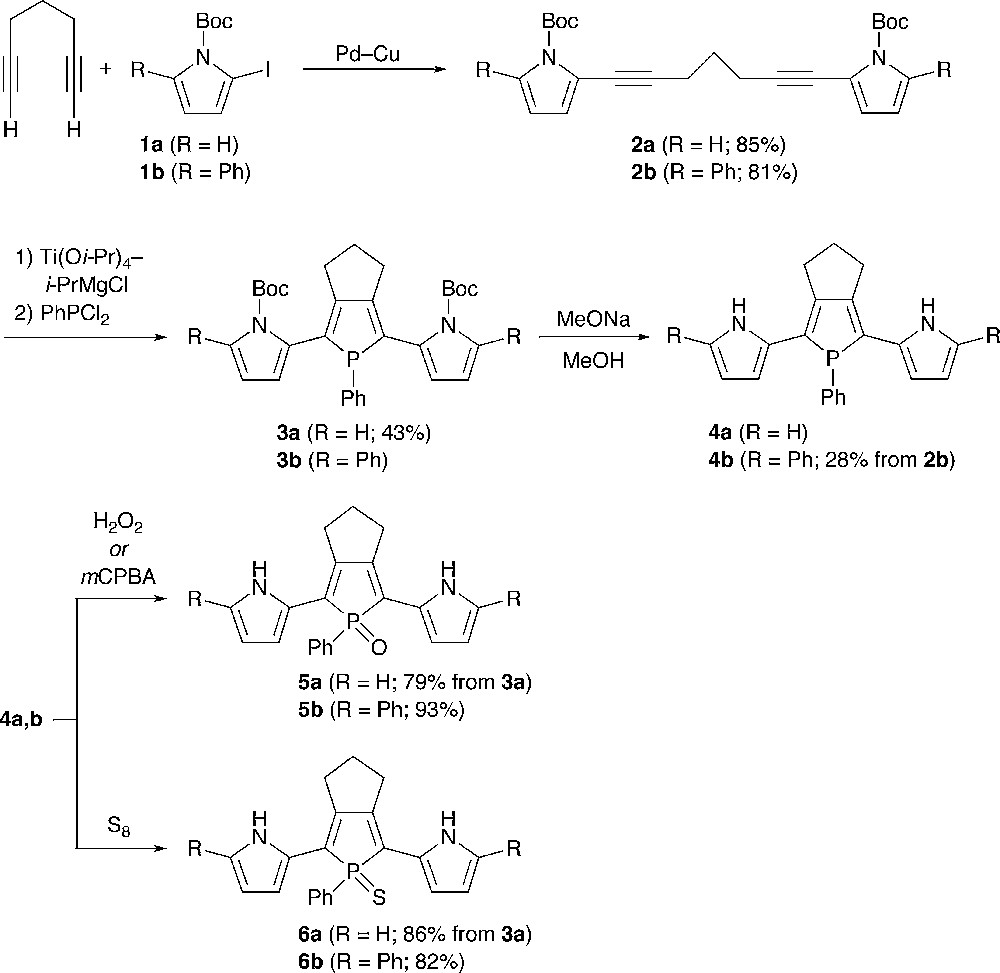
The pyrrole–phosphole–pyrrole hybrids 3–6 were characterized by conventional spectroscopic techniques (1H, 13C, 31P NMR, high-resolution MS, IR) and elemental analysis. Although 4a gradually decomposed in solution, its structure was confirmed by NMR spectroscopy and MS spectrometry. The 31P peaks of the σ4 derivatives 6a,b (δP 63.4, 63.6) and 5a,b (δP 54.3, 55.6) were observed at downfield compared to those of 4a,b (δP 28.1, 28.5), which clearly reflects differences in the electronic character and the oxidation state of phosphorus. In the 1H NMR spectra, 3–6 showed only one set of pyrrolic CH and NH protons, indicating that all of these compounds have Cs symmetry in solution. The NH protons of α,α′-free derivatives 4a, 5a, and 6a appeared at δ 8.01, 9.09, and 9.61, respectively. The same holds true for a series of the phenyl-capped derivatives 4b (δ 8.26), 5b (δ 9.28), and 6b (δ 9.96). Judging from the downfield appearance of the NH peaks of 5 and 6 relative to those of 4 and free pyrrole (δ 8.0), there may exist hydrogen-bonding interaction between the NH protons and the chalcogen atoms bound to phosphorus in 5 and 6. To get some insight into relative conformation between the pyrrole and phosphole rings in solution, we also measured NOESY spectra for 4b and 5b in CD2Cl2. In both cases, the NH proton and one of the β-CH protons of the pyrrole ring show nuclear Overhauser effects (NOE) toward allylic methylene protons of the trimethylene bridge fused to the phosphole ring. This result implies that the inter-ring Cα–Cα′ bonds between pyrrole and phosphole rings rotate rapidly in solution on the NMR timescale. In the IR spectra of 5a and 5b, PO stretching frequencies were observed at ν 1165 and 1180 cm−1, respectively. The νPO value of 5a is somewhat smaller than that (νPO = 1178 cm−1) of a similar trimethylene-fused 2,5-diphenylphosphole P–oxide [14].4
The crystal structure of 5b was elucidated by X-ray crystallography. The ORTEP diagram is shown in Fig. 1 with selected bond lengths and bond angles. The phosphorus center forms a tetrahedral geometry with average C–P–C and C–P–O angles of 101.9° and 116.4°, respectively. The C–C bond length alternation of the phosphole subunit differs considerably from that of two pyrrole subunits, which reflects the difference in aromaticity between these two kinds of heterole rings. The relatively narrow dihedral angles between the central phosphole and adjacent pyrrole rings (4.7–18.3°) indicate efficient π-conjugation through the pyrrole–phosphole–pyrrole linkage. As for the stereochemistry at the inter-ring bonds, one pyrrole ring adopts an S-trans (anti) conformation against the phosphole ring, whereas the other adopts an S-cis (syn) conformation. The N(2)–H group of the syn pyrrole ring is directed to the P–oxo group with distance of 3.20 Å, indicating that weak hydrogen-bonding interaction is present between the NH proton and the oxygen atom in 5b [15].5 The PO distance of 1.4871(11) Å is almost identical to that [1.489(1) Å] of Tanaka's 2,5-diphenyl-1-phenylphosphole P–oxide [16].
2.2 Optical and electrochemical properties of 2,5-bis(pyrrol-2-yl)phosphole derivatives
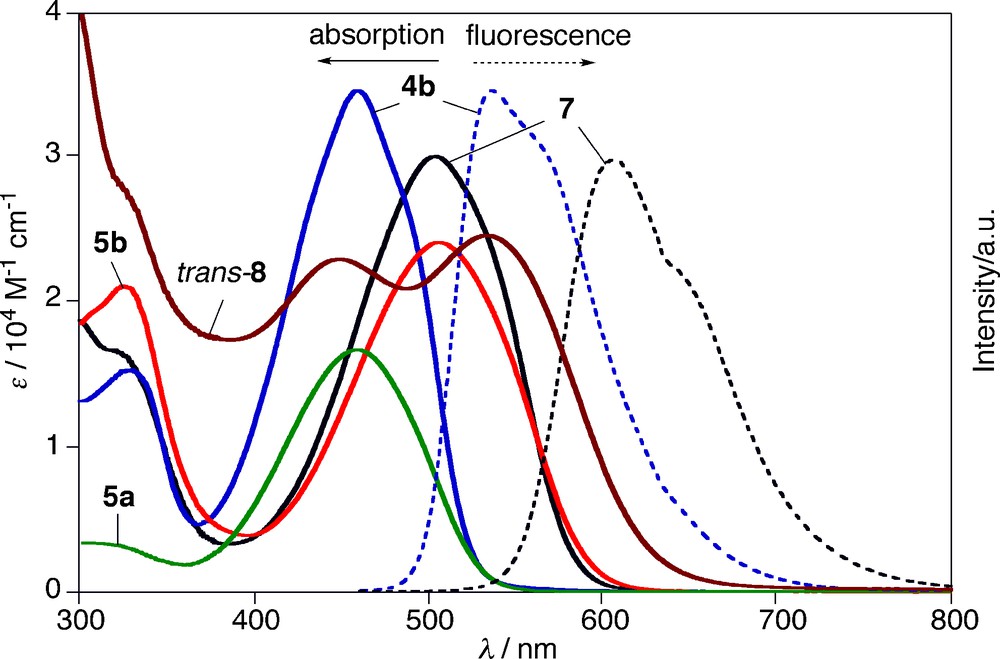
To investigate the optical and electrochemical properties of α,α′-free and α,α′-phenyl-capped pyrrole–phosphole–pyrrole π-systems, we measured UV-vis absorption and fluorescence spectra and redox potentials of 4–6 in CH2Cl2. The results are summarized in Table 1 and Fig. 2. In the absorption spectra, the α,α′-free derivatives 4a, 5a, and 6a display the lowest π–π* transitions at λab 416, 458, and 454 nm, respectively. As typically observed for phosphole derivatives, chemical functionalizations at the phosphorus center from σ3 to σ4 induce red-shifts of λab values (Δλab = 38–42 nm). The same trend was observed for the phenyl-capped series, 4b, 5b, and 6b (Δλab = 47–48 nm). The σ3 derivatives 4a,b are fluorescent, whereas the σ4 derivatives 5a,b and 6a,b are weakly fluorescent. Notably, 4b emits intense yellowish green fluorescence at λem 536 nm with fluorescence quantum yield (Φf) of 0.78. To our knowledge, this is the highest Φf value ever reported for type A 2,5-diarylphospholes.6 Nonradiative pathways from the S1 state of 4b may be suppressed and/or a radiative pathway may be accelerated by the effective π conjugation through the pyrrole–phosphole–pyrrole π-linkage.7 The absorption/emission maxima of 4a, 5a, and 6a are appreciably red-shifted compared to those of the previously reported 2,5-diarylphosphole analogs. In other words, the introduction of the pyrrolyl groups narrows HOMO–LUMO gaps of π-conjugated phospholes of type A. For instance, the optical HOMO–LUMO gaps of the respective σ3-P and σ4-P–oxo derivatives are 2.69 eV (Ar = 2-pyrrolyl) vs. 2.72–3.02 eV (2-thienyl, phenyl, 2-pyridyl, measured in THF) [4d] and 2.39 eV (Ar = 2-pyrrolyl) vs. 2.51–2.83 eV (2-thienyl, phenyl, measured in THF) [4b, 8b, 14].
Redox potentials of 4b, 5a,b, and 6a,b were measured by using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) with 0.1 M nBu4NPF6 as an electrolyte in CH2Cl2 (Table 1). Although reduction processes were not observed in the measurement window (−2.6 V to +1.0 V vs. Fc/Fc+), oxidation processes were observed for all compounds examined. The α,α′-free derivatives 5a and 6a showed irreversible voltammograms accompanied by deposition of insoluble substances, whereas the α,α′-phenyl-capped derivatives 4b, 5b, and 6b showed two reversible oxidation processes in CV measurements. In the cases of 5a and 6a, subsequent chemical reactions such as an electropolymerization may occur at the α-free pyrrole rings when electrochemically oxidized. The P-functionalizations (from 4b to 5b and 6b) make a definite impact on the first and second oxidation processes, both of which are shifted to lower potentials by 0.16–0.22 V. It is worthy to note that the electrochemical oxidations of 4, 5, and 6 occur at much lower potentials than those reported for related 2,5-diarylphospholes (aryl = 2-thienyl, phenyl, 2-pyridyl).8 For instance, the difference in the first oxidation potentials between 5a and 2,5-diphenylphosphole P–oxide [14] of the same trimethylene-fused type reaches 1 V. These experimental results demonstrate that the pyrrole components play a key role in fine-tuning of the redox properties, especially electrochemical oxidation properties, of type A phosphole derivatives.
To get more insight into electronic structures of the phosphole–pyrrole hybrid π-systems, we performed density functional theory (DFT) calculations on model compounds 4m and 5m in three forms, anti–anti, anti–syn, and syn–syn (Fig. 3). At the optimized structures, inter-ring torsion angles are small (10.1–21.7°) for all models. This indicates that the effective π-conjugation is attained over the three heterole rings. There is negligible difference in relative energies of the three conformers for 4m (ΔE < 0.3 kcal mol−1), implying that all forms are equally conceivable. In contrast, anti–syn and syn–syn conformers of 5m are more stabilized by 3.5 and 6.1 kcal mol−1, respectively, than anti–anti isomer. Considering the fact that the stabilization energy of the syn–syn conformer is about twice as that of the anti–syn one, the origin of this energetic stabilization is attributable mainly to the intramolecular hydrogen-bonding interaction. Indeed, the distance calculated for 5m (anti–syn) (3.067 Å) is within the range of this type of hydrogen bonds [15]. In each model, HOMO consists of conjugated polyene-derived π-networks, whereas LUMO possesses a significant character of central phosphole units. The P-oxygenation from 4m to 5m lowers LUMO and HOMO levels by 0.62–0.68 eV and 0.25–0.29 eV, respectively. Note that LUMOs are more stabilized than HOMOs for all conformers. This is a typical trend for phosphole derivatives and has been rationalized by considering effective σ*(P–X)–π* orbital interaction including the dienic π-systems as depicted in Fig. 3b. The LUMO levels of the P–oxo models 5m are more stabilized in the order anti–anti (−1.93 eV) > anti–syn (−2.00 eV) > syn–syn (−2.06 eV), which displays a key contribution of the hydrogen-bonds in the stabilization of LUMO. Time-dependent DFT (TD-DFT) calculation on 4m in the syn–syn form revealed that the lowest excitation band observed for the σ3-P derivatives 4 is based on a HOMO → LUMO π–π* transition.
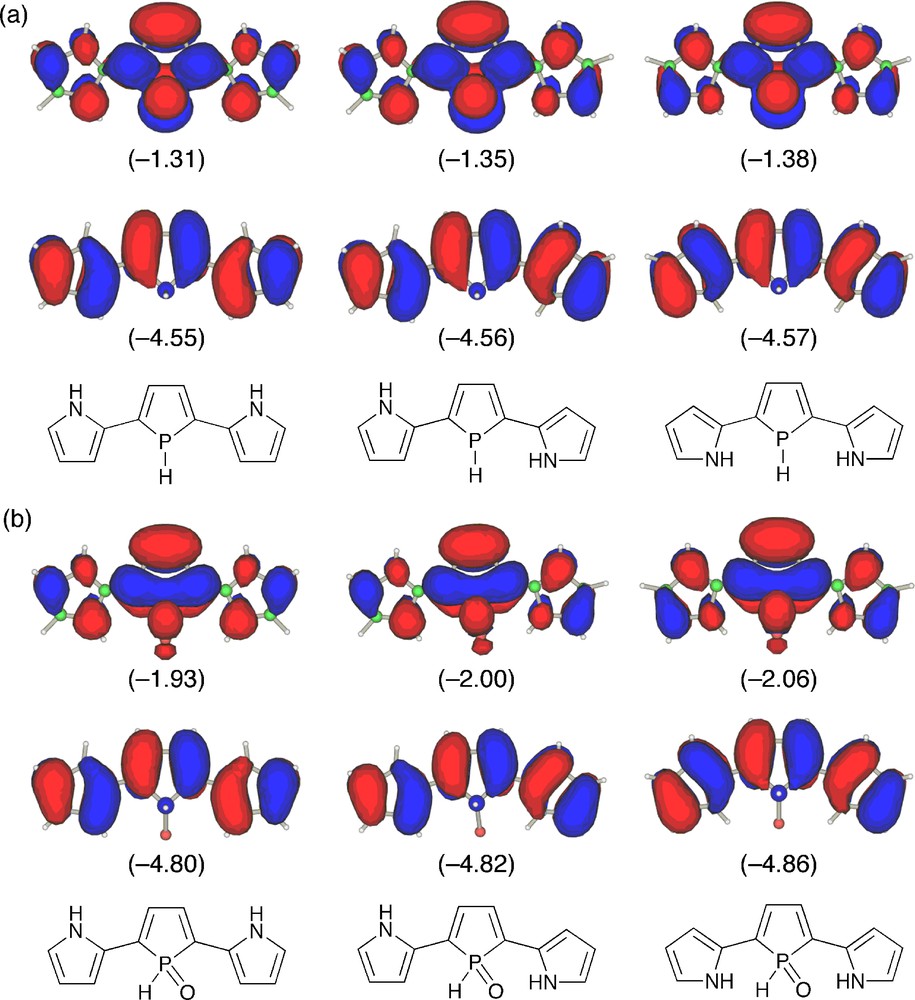
HOMO (lower) and LUMO (upper) of (a) 4m and (b) 5m: anti–anti (left), anti–syn (middle), syn–syn (right). The calculated orbital energies (in eV) are shown in parentheses.
2.3 Coordination behavior of 2,5-bis(5-phenylpyrrol-2-yl)phosphole
Recently, the structure–optical property relationship of metal-bridged phosphole-containing π-systems has received growing interest [17].9 To investigate the coordination behavior of 2,5-bis(pyrrol-2-yl)phosphole as a phosphine ligand, 4b was reacted with gold(I) and platinum(II) salts (Scheme 2). Treatment of 4b with one equiv of AuCl(SMe2) in CH2Cl2 at room temperature yielded AuCl–phosphine complex 7 in 80% yield. When treated with a half equiv of PtCl2 under the same conditions, PtCl2–bis(phosphine) complex 8 was formed as a trans/cis mixture (ca. 1:1). Metal complexes 7 and 8 were characterized by spectroscopy and X-ray crystallography. The 31P chemical shifts of 7, trans-8, and cis-8 are δP 51.4, δP 51.6 (JP–Pt = 2244 Hz), and δP 31.5 (JP–Pt = 3177 Hz), respectively. The downfield appearance of the NH protons of 8 (δ 9.73–9.79) relative to those of 7 (δ 8.48) suggests that the pyrrolic NH groups spatially interact with the chlorine atoms in 8 (vide infra). High-resolution mass spectra of 7 and trans-8 showed respective parent ion peaks (M+) at m/z 714.1267 (calcd. 714.1266) and m/z 1229.2854 (calcd. 1229.2849).
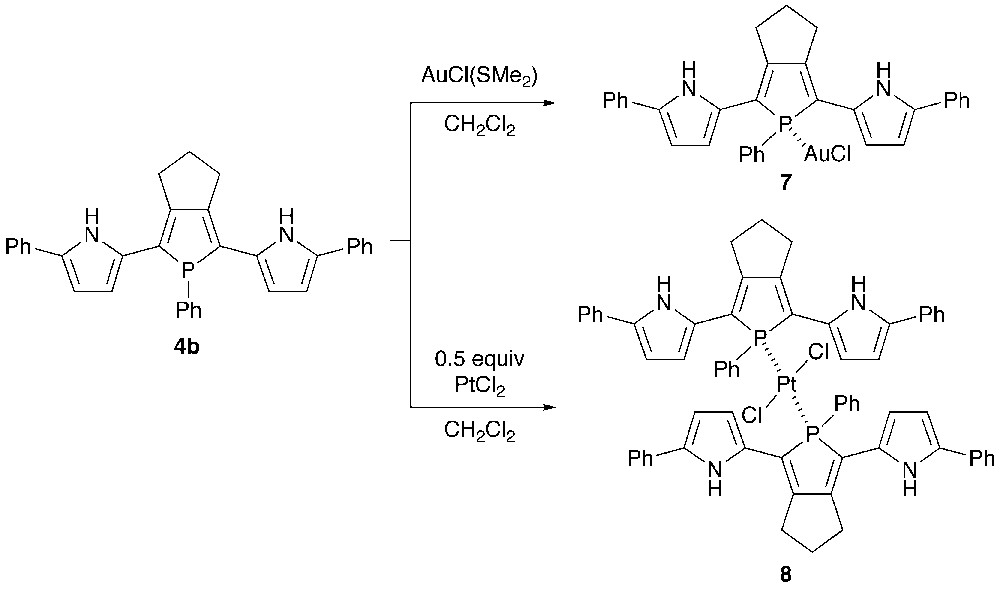
The crystal structures of 7 and trans-8 were successfully determined by X-ray crystallography. The ORTEP diagrams are shown in Figs. 4 and 5 with selected bond lengths and bond angles. In each complex, the phosphorus center forms a tetrahedral geometry with average C–P–C and C–P–M angles of 101.9–103.3° and 115.2–114.7°, respectively. The C–P–C bond angles of 7 and 8 are almost identical to those of 5b. In complex 7, the gold(I) center adopts a linear geometry with P–Au–Cl bond angle of 176.53(8)°, and both the pyrrole rings adopt anti–anti conformation against the phosphole ring with small dihedral angles between the two adjacent heterole rings (6.2–9.9°). Judging from the packing structure, neither intramolecular nor intermolecular interaction between the pyrrolic NH protons and the chlorine atoms is present in 7.
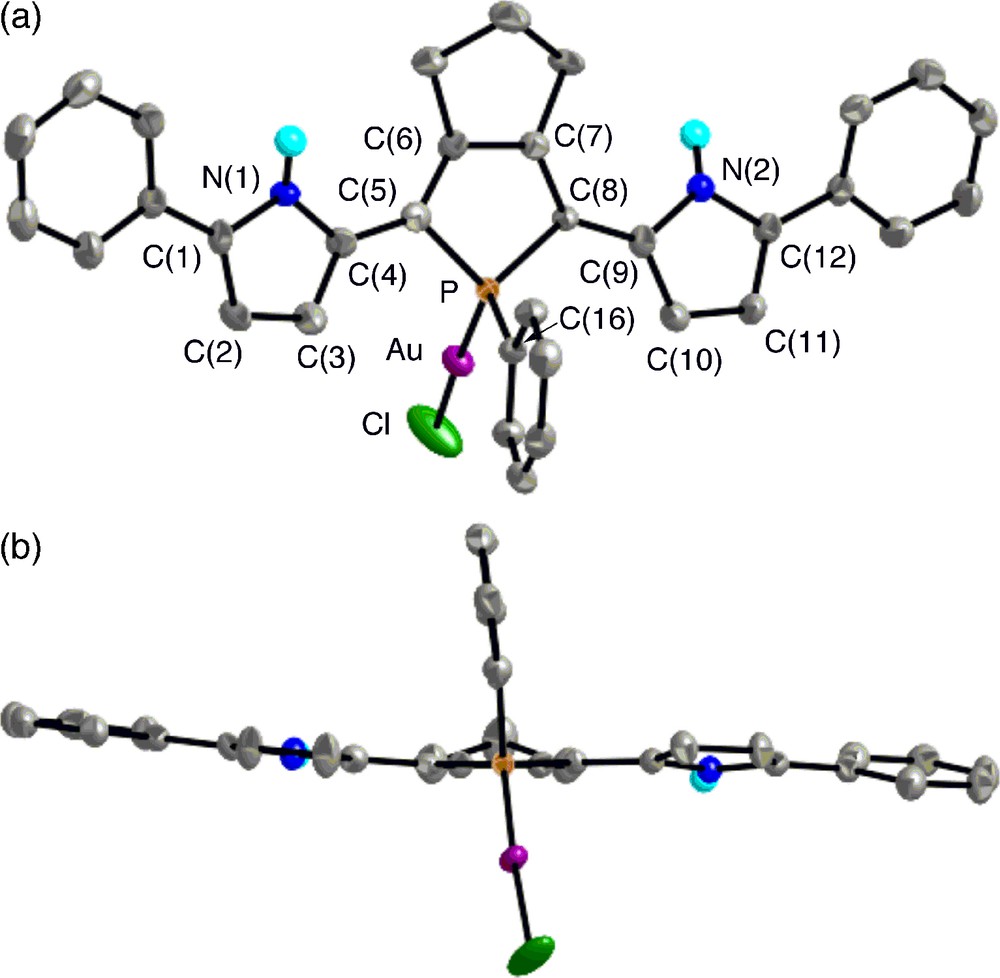
Top view (upper) and side view (lower) of 7 (30% probability ellipsoids). Hydrogen atoms except for NH are omitted for clarity. Selected bond lengths (Å) and angles (deg): C(1)–C(2), 1.383(10); C(2)–C(3), 1.403(10); C(3)–C(4), 1.377(10); C(4)–C(5), 1.439(9); C(5)–C(6), 1.365(9); C(6)–C(7), 1.458(10); C(7)–C(8), 1.351(10); C(8)–C(9), 1.426(10); C(9)–C(10), 1.380(10); C(10)–C(11), 1.394(11); C(11)–C(12), 1.371(10); P–C(5), 1.801(7); P–C(8), 1.820(7); P–C(16), 1.802(7); P–Au, 2.2302(18); Au–Cl, 2.276(2); C(5)–P–C(8), 94.6(3); C(5)–P–C(16), 109.1(3); C(8)–P–C(16), 105.6(3); C(5)–P–Au, 113.1(2); C(8)–P–Au, 119.1(2); C(16)–P–Au, 113.4(2); P–Au–Cl, 176.53(8).
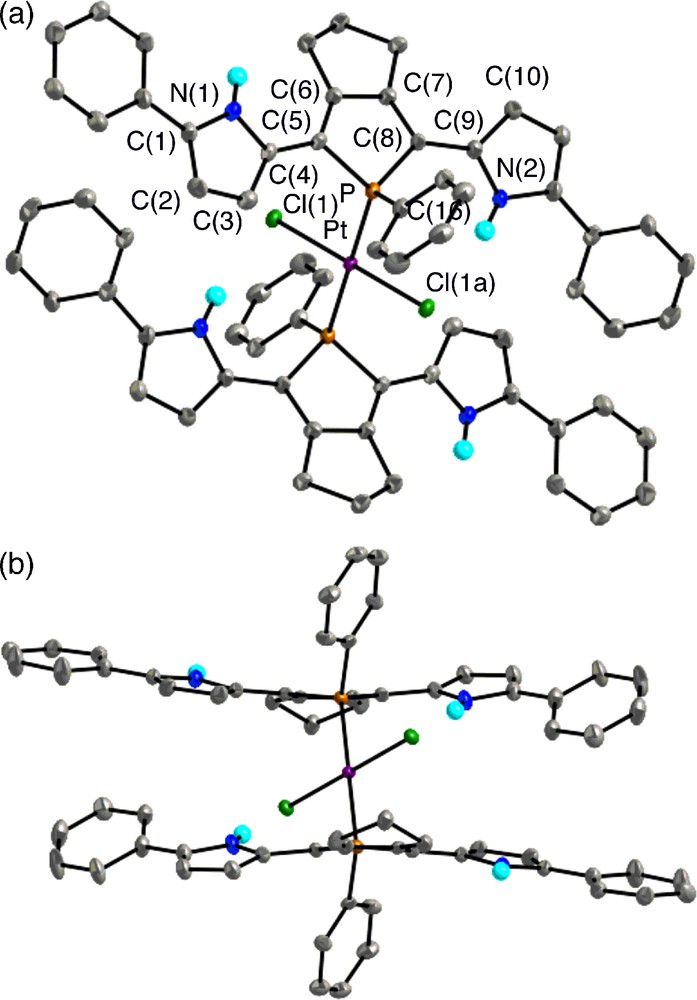
Top view (upper) and side view (lower) of 8 (30% probability ellipsoids). Hydrogen atoms except for NH are omitted for clarity. Selected bond lengths (Å) and angles (deg): C(1)–C(2), 1.368(8); C(2)–C(3), 1.406(8); C(3)–C(4), 1.389(8); C(4)–C(5), 1.442(7); C(5)–C(6), 1.360(7); C(6)–C(7), 1.442(7); C(7)–C(8), 1.357(7); C(8)–C(9), 1.454(7); C(9)–C(10), 1.387(8); C(10)–C(11), 1.417(8); C(11)–C(12), 1.374(9); P–C(5), 1.827(5); P–C(8), 1.814(5); P–C(16), 1.827(5); P–Pt, 2.3315(16); Pt–Cl, 2.3105(15); C(5)–P–C(8), 93.4(2); C(5)–P–C(16), 107.8(2); C(8)–P–C(16), 109.3(2); C(5)–P–Pt, 112.78(17); C(8)–P–Pt, 114.34(17); C(16)–P–Pt, 116.75(18); P–Pt–Cl(1), 88.68(5); P–Pt–Cl(1a), 91.32(5).
In trans-8, the platinum(II) atom is the center of Ci symmetry and forms a square planar geometry (ΣP–Pt–Cl = 180°). As shown in Fig. 5a, the Pt–P coordination bonds link two pyrrole–phosphole–pyrrole π-planes as slipped, parallel fashion, and each π system adopts anti–syn conformation with dihedral angles between the two adjacent heterole rings of 12.9–19.5°. Note that the syn pyrrole rings direct their NH groups toward the chlorine atoms bound to platinum with an intramolecular distance of 3.31 Å [18],10 indicating there exists cooperative hydrogen-bonding interaction between the NH protons and the chlorine atoms. Such a coordinating property cannot be produced by other conventional aryl substituents and shows the characteristic structural features of the pyrrole ligands. A relatively short distance (ca. 3.3 Å) between the center of anti-pyrrole ring and the nearest ortho-proton of the facing α-phenyl group implies that weak interaction is also present at least in the solid state.
2.4 Optical properties of metal complexes of 2,5-bis(1-phenylpyrrol-2-yl)phosphole
Finally, we measured absorption and fluorescence spectra of the metal complexes, 7 and trans-8.11 Fig. 2 clearly displays that these two complexes show different optical properties. The AuCl complexation (from 4b to 7) induces red-shifts of the absorption and fluorescence maxima (Δλab = 45 nm; Δλem = 70 nm) and decreases of Φf value (from 0.78 to 0.09) and fluorescence lifetime (from 3.50 ns to 1.54 ns). A small difference in the observed Stokes shift (Δλ) between 7 (Δλ = 3.34 × 103 cm−1) and 4b (Δλ = 3.13 × 103 cm−1) may suggest that the attachment of the AuCl moiety does not affect the intrinsic character of the HOMO → LUMO transition significantly. In sharp contrast, the PtCl2 complexation (from 4b to trans-8) varies optical properties of the free phosphine ligand dramatically. In the absorption spectrum of trans-8, two separated, broad bands were observed at λab 450 and 533 nm with similar intensity. The high-energy band is slightly blue-shifted (Δλab = −9 nm) relative to λab of 4b (459 nm), whereas the low-energy band is largely red-shifted (Δλab = 74 nm). It is likely that two slipped, parallel π-planes with Ci symmetry linked by the Pt–P and bonds play a crucial role in the unusual splitting observed for trans-8. No fluorescence was observed for trans-8.
To get a deep insight into the character of the observed absorption bands, we performed TD-DFT calculations on models of AuI and trans-PtII complexes (Fig. 6 and Table 2). The structures of 7m and 8m, where all the phenyl substituents were replaced by hydrogens, were optimized starting from the X-ray geometries observed for 7 and trans-8. As shown in Fig. 6a, HOMO and LUMO of 7m basically maintain the character of the respective orbitals of 4m (anti-anti) except for their relative energies; LUMO is more stabilized than HOMO by the AuCl complexation. The TD-DFT result of 7m (Table 2) implies that the intense absorption band observed for 7 is based on HOMO → LUMO excitation. Fig. 6b shows five frontier orbitals of 8m, HOMO − 1, HOMO, LUMO, LUMO + 1, and LUMO + 2, with their orbital energies. Obviously, HOMO and HOMO − 1, which are constructed by two original HOMOs of the free base, do not interact with each other. As a consequence, HOMO and HOMO − 1 have almost the same energies. On the other hand, LUMO and LUMO + 2 consist of the metal-derived d orbitals and the ligand-derived π*-orbitals. Importantly, the coordination to the platinum(II) center induces a significant split of the original LUMOs of the symmetrical π-conjugated ligands probably due to σ*(M–P)–π* orbital interaction. The TD-DFT results of 8m revealed that the two absorption bands observed for trans-8 are attributable mainly to HOMO → LUMO + 2 (2.95 eV) and HOMO − 1 → LUMO + 1 (2.64 eV) excitations. The finding that the lowest excitation state (HOMO → LUMO) is not the high-intensity transition for 8m (oscillator strength is less than 0.1) may also explain the nonfluorescent property of the Pt complex 8. These results show that the metal complexation is a promising approach to make a large impact on the optical properties of 2,5-bis(pyrrol-2-yl)phosphole derivatives.
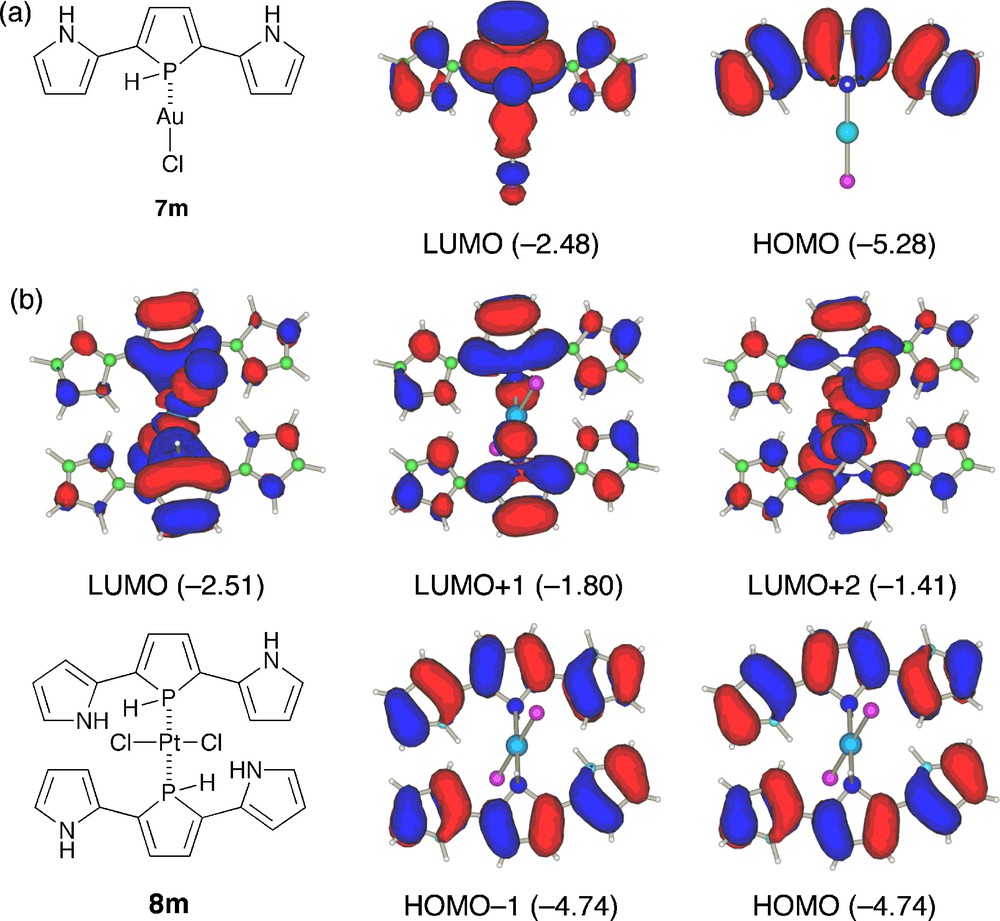
Frontier orbitals of 7m and 8m. The calculated orbital energies (in eV) are shown in parentheses.
Excitation energies and oscillator strengths of 4m (syn–syn), 7m, and 8m calculated by the TD-B3LYP methoda.
| State | Excitation energy | Oscillator strength | Excitation | Weight (%) | |
| (eV) | (nm) | ||||
| 4m (syn–syn) | |||||
| 11A″ | 2.95 | 420 | 0.5865 | HOMO → LUMO | 38 |
| 7m | |||||
| 11A″ | 2.56 | 484 | 0.4590 | HOMO → LUMO | 38 |
| 8m | |||||
| 21Au | 2.64 | 470 | 0.2346 | HOMO − 1 → LUMO + 1 | 39 |
| 31Au | 2.95 | 420 | 0.6290 | HOMO → LUMO + 2 | 38 |
a The states whose oscillator strengths are less than 0.2 are not included.
In summary, we established a convenient method for the synthesis of new π-conjugated phospholes bearing two pyrrole substituents, and revealed their optical, electrochemical, and coordinating properties. The flat π-planes, low excitation energies, and small oxidation potentials observed for the σ3-P and σ4-P derivatives represent the efficient π-conjugation through the pyrrole–phosphole–pyrrole linkage as well as electron excessive nature of the pyrrole subunits. It is also noteworthy that the gold(I) and platinum(II) complexes coordinated by the phenyl-capped σ3-P derivative exhibit quite different structural and optical properties. It is of particular interest that trans-PtII–bisphosphine complex contains slipped, parallel π-planes linked by the Pt–P bonds and the NH–Cl hydrogen bonds. This cooperative interaction gives rise to split π–π* transitions at the long-wavelength region, which can be rationalized by considering the orbital interaction between the Pt–X bonds and the hybrid π-systems. The combination of phosphole and pyrrole has great synthetic potential, since these functionalizable heteroles are readily subject to new π-framework construction. Further studies on materials chemistry of phosphole–pyrrole π-conjugated systems are now in progress.
3 Experimental section
1H, 13C, and 31P NMR spectra were recorded on a JEOL JNM-EX400 spectrometer using CDCl3 or CD2Cl2. Chemical shifts are reported in ppm as relative values vs. tetramethylsilane (internal reference for 1H and 13C) and phosphoric acid (external reference for 31P). Matrix-assisted laser desorption/ionization (MALDI) time-of-flight mass spectra (TOF) were measured on a SHIMADZU Biotech AXIMA-CFR spectrometer using 1,8-dihydroxy-9(10H)-anthracenone (dithranol) as a matrix. High-resolution mass spectra (HRMS) were obtained on a JEOL JMS-HS110 spectrometer using 3-nitrobenzyl alcohol as a matrix. IR spectra were recorded on a JASCO FT/IR-470 Plus spectrometer using KBr pellets. UV-vis absorption spectra were measured on a PerkinElmer Lambda 900 UV/vis/NIR spectrometer. Steady-state fluorescence spectra were recorded with a SPEX Fluoromax-3 spectrofluorometer (HORIBA). Electrochemical measurements were made with an ALS 630a electrochemical analyzer using a glassy carbon working electrode, a platinum wire counter electrode, and an Ag/Ag+ [0.01 M AgNO3, 0.1 M nBu4NPF6 (MeCN)] reference electrode. The potentials were calibrated with ferrocene/ferrocenium [Emid = +0.20 V vs. Ag/Ag+]. 1-Boc-2-iodopyrrole (1a) [12] and 1-Boc-2-iodo-5-phenylpyrrole (1b) [13] were prepared according to the reported procedures.
The solvents used for the reactions were distilled from sodium benzophenone ketyl (THF and Et2O) or calcium hydride (CH2Cl2) under inert atmosphere before use. Other chemicals and solvents were of reagent grade quality, purchased commercially and used without further purification. Thin-layer chromatography and flash column chromatography were performed with Alt. 5554 DC-Alufolien Kieselgel 60 F254 (Merck) and Silica-gel 60N (Kanto Chemicals), respectively. All the reactions were performed under an argon atmosphere unless otherwise noted.
3.1 Synthesis of 2a,b
A mixture of 1a (3.58 g, 12.2 mmol), CuI (212 mg, 1.11 mmol), PdCl2(PPh3)2 (390 mg, 0.55 mmol), 1,6-heptadiyne (0.64 mL, 5.6 mmol), triethylamine (12 mL), and 1,4-dioxane (8 mL) was stirred at room temperature. After 24 h, the reaction mixture was filtrated through a Celite bed, and the filtrates were concentrated under reduced pressure. The oily residue was subjected onto alumina column chromatography (hexane/CH2Cl2 = 1/1) to give 2a as a pale brown oil (Rf = 0.2, 2.0 g, 85%). 1H NMR (CDCl3, 300 MHz): δ 1.60 (s, 18H, t-Bu), 1.91 (quint, 2H, J = 7.0 Hz, CH2CH2CH2), 2.62 (t, 4H, J = 7.0 Hz, CH2CH2CH2), 6.11 (pseudo t, 2H, J = 3.3 Hz pyrrole-β), 6.43 (m, 2H, pyrrole-β), 7.21 (m, 2H, pyrrole-α); 13C{1H} NMR (CDCl3, 75 MHz): δ 18.8, 27.6, 27.7, 73.1, 83.6, 92.4, 110.5, 115.5, 119.7, 121.5, 148.2; HRMS (EI) Calculated for C25H30N2O4 (M+): 422.2206. Found: m/z 422.2203.
According to a similar procedure, compound 2b was prepared from 1b and 1,6-heptadiyne. Yield 81%. Pale brown oil. 1H NMR (CDCl3, 400 MHz): δ 1.34 (s, 18H, t-Bu), 1.94 (quint, 2H, J = 6.8 Hz, CH2CH2CH2), 2.67 (t, 4H, J = 6.8 Hz, CH2CH2CH2), 6.13 (d, 2H, J = 3.4 Hz, pyrrole-β), 6.45 (d, 2H, J = 3.4 Hz, pyrrole-β), 7.26–7.38 (m, 10H, Ph); 13C{1H} NMR (CDCl3, 100 MHz): δ 18.9, 27.17, 27.20, 27.7, 27.8, 73.0, 83.8, 93.1, 112.4, 117.4, 117.7, 127.0, 127.7, 128.1, 133.9, 135.6, 148.7; MS (EI): m/z 374 ([M − 2Boc]+).
3.2 Synthesis of 3a,b
To a mixture of 2a (6.3 g, 15 mmol), Ti(Oi-Pr)4 (4.4 mL, 15 mmol), and Et2O (210 mL) was slowly added an ether solution of i-PrMgCl (2.0 M × 15 mL, 30 mmol) at −78 °C, and the resulting mixture was stirred for 2 h at −50 °C. Dichloro(phenyl)phosphine (2.0 mL, 15 mmol) was then added to the mixture at this temperature, and the resulting suspension was allowed to warm to 0 °C and stirred for 1 h at this temperature. After stirring for an additional 2 h at room temperature, a saturated aq NH4Cl solution was poured into the reaction mixture, and insoluble substances were filtrated off through a Celite bed. The Celite bed was washed with AcOEt repeatedly, and the filtrate was washed with brine. The aqueous phase was extracted with AcOEt, and the combined organic extracts were dried over Na2SO4 and evaporated under reduced pressure. The oily residue was subjected onto alumina column chromatography (hexane/CH2Cl2 = 2/1). The yellow fraction (Rf = 0.4) was collected and evaporated to give a yellow solid (3.4 g, 43%). Mp 46–47 °C; 1H NMR (CDCl3, 400 MHz): δ 1.52 (s, 18H, t-Bu), 2.13–2.22 (m, 2H, CH2CH2CH2), 2.31–2.43 (m, 2H, CH2CH2CH2), 2.50–2.69 (m, 2H, CH2CH2CH2), 5.95 (broad s, 2H, pyrrole-β), 6.07 (pseudo t, 2H, J = 3.4 Hz, pyrrole-β), 7.22–7.25 (m, 2H, pyrrole-α), 7.15–7.25 (m, 3H, P-Ph-m,p), 7.34–7.40 (m, 2H, P-Ph-o); 13C{1H} NMR (CDCl3, 100 MHz): δ 27.9, 28.8, 29.0, 83.2, 110.6, 114.6 (d, JP–C = 6.6 Hz), 122.5, 128.1 (d, JP–C = 7.4 Hz), 128.7, 129.1, 129.7 (d, JP–C = 24.0 Hz), 133.0 (d, JP–C = 15.7 Hz), 133.5 (d, JP–C = 19.0 Hz), 149.2, 155.5 (d, JP–C = 11.6 Hz); 31P{1H} NMR (CDCl3, 162 MHz): δ + 45.9; UV-vis (CH2Cl2): λmax (ɛ) 371 nm (10600); HRMS (FAB) calculated for C31H35N2O4P (M+): 530.2334. Found: m/z 530.2353.
According to a similar procedure, compound 3b was prepared from 2b. However, 3b could not be purified completely, as the remaining 2b was difficult to separate from the crude product. 1H NMR (CD2Cl2, 400 MHz): δ 1.21 (s, 18H, t-Bu), 2.17–2.27 (m, 2H, CH2CH2CH2), 2.38–2.50 (m, 2H, CH2CH2CH2), 2.68–2.80 (m, 2H, CH2CH2CH2), 6.01–6.13 (m, 4H, pyrrole-β), 7.20–7.40 (m, 15H, Ph); 31P{1H} NMR (CDCl3, 162 MHz): δ + 45.6; HRMS (FAB) calculated for C43H43N2O4P (M+): 682.2960. Found: m/z 682.2989.
3.3 Synthesis of 4a,b
To a THF solution (1.5 mL) of 3a (100 mg, 0.188 mmol) was added a MeOH solution (3 mL) of NaOMe (102 mg, 1.88 mmol) at room temperature. After stirring at 70 °C for 16 h, water (10 mL) and diethyl ether (10 mL) were added. The aqueous phase was extracted with diethyl ether and the combined organic extracts were dried over Na2SO4. After removal of solvents under reduced pressure, the resulting mixture was subjected to alumina column chromatography (hexane/CH2Cl2 = 2/1 to CH2Cl2/acetone = 6/1). The yellow fraction (Rf = 0.5; hexane/CH2Cl2 = 2/1) was collected, evaporated, and reprepicipated from CH2Cl2/hexane to give 4a (47 mg, 76%) as a brownish red solid. 1H NMR (CDCl3, 400 MHz): δ 2.33–2.49 (m, 2H, CH2CH2CH2), 2.63–2.72 (m, 2H, CH2CH2CH2), 2.78–2.87 (m, 2H, CH2CH2CH2), 6.17–6.18 (m, 4H, pyrrole-β), 6.65–6.66 (m, 2H, pyrrole-α), 7.26–7.34 (m, 3H, P-Ph-m,p), 7.53–7.57 (m, 2H, P-Ph-o), 8.01 (broad s, 2H, NH); 13C{1H} NMR (CDCl3, 100 MHz): δ 28.6, 29.2, 107.3 (d, JP–C = 5.0 Hz), 109.8, 118.1, 125.7, 129.1 (d, JP–C = 7.4 Hz), 129.5 (d, JP–C = 22.3 Hz), 130.0, 133.3 (d, JP–C = 14.1 Hz), 133.6 (d, JP–C = 19.8 Hz), 150.7 (d, JP–C = 8.3 Hz); 31P{1H} NMR (CDCl3, 162 MHz): δ + 28.1; UV-vis (CH2Cl2): λmax: 416 nm; HRMS (EI) calcd for C21H19N2P (M+): 330.1286. Found: m/z 330.1292.
According to a similar procedure, compound 4b was prepared from 3b. Yield: 28% (based on 2b). Red solid. Mp 194–195 °C; 1H NMR (CD2Cl2, 400 MHz): δ 2.40–2.55 (m, 2H, CH2CH2CH2), 2.70–2.80 (m, 2H, CH2CH2CH2), 2.85–2.94 (m, 2H, CH2CH2CH2), 6.25 (pseudo t, 2H, J = 2.9 Hz, pyrrole-β), 6.47 (pseudo t, 2H, J = 2.9 Hz, pyrrole-β), 7.15–7.19 (m, 2H, P-Ph-m), 7.31–7.37 (m, 11H, Ph), 7.61–7.65 (m, 2H, P-Ph-o), 8.26 (broad s, 2H, NH); 13C{1H} NMR (CDCl3, 75 MHz): δ 28.6, 29.1, 107.8, 109.6 (d, JP–C = 5.6 Hz), 118.2, 123.2, 125.5, 126.0, 128.8, 129.2 (d, JP–C = 8.1 Hz), 130.2 (d, JP–C = 1.9 Hz), 130.7 (d, JP–C = 22.3 Hz), 132.02, 132.05, 133.4 (d, JP–C = 14.3 Hz), 133.5 (d, JP–C = 19.8 Hz), 151.1 (d, JP–C = 9.3 Hz), One of the carbon atoms could not be detected clearly; 31P{1H} NMR (CD2Cl2, 162 MHz): δ + 28.5; UV-vis (CH2Cl2): λmax (ɛ) 330 (15200), 459 nm (34400); HRMS (EI) calcd for C33H27N2P (M+): 482.1912. Found: m/z 482.1905. Anal. calculated for C33H27N2P: C, 82.14; H, 5.64; N, 5.81. Found: C, 81.88; H, 5.63; N, 5.75.
3.4 Synthesis of 5a,b
To a THF solution (1.5 mL) of 3a (71.5 mg, 0.135 mmol) was added MeOH (3 mL) solution of NaOMe (72.8 mg, 1.35 mmol) at room temperature. After stirring at 70 °C for 16 h, water (10 mL) and diethyl ether (10 mL) were added. The aqueous phase was extracted with diethyl ether and the combined organic extracts were dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected onto alumina column chromatography (hexane/CH2Cl2 = 2/1 to CH2Cl2/acetone = 6/1). The yellow fraction (Rf = 0.4; hexane/CH2Cl2 = 2/1) was collected. After removal of solvents under reduced pressure, the resulting mixture was resolved in CH2Cl2. To the resulting solution was added hydrogen peroxide (30% solution in H2O, 0.1 mL). After stirring 1 h, the mixture was concentrated under reduced pressure, and the solid residue was washed with diethyl ether to give 5a as a red solid (37.1 mg, 79%). Mp 255 °C (dec); 1H NMR (CDCl3, 400 MHz): δ 2.00–2.10 (m, 1H, CH2CH2CH2), 2.28–2.40 (m, 1H, CH2CH2CH2), 2.66–2.80 (m, 2H, CH2CH2CH2), 2.81–2.90 (m, 2H, CH2CH2CH2), 6.25 (s, 2H, pyrrole-β), 6.32 (s, 2H, pyrrole-β), 6.80 (s, 2H, pyrrole-α), 7.30–7.45 (m, 3H, P-Ph-m,p), 7.65–7.78 (m, 2H, P-Ph-o), 9.09 (broad s, 2H, NH); 13C{1H} NMR (CDCl3, 100 MHz): δ 25.8, 29.3 (d, JP–C = 11.6 Hz), 109.2 (d, JP–C = 7.4 Hz), 110.1, 116.0, 117.0, 119.9, 126.2 (d, JP–C = 11.6 Hz), 128.9 (d, JP–C = 12.4 Hz), 130.4 (d, JP–C = 10.7 Hz), 132.1 (d, JP–C = 3.3 Hz), 149.6 (d, JP–C = 24.8 Hz); 31P{1H} NMR (CDCl3, 162 MHz): δ + 54.3; IR (KBr): ν = 1165 (PO) cm−1; UV-vis (CH2Cl2): λmax (ɛ) 458 nm (16700); HRMS (EI) calculated for C21H19N2OP (M+): 346.1235; Found: m/z 346.1241.
Compound 5b was prepared from 4b and mCPBA. Yield, 93%. Reddish brown solid. Mp 260 °C (dec); 1H NMR (CDCl3, 400 MHz): δ 2.00–2.15 (m, 1H, CH2CH2CH2), 2.31–2.42 (m, 1H, CH2CH2CH2), 2.69–2.80 (m, 2H, CH2CH2CH2), 2.80–2.94 (m, 2H, CH2CH2CH2), 6.38 (pseudo t, 2H, J = 2.9 Hz, pyrrole-β), 6.55 (s, 2H, pyrrole-β), 7.18–7.21 (m, 2H, P-Ph-m), 7.27–7.49 (m, 11H, Ph), 7.79–7.84 (m, 2H, P-Ph-o), 9.28 (broad s, 2H, NH); 13C{1H} NMR (CDCl3, 100 MHz): δ 25.8, 29.3 (d, JP–C = 11.6 Hz), 108.2, 111.4 (d, JP–C = 7.4 Hz), 115.8, 116.7, 123.8, 126.6, 127.6 (d, JP–C = 12.4 Hz), 128.9, 129.1 (d, JP–C = 12.4 Hz), 130.0, 130.3 (d, JP–C = 10.7 Hz), 130.9, 131.7, 132.2 (d, JP–C = 2.5 Hz), 133.8, 149.8 (d, JP–C = 25.6 Hz); 31P{1H} NMR (CDCl3, 162 MHz): δ + 55.6; IR (KBr): ν = 1180 (PO) cm−1; UV-vis (CH2Cl2): λmax (ɛ) 322 (21000), 506 nm (24100); HRMS (EI) calculated for C33H27N2OP (M+): 498.1861; Found: m/z 498.1858.
3.5 Synthesis of 6a,b
To a THF solution (3 mL) of 4a (198 mg, 0.373 mmol) was added MeOH solution (6 mL) of NaOMe (204 mg, 3.77 mmol) at room temperature. After stirring at 70 °C for 16 h, water (10 mL) and diethyl ether (10 mL) were added. The aqueous phase was extracted with diethyl ether and the combined organic extracts were dried over Na2SO4. After removal of solvents under reduced pressure, the resulting mixture was resolved in CH2Cl2. To the resulting solution was added elemental sulfur (60.3 mg, 1.88 mmol). After stirring 3 h, the mixture was concentrated under reduced pressure, and the solid residue was subjected to alumina column chromatography (hexane/CH2Cl2 = 2/1). The orange fraction (Rf = 0.2) was collected, evaporated, and reprecipitated from CH2Cl2/hexane to give 6a as an orange solid (117 mg, 86%). Mp 220 °C (dec); 1H NMR (CDCl3, 400 MHz): δ 2.13–2.25 (m, 1H, CH2CH2CH2), 2.33–2.47 (m, 1H, CH2CH2CH2), 3.20–3.40 (m, 4H, CH2CH2CH2), 6.21–6.23 (m, 2H, pyrrole-β), 6.30 (s, 2H, pyrrole-β), 6.76–6.78 (m, 2H, pyrrole-α), 7.33–7.44 (m, 3H, P-Ph-m,p), 7.84–7.90 (m, 2H, P-Ph-o), 9.61 (broad s, 2H, NH); 13C{1H} NMR (CDCl3, 100 MHz): δ 26.9, 29.3 (d, JP–C = 10.7 Hz), 109.7 (d, JP–C = 8.3 Hz), 109.8, 117.8, 118.6, 119.6, 126.3 (d, JP–C = 12.4 Hz), 128.9 (d, J P–C = 12.4 Hz), 130.2 (d, JP–C = 11.6 Hz), 132.0 (d, JP–C = 3.3 Hz), 151.0 (d, J P–C = 22.3 Hz); 31P{1H} NMR (CDCl3, 162 MHz): δ + 63.4; UV-vis (CH2Cl2): λmax (ɛ) 454 nm (17,500); HRMS (EI) calculated for C21H19N2PS (M+): 362.1007; Found: m/z 362.1011.
According to a similar procedure, compound 6b was prepared from 4b and elemental sulfur. Yield, 82%. Brown solid. Mp 286 °C (dec); 1H NMR (CD2Cl2, 400 MHz): δ 2.20–2.35 (m, 1H, CH2CH2CH2), 2.35–2.52 (m, 1H, CH2CH2CH2), 2.75–2.90 (m, 4H, CH2CH2CH2), 6.41 (s, 2H, pyrrole-β), 6.57 (s, 2H, pyrrole-β), 7.20–7.25 (m, 2H, P-Ph-m), 7.30–7.52 (m, 11H, Ph), 7.90–8.05 (m, 2H, P-Ph-o), 9.96 (broad s, 2H, NH); 13C{1H} NMR (CDCl3, 100 MHz): δ 27.0, 29.4 (d, JP–C = 11.6 Hz), 107.7, 111.7 (d, JP–C = 8.3 Hz), 117.4, 118.2, 123.8, 126.6, 127.6 (d, JP–C = 12.4 Hz), 128.9, 129.0, 129.1 (d, JP–C = 12.4 Hz), 130.2 (d, JP–C = 11.6 Hz), 131.8, 132.1 (d, JP–C = 3.3 Hz), 133.6, 151.3 (d, JP–C = 21.5 Hz), Two of the carbon atoms could not be detected clearly; 31P{1H} NMR (CD2Cl2, 162 MHz): δ + 63.6; UV-vis (CH2Cl2): λmax (ɛ) 320 (20300), 507 nm (22700); HRMS (EI) calculated for C33H27N2PS (M+): m/z 514.1633; Found: m/z 514.1630.
3.6 Synthesis of 7
A mixture of 4b (10 mg, 0.021 mmol), (6.1 mg, 0.021 mmol), and CH2Cl2 (1 mL) was stirred for 30 min at room temperature. The mixture was then concentrated under reduced pressure and subjected to silica gel column chromatography (CH2Cl2). The red fraction (Rf = 0.7) was collected, evaporated, and reprecipitated from CH2Cl2/hexane to give 7 as a dark brown solid (12.0 mg, 80%). Mp 215–216 °C; 1H NMR (CD2Cl2, 400 MHz): δ 2.47–2.55 (m, 2H, CH2CH2CH2), 2.88–3.02 (m, 4H, CH2CH2CH2), 6.46 (broad s, 2H, pyrrole-β), 6.50 (broad s, 2H, pyrrole-β), 7.20–7.26 (m, 2H, P-Ph-m), 7.33–7.58 (m, 11H, Ph), 7.85–7.91 (m, 2H, P-Ph-o), 8.48 (broad s, 2H, NH); 31P{1H} NMR (CD2Cl2, 162 MHz): δ + 51.4; UV-vis (CH2Cl2): λmax (ɛ) 504 nm (29900); HRMS (FAB) calculated for C33H27AuClN2P (M+): 714.1266; Found: m/z 714.1267.
3.7 Synthesis of 8
A mixture of 4b (10.8 mg, 0.022 mmol), PtCl2 (3.0 mg, 0.010 mmol), and CH2Cl2/MeOH (2:1, 1.5 mL) was stirred at room temperature. After 3 h, the mixture was concentrated under reduced pressure and then filtered off through a Celite bed. The filtrate was reprecipitated from CH2Cl2/hexane to afford 8 (8.3 mg, 67%) as a trans/cis mixture (ca. 1:1). Recrystallization of trans-rich 8 from CH2Cl2–hexane by a slow diffusion method afforded single crystals of trans-isomer as a dark brown solid. trans-8: 1H NMR (CD2Cl2, 400 MHz): δ 2.15–2.44 (m, 4H, CH2CH2CH2), 2.71–2.74 (m, 8H, CH2CH2CH2), 6.43 (broad s, 2H, pyrrole-β), 6.55 (broad s, 2H, pyrrole-β), 7.12–7.25 (m, 18H, Ph and P-Ph-m,p), 7.39–7.42 (m, 8H, Ph), 7.55–7.71 (m, 4H, P-Ph-o), 9.73 (broad s, 4H, NH); 31P{1H} NMR (CD2Cl2, 162 MHz): δ + 51.6 (JPt–P = 2244 Hz); UV-vis (CH2Cl2): λmax (ɛ) 450 (23000), 533 nm (25000); HRMS (FAB) calculated for C66H54N4Cl2P2Pt (M+): 1229.2849. Found: m/z 1229.2854. cis-8 (cis/trans > 95/5): 1H NMR (CDCl3, 400 MHz): δ 2.05–2.30 (m, 4H, CH2CH2CH2), 2.30–2.61 (m, 8H, CH2CH2CH2), 6.19 (broad s, 2H, pyrrole-β), 6.31 (broad s, 2H, pyrrole-β), 7.05–7.34 (m, 18H, Ph and P-Ph-m,p), 7.40–7.50 (m, 8H, Ph), 7.61–7.75 (m, 4H, P-Ph-o), 9.79 (broad s, 4H, NH); 31P{1H} NMR (CD2Cl2, 162 MHz): δ + 31.5 (JPt–P = 3177 Hz); UV-vis (CH2Cl2): λmax (ɛ) 541 nm; HRMS (FAB) calculated for C66H54N4Cl2P2Pt (M+): 1229.2849. Found: m/z 1229.2837.
3.8 X-ray structural determination
Single crystals of 5b, 7, and trans-8 were grown from CH2Cl2–MeOH (for 5b and 7) or CH2Cl2–hexane (for 8). All measurements were made on a Rigaku Mercury-8 CCD area detector with graphite monochromated Mo–Kα radiation at 143 K. The structures were solved by a direct method (SIR92) [19] and expanded using Fourier techniques [20]. Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were refined using the rigid model. All calculations were performed using CrystalStructure12 crystallographic software package except for refinement, which was performed using SHELXL-97 [21]. Crystal data and structural refinement parameters are as follows. 5b: C33H27N2OP, red block, 0.30 × 0.20 × 0.08 mm, MW = 498.54, monoclinic, space group = P21/n, a = 11.092(2) Å, b = 18.861(3) Å, c = 11.952(2) Å, β = 98.244(2)°, V = 2474.5(8) Å3, Z = 4, Dc = 1.338 g cm−3, μ = 1.42 cm−1, 28128 observed, 5653 unique, 335 variables, Rw = 0.1239, R = 0.0495 (I > 2.0σ(I)), GOF = 1.152. 7: C33H27AuClN2P, red block, 0.25 × 0.20 × 0.08 mm, MW = 714.95, monoclinic, space group = P21/c, a = 11.116(3) Å, b = 19.045(5) Å, c = 13.303(4) Å, β = 97.365(5)°, V = 2793.1(14) Å3, Z = 4, Dc = 1.700 g cm−3, μ = 54.64 cm−1, 31812 observed, 6257 unique, 344 variables, Rw = 0.1311, R = 0.0533 (I > 2.0σ(I)), GOF = 1.175. trans-8: C66H54Cl2N4P2Pt, dark brown block, 0.25 × 0.20 × 0.10 mm, MW = 1231.06, monoclinic, space group = P21/c, a = 15.424(9) Å, b = 11.803(6) Å, c = 18.046(10) Å, β = 114.382(8)°, V = 2992(3) Å3, Z = 2, Dc = 1.366 g cm−3, μ = 25.30 cm−1, 19757 observed, 6716 unique, 341 variables, Rw = 0.1761, R = 0.0653 (I > 2.0σ(I)), GOF = 1.087. CCDC-762249 (5b), CCDC-762250 (7), and CCDC-763570 (trans-8) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request.cif.
3.9 Fluorescence lifetime measurements
The time profiles of fluorescence in CH2Cl2 at 21 °C were measured by a streak camera, using the second harmonic output (400 nm, 150 fs) of a Ti:Sapphire regenerative amplifier system as an excitation source. The fluorescence decays of 4b and 7 were well fitted by the single exponential component.
3.10 Computational details
The geometry optimization was performed by the B3LYP method [22] with basis sets of 6-31G* [23] for C, H, N, O, P, and Cl atoms and LANL2DZ [24] for Pt and Au atoms. X-ray structures are used as initial geometries for 7m and 8m. We carried out vibration analysis with the B3LYP method to ascertain that each optimized geometry was not in saddle but in equilibrium points. The excited states and oscillator strengths are evaluated by the TD-B3LYP method, where forty excited states were solved. In Table 2, the states whose oscillator strengths are less than 0.2 are not included. All calculations were carried out with the Gaussian 03 package [25].
Acknowledgment
We thank Dr. Yoshihide Nakao (Kyoto University) for his valuable suggestions about DFT calculations. We also thank Prof. Hiroko Yamada and Mr. Daiki Kuzuhara (Ehime University) for the measurement of absolute fluorescence quantum yield of 4b and Prof. Yoshifumi Kimura (Kyoto University) for the measurement of fluorescence lifetimes of 4b and 7. This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (No. 21108511, “π-Space”) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
1 HOMO energies of pyrrole, benzene, thiophene, pyridine, and phosphole calculated at B3LYP/6-31G* level are −5.48, −6.70, −6.33, −6.87, and −6.25 eV, respectively.
2 In 1998, Mathey et al. reported the synthesis of 2-(1-methyl-2-pyrrolyl)phospholes via a base-promoted [1,5]-sigmatropic shift of the pyrrolyl group from the phosphorus atom to the α carbon.
3 Tomita et al. and Tanaka et al. used a similar protocol for the synthesis of phosphole–phenylene copolymers and phosphole-cored dendrimers, respectively.
4 Optical data (λab/nm, λem/nm, and Φf; measured in CH2Cl2) for 3,4-trimethylene-1,2,5-triphenylphosphole P–oxide are 386, 491, and 0.19, respectively.
5 The N–O distances for typical NH–O hydrogen bonds were reported to be ca. 3 Å (max 3.25 Å).
6 Optical data (λab/nm, λem/nm, Φf; measured in THF) for selected 2,5-diaryl-3,4-tetramethylene-1-phenylphosphole derivatives reported by Réau's group [4d] and Tanaka's group [8b]: The O3-P series: Ar = phenyl (354, 466, 0.143); Ar = 2-thienyl (412, 501, 0.050); Ar = 2-pyridyl (390, 463, 0.011). The O4-PO series: Ar = phenyl (370, 505, 0.0012); Ar = 2-thienyl (434, 556, 0.0069). The O4-PS series: Ar = 2-thienyl (432, 548, 0.046); Ar = 2-pyridyl (364, 470, 0.00004). For details on the measurement conditions, see the original references.
| Compound | λab/nmb (log ɛ) | λem/nm (Φf) | Eox/Ve,f |
| 4a | 416 (n.d.)g | 506 (n.d.)g | n.d.g |
| 4b | 459 (4.54) | 536 (0.78)c | −0.07 (r), +0.22 (r) |
| 5a | 458 (4.22) | 581 (0.002)d | +0.20 (ir) |
| 5b | 506 (4.38) | 620 (0.002)d | +0.12 (r), +0.44 (r) |
| 6a | 454 (4.24) | 576 (0.01)d | +0.22 (ir) |
| 6b | 507 (4.36) | 607 (0.02)d | +0.14 (r), +0.38 (r) |
| 7 | 504 (4.48) | 606 (0.09)d | n.d.g |
| trans-8 | 450 (4.36), 533 (4.40) | Non-fluorescent | n.d.g |
a Measured in CH2Cl2.
b The longest absorption maxima.
c Absolute fluorescence quantum yield.
d Fluorescence quantum yield relative to 4b.
e Redox potentials (vs. Fc/Fc+) determined by DPV. For details, see Experimental section.
f r: reversible; ir: irreversible.
g Not determined.
7 The fluorescent lifetimes (τf) of 4b and 7 in CH2Cl2 were determined to be 3.50 ns and 1.54 ns, respectively. With the τf and Φf values (Table 1), radiative rate constants from the S1 states of 4b and 7 were determined as 2.2 × 108 s−1 and 5.8 × 107 s−1, respectively.
8 Redox potentials (Epa /V, Epc/V; measured in THF) for selected 2,5-diaryl-3,4-tetramethylene-1-phenylphosphole derivatives reported by Réau's group [4d]: The σ3-P series: Ar = phenyl (+0.69, −2.88); Ar = 2-thienyl (+0.40, −); Ar = 2-pyridyl (+0.83, −2.45). The σ4-PO series: Ar = 2-thienyl (+0.63, −2.03). The σ4-PS series: Ar = 2-thienyl (+0.68, −1.95); Ar = 2-pyridyl (+1.43, −1.88). For details on the measurement conditions, see the original references.
9 Réau et al. and Baumgartner et al. investigated this topic extensively.
10 The distances of trans-PtCl2 complexes of phosphole-containing calix[4]pyrroles having hydrogen bonds are 3.18–3.52 Å.
11 We could not isolate cis-8 in a completely pure form and hence its spectrum is not included in Fig. 2. It should be mentioned, however, that cis-rich 8 (> 95%, determined by 1H NMR) showed one broad absorption band at λab 541 nm in CH2Cl2. In this paper, we do not discuss the observed difference in spectral shape between cis-8 and trans-8 because no structural information is available for the former complex at present.
12 Crystal Structure 3.8.2: Crystal Structure Analysis Package, Rigaku and Rigaku/MSC (2000–2006). 9009 New Trails Dr. The Woodlands TX 77381, USA.