

1 Introduction
The continued expansion of the burgeoning field of glycoscience depends critically on the development of improved glycochemistry for the supply of natural and artificial oligosaccharides and their conjugates. The central reaction in glycochemistry is that of glycosidic bond formation, or glycosylation, but surprisingly the details of this critical reaction remain relatively poorly understood [1]. For example, the glycosyl oxocarbenium ion, considered by most to play a key role in glycosylation, has yet to be observed experimentally. In this review we begin by exploring the evidence for and against the existence of glycosyl oxocarbenium ions before continuing with a broader look at the glycosylation reaction in general and factors affecting its mechanism. The intention is to present an overview of the current state of the art from a mechanistic perspective and, hopefully, to stimulate further physical organic work to serve as the basis for future rational reaction development.
2 Carbenium ions and oxocarbenium ions
The “embryonic concept” of carbocations dates to the beginning of the 20th century and conductivity measurements on solutions of the triphenylmethyl halides, which revealed the salt-like character of the solute [2–4]. Coupled with the observations that colorless pure triphenylmethanol gave colored solutions in concentrated sulfuric acid, and that triphenylmethyl chloride similarly formed orange complexes with aluminum and tin chlorides, these findings suggested salt formation [5,6]. Almost two decades later, in a seminal contribution to the field, Meerwein and van Emster observed that the rate of rearrangement of camphene hydrochloride into isobornyl chloride increased with the dielectric constant of the solvent and with the addition of Lewis acids, and suggested that carbenium ions may be intermediates in many reactions involving covalent reactants in solution [7]. Subsequently, the major work on the kinetics [8,9] and stereochemistry [10–12] of nucleophilic substitution and elimination reactions, by Hughes and Ingold, greatly contributed to the establishment of the concept of aliphatic carbenium ions as intermediates in what they termed SN1 and E1 reactions.
Nevertheless, until Olah reported his pioneering characterizations of simple alkyl cations by NMR spectroscopy [13–15], made possible by the use superacids [16] in the early 1960s, alkyl cations were considered only to be transient species. His main idea of suppressing bases and nucleophiles to avoid either cation-deprotonation or quenching was essential to the obtention of long-lived carbenium ions. The significant deshielding in their NMR spectra of the 13C signals when compared with the covalent precursors highlighted the cationic nature of the generated species. The case of the t-butyl cation generated from t-butyl chloride is an illustrative example in which the quaternary carbon atom of t-butyl chloride was shifted downfield in antimony pentafluoride solution by 273 ppm. Such significant deshielding excluded a donor-acceptor complex and established that the carbon atom had undergone rehybridization to sp2 and carried a substantial positive charge [15].
Secondary and tertiary alkoxycarbenium ions were first prepared by Meerwein et al. [17], and it was confirmed shortly after that they are, as anticipated [17], highly stabilized in comparison to alkyl cations. Direct observations of these stabilized cations by NMR spectroscopy were reported (Table 1) [18–23], and Olah prepared the first long-lived primary alkoxycarbenium ions and reported their NMR data in superacid media (Table 1) [24,25].
Selected oxocarbenium ions characterized by NMR spectroscopy.
| Entry | Oxocarbenium ion | 13C NMR (field, solvent, temperature): δ | Refs. |
| 1 | (20 MHz, SO2, −80 °C): 220.3 (C1), 83.3 (C1′) | [25] | |
| 2 | (150 MHz, CH2Cl2/CD2Cl2 (10:1), −80 °C): 230.6 (C1), 75.6 (C1′), 40.3 (C2) (125 MHz, CD2Cl2, −80 °C): 230.9 (C1), 76.1 (C1′), 40.7 (C2) | [29] | |
| 3 | (150 MHz, CH2Cl2/CD2Cl2 (10:1), −80 °C): 227.8 (C1), 82.5 (C2), 35.4 (C3), 19.5 (C4), 12.1 (C5) | [29] | |
| 4 | (25 MHz, SO2ClF, −40 °C): 245.1 (C1), 68.8 (C1′), 32.2 (C2), 27.0 (C3) | [23] | |
| 5 | (25 MHz, SO2ClF, −40 °C): 243.1 (C1), 82.5 (C1′), 32.4 (C2), 27.3 (C3), 13.0 (C2′) | [23] | |
| 6 | (25 MHz, SO2ClF, −40 °C): 240.72 (C1), 95.1 (C1′), 32.7 (C2), 26.7 (C3), 20.8 (C2′) | [23] | |
| 7 | (25 MHz, SO2ClF, −40 °C): 247.9 (C1), 80.7 (C1′), 34.3 (C2), 28.7 (C3), 13.0 (C2′), 11.0 (C4), 8.0 (C5) | [23] | |
| 8 | (25 MHz, SO2ClF, −40 °C): 241.8 (C1), 84.6 (C2), 37.7 (C3), 30.2 (C4), 20.5 (C5), 14.7 (C6) | [23] | |
| 9 | (25 MHz, SO2ClF, −78 °C): 248.3 (C1), 154.2 (C1′), 131.9 (C3′), 131.1 (C4′), 118.0 (C2′), 32.2 (C2), 29.1 (C3) | [18] |
Many years later long-lived alkoxycarbenium pools were generated by the Yoshida laboratory from α-silyl ethers or thioacetals by low temperature electrochemical oxidation in deuterated dichloromethane, using tetrabutylammonium tetrafluoroborate as electrolyte. Low temperature NMR spectroscopic analysis of the solution indicated the carbocationic character of the species so formed [26,27]. A sequential one-pot indirect method has also been developed in which an active thiophilic reagent is first generated electrochemically and then allowed to react with a thioacetal to generate an alkoxycarbenium pool. In this version, the time needed for carbocation generation is strongly reduced [28,29]. This methodology enabled Yoshida et al. to spectroscopically characterize the tetrahydropyranyl cation by NMR spectroscopy (Table 1, entry 3), but not a glycosyl cation [26].
More recently, Woerpel et al. have studied the conformational preferences of polysubstituted dioxocarbenium ions [30] by comparing experimental 1H NMR coupling constants with those predicted by computational methods (Fig. 1) [31]. This study provided insight into the three-dimensional structure of highly substituted oxocarbenium ions, which are related to the glycosyl cation. It was concluded that electrostatic forces [32] dictate the conformational preferences in the absence of severe steric interactions, but the importance of considering both factors when predicting favored conformations of cyclic oxocarbenium ions and hence those of glycosyl cations was underlined. Despite all of this excellent and painstaking work the glycosyl oxocarbenium ion itself has yet to be observed experimentally, and one might consider, therefore, that the field remains at the level at which carbenium ion chemistry in general found itself in the 1950s.
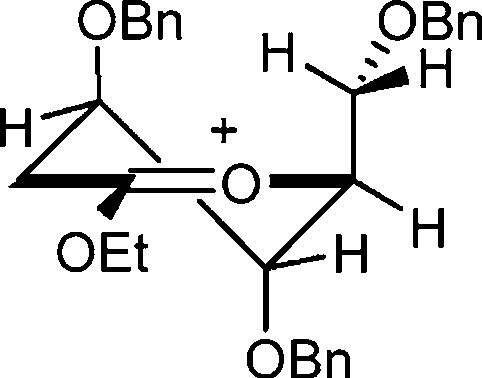
Cyclic dioxocarbenium ion studied by Woerpel.
In the absence of direct observation of glycosyl oxocarbenium ions themselves, computational studies have been advanced to predict their conformations [33], and indirect methods have been employed to estimate their lifetimes. Most pertinently, Amyes and Jencks employed the diffusion-limited reaction of azide anion with alkoxycarbenium ions as a clock with which to peg the rate of reaction of 55 M water with methoxymethyl cation at 1012 s−1 [34]. Extrapolating considerably from these values, and taking into account the electron-withdrawing effect of the additional C-O bonds in pyranosyl cations they arrived at the analogous figure of 1012 s−1 for the estimated lifetime of the glucosyl cation in aqueous solution at ambient temperature [34]. They considered the stability of a glycosyl cation as similar or larger than that of the methoxymethyl cation in water at room temperature but pointed out the absence of a significant lifetime for the glycosyl cation in the presence of a stronger nucleophile [34]. Further reflection led them to the conclusion that very rapid internal return is likely to preclude the existence of glycosyl cations within contact ion pairs and consequently, that if such cations have a real existence then it must necessarily be in the form of solvent separated ion pairs [34,35]. These conclusions have been widely adopted by the physical organic and mechanistic enzymology communities in terms of a “borderline existence” of glycosyl cations and, in the words of Sinnott [36], indicate that “… if intimate ion pairs of glycosyl cations and anions are too unstable to exist in water, a fortiori they have no real existence in organic solvents and mechanistic proposals, which invoke them are simply in error”.
The preparative carbohydrate chemistry community on the other hand, apparently unaware of, or at least in disagreement with, the above position largely continue to draw glycosyl cations, frequently with complete disregard for any counter ions, in their mechanistic schemes as is evident from perusal of the enormous majority of papers dealing with synthetic carbohydrate chemistry. It is clearly appropriate to ask “how relevant are Jencks’ conclusions to glycosylation reactions conducted in organic solution?” Yes, dichloromethane – currently a favorite solvent for many glycosylation reactions with its dielectric constant of 8.93 at 25 °C – is far less polar than water (ɛ = 78.30 at 25 °C), and is thus far less able to support the separation of charge needed to sustain an ion pair, but on the other hand the concentration of nucleophiles in a glycosylation reaction never approaches the 55 M of the Jencks’ conditions. Clues can be derived from the seminal work of Yoshida et al. on the generation of alkoxycarbenium ions from alkoxysilanes and monothioacetals [26–29]. Working at –80 °C in CD2Cl2 in the presence of tetrabutylammonium tetrafluoroborate as supporting electrolyte these workers obtained a solution of a species exhibiting a resonance with a chemical shift of δ ∼ 231 ppm, which they attributed to the sp2 carbon of the 1-methoxynonyl cation (Table 1, entry 2). This figure falls between the chemical shift determined by Olah for the methoxymethyl cation (Table 1, entry 1) and more substituted alkoxycarbenium ions (Table 1, entries 4 to 9) in superacid media, and it is clear that the species observed by Yoshida and coworkers very closely resembles an alkoxycarbenium ion. At worse, owing to possible solvent effects this species might be viewed as an alkoxycarbenium ion very loosely covalently bound to the counterion; alternatively it can be envisaged as a dynamic equilibrium, rapid on the NMR timescale, between the alkoxycarbenium ion and the covalent species in which the equilibrium heavily favors the alkoxycarbenium ion. Whichever postulate is correct, it is clear that a species that closely resembles a simple alkoxycarbenium has an existence at least of minutes in the absence of strong nucleophiles in dichloromethane solution at −80 °C, which is a far cry from the lifetime estimated by Amyes and Jencks for the methoxymethyl and the glycosyl oxocarbenium ions in water at ambient temperature [34]. Obviously, temperature is an important factor here and bearing this in mind Yoshida and coworkers examined their alkoxycarbenium ion by variable temperature NMR spectroscopy and found it to be generally stable to −50 °C above which decomposition rapidly sets in [26–29]. Further experiments using the indirect cation pool method [29] enabled the same group to observe the formation of the oxocyclohexenium ion by NMR (Table 1, entry 3). However, attempts at generation and observation of the tetramethyl glucopyranosyl cation by the direct cation pool method under similar conditions resulted only in generation of a glycosyl fluoride by presumed fluoride abstraction from tetrafluoroborate by the presumed oxocarbenium ion [26]. Comparable results were observed in the presence of trifluoromethanesulfonate (triflate) [37] and bis(trifluoromethanesulfonyl)imide (triflimide) [38] as counter ion. Perchlorate anion is also known to bind covalently to the glycosyl cation in organic solution [39], and the triflate anion has been found to bind covalently even in ionic liquids at low temperature [40].
The apparent higher reactivity of glycosyl oxocarbenium ions toward nucleophiles, compared to that of simple open-chain alkoxycarbenium ions, may perhaps be rationalized in terms of thermodynamic and kinetic factors that play a major role in the quenching reaction leading to a covalent adduct. On the one hand the inductive effect of the extra OR groups increases the energy of the cation (ground state destabilization), while on the other hand, the anomeric effect that develops on going from the reactant (oxocarbenium ion) to the transition state lowers the energy of the latter. The synergy of both factors should lower the activation energy barrier for the nucleophilic quenching of glycosyl oxocarbenium ions.
Whatever the reasons, one is left with the distinct impression that glycosyl cations are likely to have a real existence in dichloromethane solution at low temperature and that their observation only awaits the development of a suitable “flash” technique for their generation in the probe of an NMR spectrometer. Overall, we are forced to the conclusion that the intuition of preparative carbohydrate chemists in writing glycosyl cation intermediates may not be entirely wrong in certain circumstances, although the complete mechanistic picture is a complex one and many factors have to be weighed as we discuss below.
3 General Glycosylation Mechanism
Glycosylation is a complex phenomenon that cannot be considered simply in terms of the oxocarbenium ion. It involves a glycosyl donor, a glycosyl acceptor, a promoter and, until relatively recently, commonly a heterogeneous one. In this respect it is perhaps not too surprising that the extent of careful physical organic work in the area is extremely limited [41–46] and that, in the words of Green and Ley [47], “much of the evidence used to substantiate proposed inter-glycosyl coupling mechanisms is anecdotal or circumstantial”. A very large amount of painstaking work has been conducted on the somewhat parallel but simpler hydrolysis of glycosidic bonds and the conclusions drawn from this work are frequently extrapolated to glycosylation reactions. The disparity between the extents of work on glycoside hydrolysis and glycosylation is evident from the excellent 1969 review of the area by Capon [41] as well as from the more recent review by Horenstein [46] and from the critical compendium of Sinnott [36]. The mechanistic enzymology of glycosidases and glycosyl transferases has been the subject of a great deal of careful investigation and forms the basis of the recent spectacular advances in, inter alia, the rational design of potent glycosidase inhibitors and of functioning “glycosynthases” [48–53]. In view of the plethora of recipes available [54,55] it is difficult to escape the conclusion that, relatively speaking, chemical glycosylation suffers greatly from the lack of an extensive physical organic underpinning and, accordingly, would benefit considerably from the application of modern methods and instrumentation.
Most mechanistic thinking in the area is shaped around the seminal concept of a series of equilibrating oxocarbeniun ion/counterion ion pairs, of the contact, solvent separated and free varieties as advocated by Winstein for solvoysis reactions in general [56,57], first applied to glycosylation by Rhind-Tutt and Vernon [58], and as refined and extended by Lucas and Schuerch [59], and by Lemieux et al. [60].
The very short lifetimes predicted by Jencks for glycosyl cations in aqueous solution has led to the widespread adoption by mechanistic enzymologists of the concept of “exploded transition states” [36,50] (Fig. 2) to describe substitution reactions at the anomeric center. Geometrically, such an exploded transition state resembles the familiar trigonal bipyramid of an SN2 reaction that has been stretched about its long axis such that the interactions between both the incoming nucleophile and the departing nucleofuge are very weak and such that the anomeric carbon bears a substantial positive charge. Such an exploded transition state clearly bears a close resemblance to a stereoselective attack on a contact ion pair from the face opposite to that shielded by the counterion.
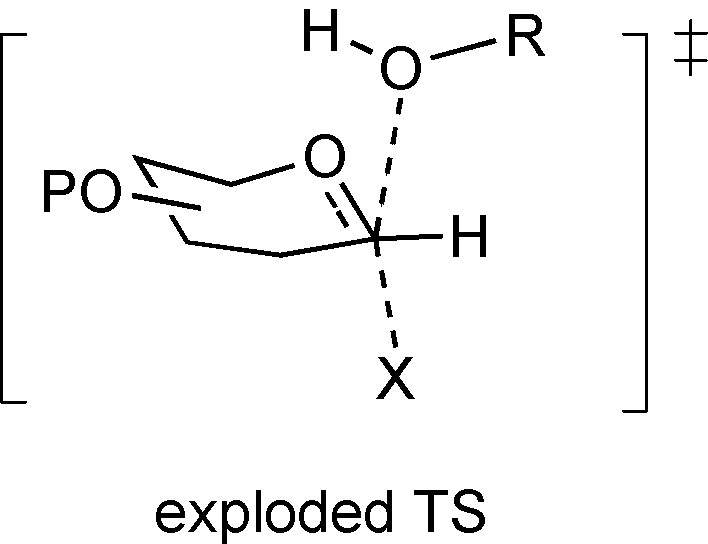
Exploded transition state for glycosylation.
Overall, one is left with a continuum of mechanisms possibly spanning the entire range between the pure SN2 mechanism at the one extreme and the SN1 mechanism with free oxocarbenium ions at the other (Scheme 1). On to this relatively straightforward picture must be layered the concepts of neighboring group participation and anchimeric assistance, those of the solvent and/or other additives and obviously the effect of the promoter.
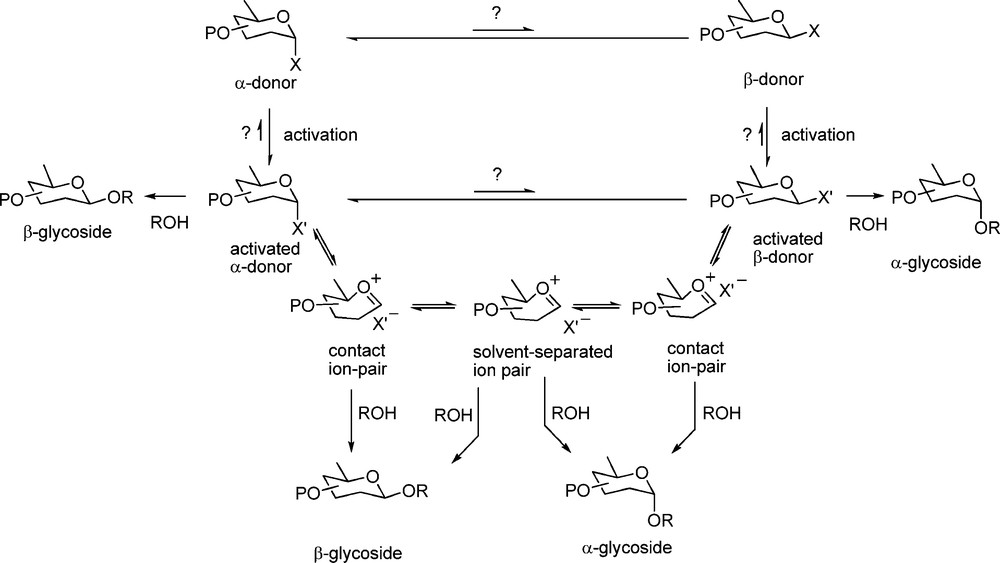
Full spectrum of mechanisms from SN2 all the way to pure SN1, going via the “Lemieux” ion pairs.
The SN2 extreme of the mechanistic spectrum can be represented by the reaction of sodium thiophenoxide with 2,3,4,6-tetra-O-methyl-α-D-glucopyranosyl chloride in propanol solution for which clean second-order kinetics were observed and for which the product of inversion of configuration phenyl 2,3,4,6-tetra-O-methyl-1-thio-β-D-glucopyranoside was obtained in almost quantitative yield [58]. An example of a dissociative reaction, that tends toward the SN1 mechanism but which nicely illustrates the complexities involved in the ion pair manifold, is provided by the reaction of 2,3,4,6-tetra-O-methyl-α-D-glucopyranosyl bromide (1) with cyclohexanol in a benzene/nitromethane mixture in the presence of the soluble promoter mercuric cyanide (Scheme 2) [44]. Kinetically, this reaction was shown to be cleanly first-order in the bromide and in the promoter but zero-order in cyclohexanol thereby fulfilling the requirement that the rate-determining step to be dissociative. Importantly however, the stereochemical outcome of the reaction was found to be dependent on the concentration of the alcohol. Thus, a higher concentration of cyclohexanol gave a greater proportion of the β-anomeric product (2β) than a lower concentration, which was interpreted as reflecting an increased rate of trapping of the α-ion pair (CIP) with the higher alcohol concentration. The α-anomeric product (2α) was viewed as arising from reaction of the alcohol with the free anomeric oxocarbenium ion (SSIP) [44].
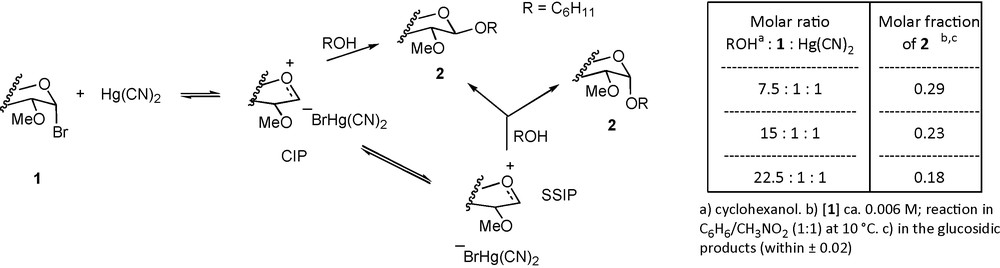
Selectivity dependence on alcohol concentration.
More recently, work from our laboratory using kinetic isotope methods has established that even the highly β-selective reaction of the spectroscopically demonstrated 4,6-O-benzylidene protected α-mannopyranosyl triflates [61] proceeds by a glycosyl oxocarbenium ion or at least an exploded transition state that closely resembles it [62].
4 Solvents, additives, and promoters
The choice of solvent for a glycosylation reaction can and frequently does have a major effect on its outcome. This effect arises most obviously through variation of the polarity of the reaction medium and its ability to support charge separation. Other factors that have to be considered include the extent of hydrogen bonding with the acceptor alcohol [33] that obviously affects the degree of aggregation and the effective steric bulk of this essential reaction partner. Solvents can, however, participate in a much more direct manner by adduct formation with the glycosyl donor. The most common effect of this kind is the nitrile effect whereby, as first proposed by Lemieux and Ratcliffe [63], ether-protected glycosyl cations are trapped by the solvent to afford intermediate α-glycopyranosyl nitrilium ions that are then displaced in a highly stereoselective manner by the acceptor to give β-glycosides (Scheme 3) [64,65].
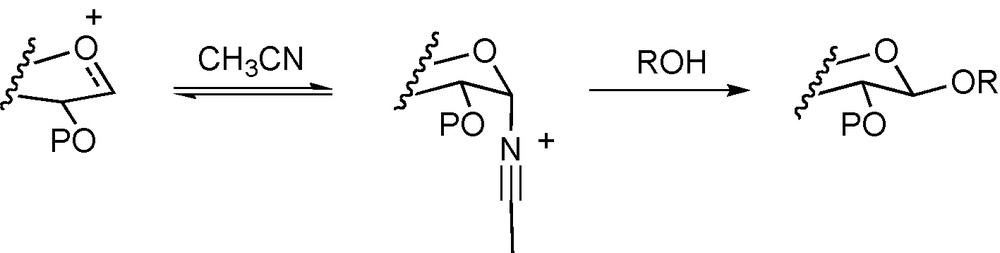
Nitrile participation.
The method provides a convenient means of entry to the 1,2-trans-equatorial β-glycosides in the absence of neighboring group participation, continues to be optimized with low concentrations of donor apparently giving the best results [66], and is widely employed in synthesis. Curiously, attempted application to the synthesis of the 1,2-cis-equatorial glycosides as in the β-mannopyranosides and β-rhamnopyranosides fails [67]. In the sialic acid field the nitrile effect again comes into its own and is frequently the method of choice for the synthesis of the α-sialosides [68,69]. Interestingly, despite the widespread application of the acetonitrile method there are no reports in the literature of the direct observation of the proposed intermediate glycosyl nitrilium ions by, e.g., NMR spectroscopy although chemical trapping experiments have more than adequately demonstrated their existence (Scheme 4) [70–72]. Acetonitrile also serves to illustrate yet another mode of interaction of the solvent in glycosylation reactions, namely interaction with the promoter. Thus, in order to increase the stereoselectivity of sialylation reactions conducted in acetonitrile, or its lower freezing cousin propionitrile, or in nitrile-containing solvent mixtures it is frequently desirable to lower the reaction temperature. Unfortunately, with sialoside thioglycosides as donors and the typical mixtures of N-iodosuccinimide and triflic acid as promoter the reactions are prohibitively slow much below –30 °C. This is presumably due to the stabilization of the iodonium ion by the nitrile group and it is only with the recent advent of the more reactive tertiary adamantanyl thiosialosides and the like that this problem has been circumvented [73,74].
Trapping of a glycosyl nitrilium ion.
Adduct formation is not limited to the use of acetonitrile but may also be induced by the deliberate introduction of nucleophilic catalysts into the reaction mixture. Perhaps the earliest, and certainly best known, such protocol is the use of tetraalkylammonium bromides by Lemieux and coworkers, dubbed the halide ion catalyzed method [60]. In this Curtin-Hammet [75] type protocol, the added bromide ion displaces bromide from a 2-O-benzyl protected α-glucopyranosyl bromide to give the corresponding β-bromide which, being inherently more reactive, is then displaced by the acceptor to give predominantly the α-glycoside (Scheme 5).

Bromide ion catalysis.
Diaryl sulfoxides were introduced as adduct forming additives by Gin and coworkers in their work on the activation of anomeric hemiacetals by the combination of diaryl sulfoxides and triflic anhydride [76,77]. Importantly, the adducts formed were shown to be more stable than the glycosyl triflates [77] and, following the reactivity selectivity principle, therefore necessarily less reactive (Scheme 6). When applying the dehydrative method to the formation of sialic acid glycosides Haberman and Gin demonstrated that variation of the substituents on the diaryl sulfoxide had a direct effect on the stereochemical outcome of the process thereby strongly implicating the sulfoxide in the actual glycosylation step in addition to the activation [78]. We subsequently examined the use of diphenyl sulfoxide as additive in sialylation reactions employing thiosialosides as donors and were able to identify two diastereomeric adducts by NMR spectroscopy, and to demonstrate thereby a rare example of stereocontrol in sialylation in the absence of acetonitrile [79].
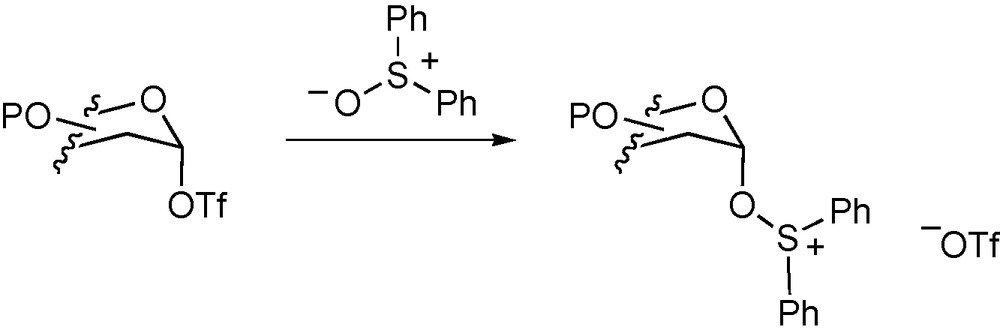
Displacement of a triflate by a sulfoxide.
Pyridine forms very stable salts when allowed to react with glycosyl donors [80,81]. These adducts can be isolated, crystallized, and undergo only slow hydrolysis in aqueous solution. For this reason all but sterically hindered heterocyclic bases are always avoided in glycosylation reactions. It is perhaps not surprising therefore that when rendered intramolecular, as in the work of Demchenko et al. [82], they react in a highly stereoselective fashion with nucleophiles but only slowly and under forcing conditions. In this light, the recent observations by Yu and coworkers on the directing effect of 4-dimethylaminopyridine and the like in dichloromethane at room temperature, and suggested to involve the intermediacy of glycosyl (4-dimethylaminopyridinium) salts, are interesting and noteworthy [83]. Of course, reactions that proceed with destruction of charge, as in the Yu alkoxide reaction with glycosyl pyridinium salts, should be accelerated in non-polar solvents.
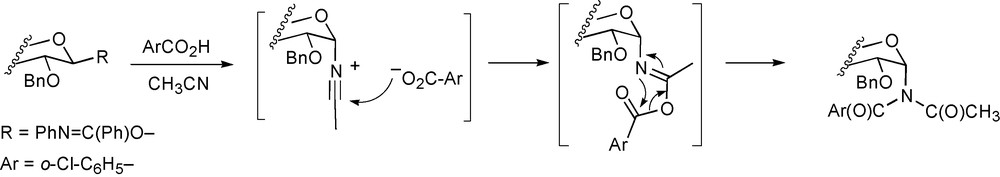
Added thioethers have been shown recently to form covalent sulfonium ion-type adducts with glycosyl cations by two groups. Thus, Boons and coworkers, working in the 3,4,6-tri-O-acetyl-2-azido-2-deoxyglucopyranosyl series demonstrated the formation of two diastereomeric β-sulfonium salts on activation of a trichloroacetimidate in the presence of ethyl phenyl sulfide (Fig. 3). These sulfonium salts, and the corresponding salt derived from the addition of thiophene, then took part in α-selective reactions in dichloromethane solution at 0 °C [84]. In contrast, Yoshida and coworkers found that the addition of dimethyl sulfide to an electrochemically generated 3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-glucopyranosyl triflate gave a mixture of α- and β-glycosyl dimethyl sulfonium salts albeit favoring the more stable, less hindered β-isomer (Fig. 3) [85]. On addition of methanol at 0 °C only the α-sulfonium salt was consumed but the product was formed as a mixture of anomers indicating a dissociative mechanism. While the different stereochemical mix of sulfonium salts observed by the two groups for a common glycopyranosyl moiety can be attributed to the use of sulfides of different bulk, the apparently differing reactivities of the glycosyl sulfonium salts observed requires an alternative explanation. Presumably the use of different entries into the glycosyl sulfonium salts is at the heart of this dichotomy with the Boons group employing a glycosyl trichloroacetimidate as donor with activation by TMS triflate, while the Yoshida laboratory made use of electrochemical activation of a thioglycoside in the presence of tetrabutylammonium triflate as supporting electrolyte. Clearly, the 0.1 M ammonium salt is not an innocuous additive in the electrochemical protocol and substantially increases the polarity of the reaction medium, thereby presumably stabilizing the glycosyl sulfonium salts. The effect of supporting electrolytes on the outcomes of glycosylation reactions can also be observed in the stereochemical outcome of the reactions of various electrochemically generated glycosyl triflates when contrasted with those of the same species generated chemically [37].
Sulfonium adducts observed by Boons (left) and Yoshida (center and right).
Molecular sieves are frequently employed by preparative carbohydrate chemists directly in glycosylation reaction mixtures in an attempt to suppress competing hydrolysis reactions by adventitious water. It must be recalled, however, that molecular sieves are highly reactive surfaces that can influence reactions in many ways. One such example is the use of basic aluminosilicates, which can quite simply prevent the activation of thioglycosides by the well-known mixture of N-iodosuccinimide with triflic acid. In such cases, and when the addition of sieves is considered necessary, the acid washed variety are greatly to be preferred [86].
Promoters clearly are very important parts of almost all glycosylation reactions serving the essential function of activating the glycosyl donor. Beyond this obvious function though promoters introduce other species into the reaction mixture and complicate the mechanistic picture. For example, the use of the highly reactive silver triflate to activate thioglycosides and/or glycosyl halides must invoke the formation of glycosyl triflates as intermediates in the reaction mixture. Classically, many promoters were insoluble and this led to glycosylations frequently being surface phenomena with the attendant difficulties in scale-up and reproducibility. An extreme example of the influence of an effect on an insoluble promoter surface arises from the use of the cis-fused 2,3-O-carbonate protected donors in the manno- and rhamnopyranosyl series. With glycosyl halides and insoluble silver(I) promoters, donors protected in this manner were found to be highly β-selective, which was attributed in part to the effect of the surface and in part to the strongly electron-withdrawing effect of the cyclic carbonate [87,88]. Many years later it was found that homogeneous solution phase glycosylations employing 2,3-O-carbonate protected manno- and rhamnopyranosyl donors are very highly α-selective (Scheme 7) [89]. This switch in selectivity led to the realization that the cis-fused 2,3-O-carbonate, far from being disarming as originally supposed, is in fact arming. This effect is achieved by virtue of the ground state destabilization of the donor through the half-chair conformation the carbonate imposes on it [90,91]. The previous β-selective chemistry is therefore entirely a surface phenomenon due to the selective absorption of the α-face of the donor onto the insoluble promoter.
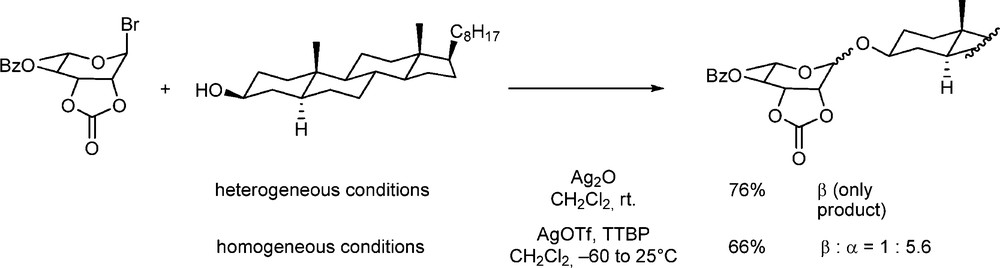
Change of selectivity with a rhamnosyl 2,3-O-carbonate in going from an insoluble to a soluble promoter.
5 The role of the acceptor in the glycosylation mechanism
The role that the acceptor alcohol plays in determining the outcome of a glycosylation reaction can be expected to depend on where the particular reaction lies on the mechanistic continuum, and so, for a given disaccharide product, to be a function of leaving group, promoter, and solvent. The extent to which the acceptor alcohol plays a determining role in the stereochemical outcome of the glycosylation process might reasonably be expected to diminish across the continuum as the transition state becomes looser and closer to the intermediacy of a solvent separated ion pairs and free oxocarbenium ions. However, as is clear from the early work of Wallace and Schroeder (Scheme 2) it is apparent that even for systems displaying established zero-order kinetics in the acceptor that the concentration of the acceptor has a role to play in the stereochemical outcome of the process. More recently, and building on his work on the mechanisms of C-glycosylation, and referring to acetal substitution reactions in general, Woerpel et al. have stated that “Stereoselective SN1-type mechanisms occur with weak and moderate nucleophiles and poor leaving groups, and unselective diffusion-limited SN1 mechanisms and stereoselective SN2 reaction pathways emerge with strong nucleophiles.” [92]. In other words there is no escaping the fact that the alcohol is perforce involved in the product-determining step, however exploded the transition state.
Probing of the existence of diastereomeric matching and mismatching, through the use of an enantiomeric pair of acceptors with an enantiomerically pure donor or the converse [93], might be considered as another possible entry into determining the extent of involvement of the acceptor in a glycosylation reaction. For example, a pair of coupling reactions exhibiting a high degree of diastereomeric discrimination as in the coupling of perbenzoyl D- and L-fucosyl bromides (D-3 and L-3) with the D-glucosamine based acceptor D-4 resulting in two sets of two anomers (Scheme 8) [93] must be considered as having a relatively tight transition state, whereas one with little or no such discrimination, as in the coupling of the enantiomeric chiro-inositol acceptors D- and L-6 with the D-2-azido-2-deoxy glucopyranosyl trichloroacetimidate donor D-7 (Scheme 9) [94], necessarily is highly exploded.
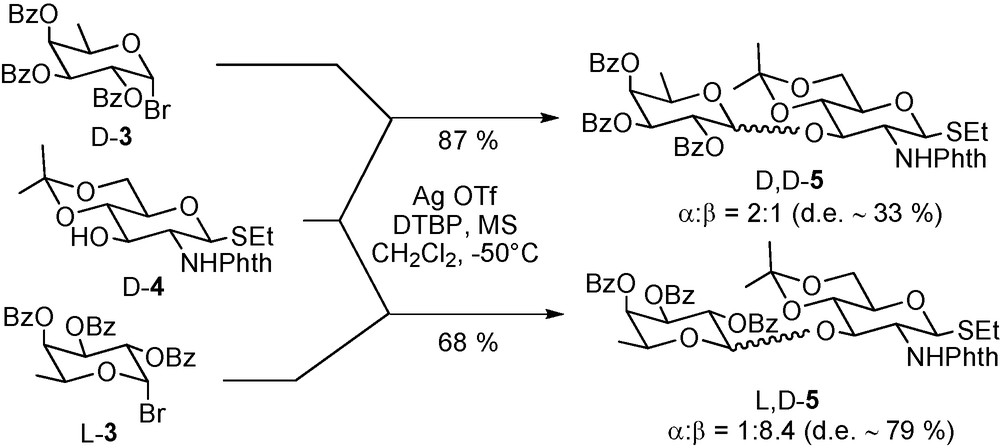
Donor-acceptor pair exhibiting high diastereoisomeric discrimination.
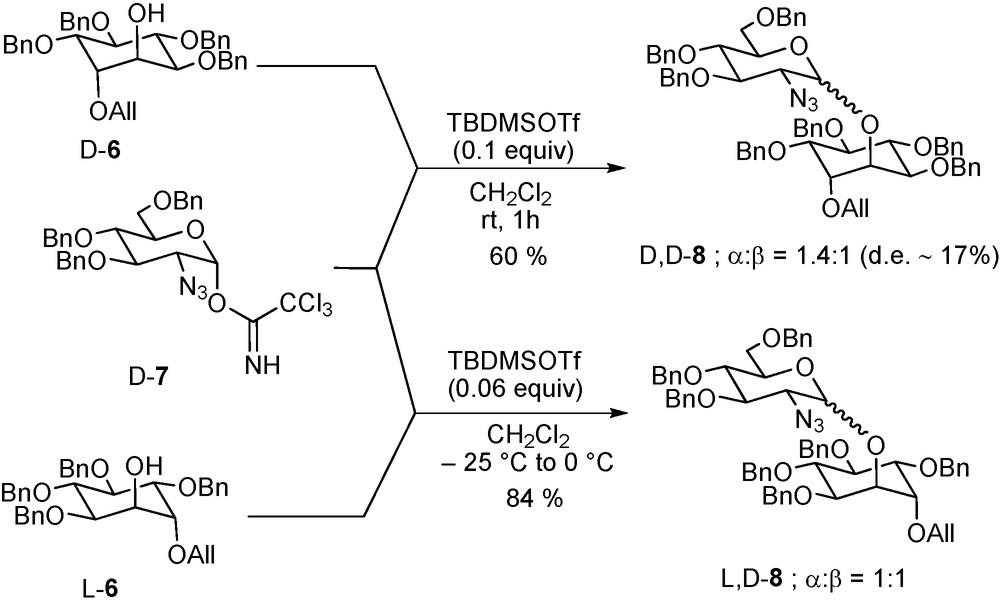
Donor-acceptor pair exhibiting small diastereoisomeric discrimination.
6 Intramolecular participation by protecting groups
Thus far we have considered glycosylation in terms of the continuum of mechanisms represented in Scheme 1, with the use of additives presenting a relatively minor excursion from the main highway. We now consider the more major deviation presented by the widespread case of neighboring group participation. The rationalization of the preferential formation of the 1,2-trans-glycosides, axial or equatorial, from donors bearing a 2-O-carboxylate ester as being the result of formation of an intermediate cyclic dioxalenium ion is due to Frush and Isbell [95]. While the stereochemical benefits of neighboring group-directed glycosylation are extremely widely appreciated, the kinetic aspects are often overlooked. This is due in no small measure to uncritical application of Fraser-Reid's seminal concept [96] of armed and disarmed protecting groups. Regarding the use of donors in which the 2-O-carboxylate and the anomeric-leaving group have the cis-configuration there can be no doubt as to the disarming nature of the ester group. However the situation is much less clear for the trans-isomers when the departure of the leaving group may be assisted by the ester [56,97], and when the latter is thus necessarily armed. The extent of kinetic assistance by a trans-ester in this manner is a function of the leaving group and the promoter as was recognized by Paulsen and Herold who found that β-glucopyranosyl pentaacetate underwent reaction with antimony pentachloride under conditions in which the corresponding α-anomer did not. Under the same conditions, however, the more reactive chlorides were consumed irrespective of configuration (Scheme 10) [98].
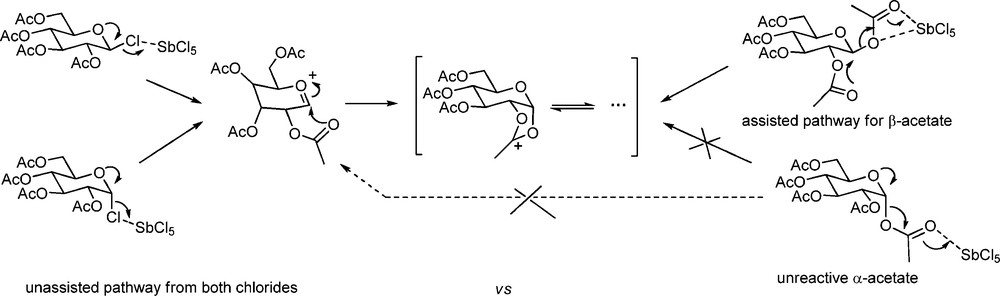
Differential reactivity of α- and β-glucopyranosyl donors as a function of leaving group.
Similar observations were made by Konradsson who noted only a 1.7-fold difference in reactivity between the two anomers of the pentenyl per-O-benzyl glucopyranosides, but found a 5.2-fold difference for the corresponding peracetyl series, in both cases favoring the β-anomer [99]. More recently, Demchenko's observation [100,101] that a pair of 2-O-acetyl and 2-O-benzoyl S-benzoxazolyl 3,4,6-tri-O-benzoyl-β-D-glucopyranosyl thioglycosides were more reactive under promotion by copper triflate than the corresponding 2-O-benzyl analog, was found not to hold in the corresponding α-anomeric series, thereby again revealing the role of the esters in augmenting the reactivity of the β-series (Scheme 11) [102].
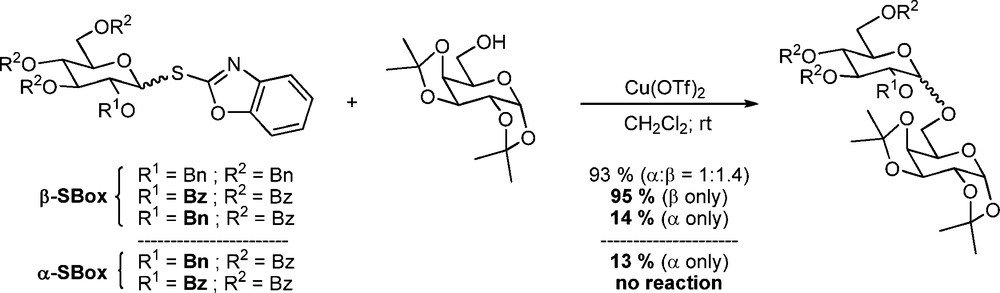
Dependence of donor reactivity on anomeric stereochemistry as a function of protecting group.
Overall, it is apparent that the more weakly activated a system – in terms of the strength of the leaving group-promoter interaction – the more likely it is to benefit from the additional push of an antiperiplanar vicinal ester group and, thus, the less likely such an ester is to be disarming.
The mechanism of neighboring group participation-directed glycosylation by vicinal esters proceeds through the intermediacy of cyclic dioxalenium ions and, when formed in the absence of nucleophiles, these may be isolated from the reaction mixture by crystallization [39,98] or studied in the reaction mixture by low temperature NMR spectroscopy [103]. In the presence of a hindered base the cyclic dioxalenium ion reacts with the acceptor alcohol to give isolable orthoesters, which form one starting point for the well-known orthoester glycosylation method [104–108]. In the presence of Brønsted or Lewis acid catalysts the orthoesters undergo rearrangement to the glycosidic bond with inversion of configuration at the anomeric center (Scheme 12). It is generally agreed that, even in the absence of base, the orthoester is the kinetic product of the reaction, no doubt owing to the more rapid attack of the acceptor alcohol at the site of highest charge density [103,109–111].
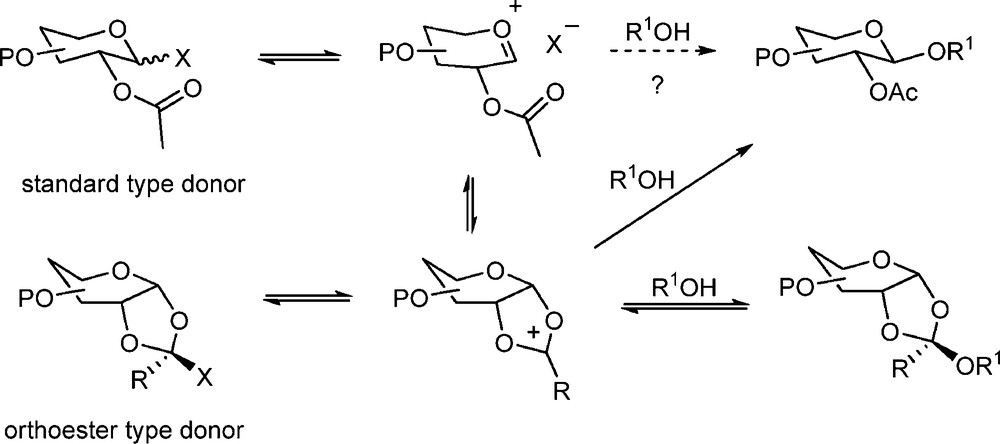
Basic neighboring group participation mechanism with intermediate dioxalenium ion.
The main debate therefore revolves around the mechanism of the acid-catalyzed rearrangement of the orthoester to the glycoside, a subject, which has been the focus of surprisingly little experimental work. Banoub and coworkers determined the rearrangement of a rhamnopyranosyl orthoester to the corresponding α-rhamnoside to follow first-order kinetics on catalysis with triflic acid in the presence of tetramethylurea and, on the basis of the large negative value of the free entropy of activation, concluded that the mechanism was intramolecular and involves a tight ion pair intermediate [112]. Somewhat remarkably in view of the extremely widespread use of neighboring group directed glycosylation, crossover experiments were not reported in the literature until 2000. This seminal work from the Yu group employing a range of concentrations of TMS triflate as promoter showed a very high degree of exchange and therefore strongly indicates the rearrangement of the orthoesters to be intermolecular (Scheme 13) [113].
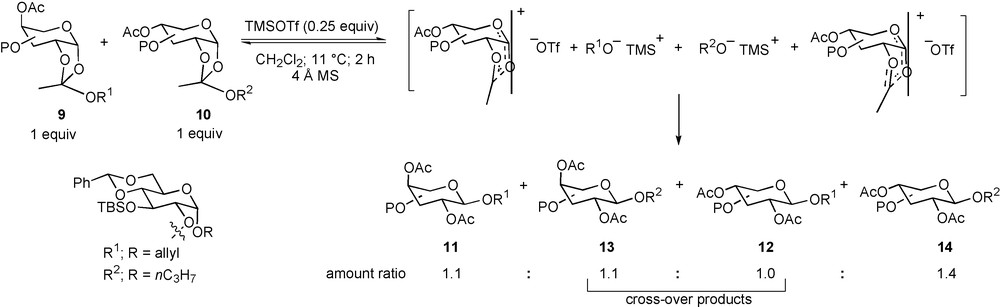
Crossover experimental pertinent to the mechanism of neighboring group participation.
Interestingly, and consistent with earlier indications from our laboratory [61], Huang and coworkers recently reported that cyclic dioxalenium ions are not necessarily the most stable intermediates in glycosylation processes directed by vicinal esters [114]. Thus, activation of a perbenzoyl galactopyranosyl thioglycoside with an arenesulfenyl triflate in CDCl3 at −60 °C resulted in the formation of a single observable species, namely the glycosyl triflate (Scheme 14). On the other hand, the analogous 2-O-benzoyl-3,4,6-tri-O-benzyl protected system afforded only the cyclic dioxalenium ion under the same conditions. Intermediate between these two extremes was the case of a per-O-acetyl glucopyranosyl thioglycoside, which afforded a 1:1 mixture of an anomeric triflate and a cyclic dioxalenium ion (Scheme 14). On the addition of an alcohol to the latter reaction mixture the two intermediates were consumed at the same rate, thereby strongly suggesting their rapid interconversion, and implicating the dioxalenium ion in the actual glycosylation process.
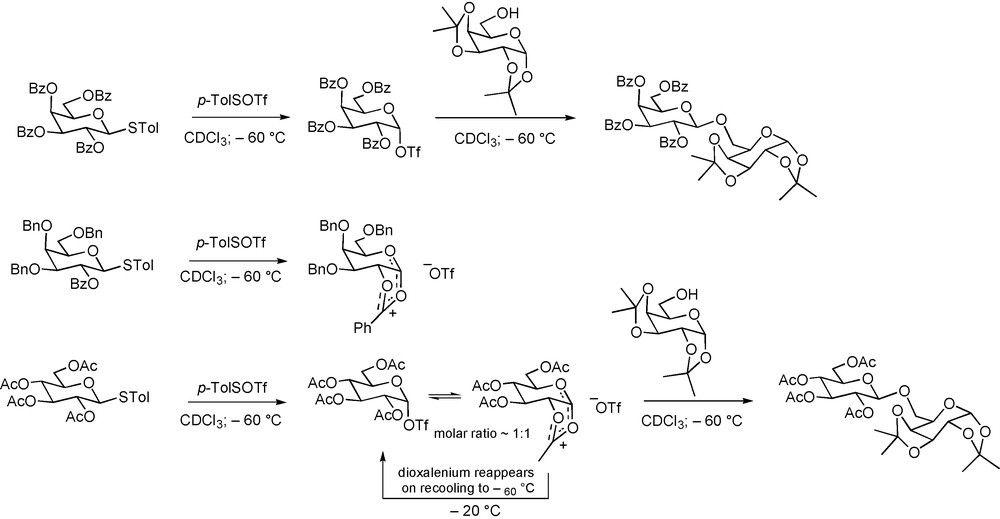
Observation of glycosyl triflates in the presence of 2-O-acyl esters.
If the situation regarding participation by vicinal carboxylate esters is relatively well established albeit lacking in appreciation of the finer points that concerning participation by more remote esters is at best turgid. There is a considerable amount of speculation in the literature as to participation by esters from the three- [115–120], four-[115,121–126] (axial and equatorial), and six-positions [124,125,127,128] that is almost entirely based on the observation of often very modest changes in stereoselectivity on replacing esters by ethers at those positions. Using the t-butyloxycarbonyl group as a replacement for an ester group and as a probe for participation through decomposition of any intermediate cyclic dioxalenium ion to a cyclic carbonate, we were recently able to confirm participation by axial esters on the three-position of glycopyranosyl donors (Scheme 15). However, we could find no evidence for the intermediacy of cyclic ions derived from equatorial esters at the three-position, by equatorial or axial esters at the four-position, or by esters at the six-position [129]. Very recent work by Kim et al. supporting participation by equatorial 3-O-esters and especially esters at the six-position uses the much more nucleophilic imidate esters as probes that bears little relevance to the issue of participation by esters and is therefore of questionable relevance [130].
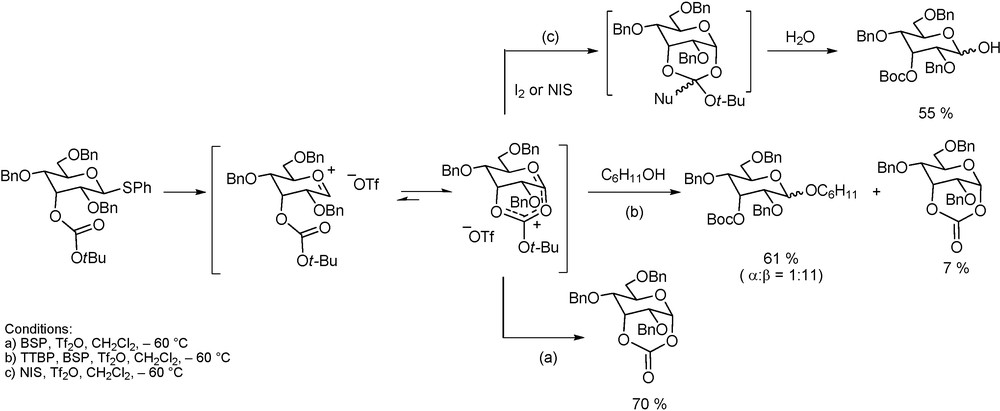
Use of a Carbonate ester to probe participation.
The dominant position of carboxylate esters as participating groups par excellence has recently been challenged by the introduction of a cleverly conceived system of diastereomeric thiophenyl benzyl ethers by the Boons group. Both diastereoisomers were shown to form six-membered cyclic glycosyl sulfonium ions on activation by NMR spectroscopy but, owing to the minimization of steric constraints, one gave a trans-fused ion with an equatorial C1-S bond and the other a cis-fused system with an axial C1-S bond. The trans-fused salt was strongly α-directing whereas the less stable cis-fused system gave rise to a mixture of isomers (Scheme 16) [131].
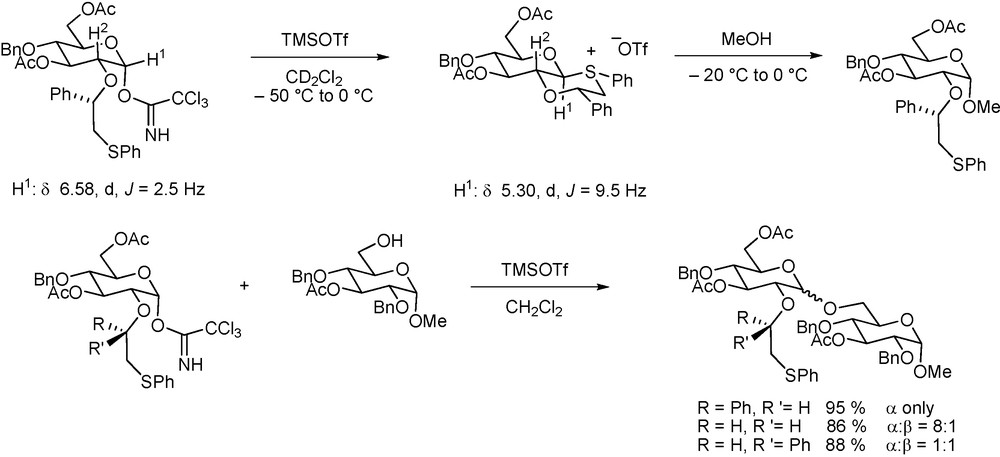
Stereodirecting effect of a pair of enantiomeric ethers.
In a related vein Smoot et al. introduced the 2-O-(2-picolinyl) ethers as arming but participating ethers [82]. As discussed above, cyclic pyridinyl salts were indeed formed on activation but their stability led to prolonged reaction times at elevated temperatures, thereby diminishing their utility and arguing strongly against the claim of an armed system (Scheme 17). The concept was subsequently extended to the use of 2-O-(2-thienylmethyl) ethers by Cox and Fairbanks, but no evidence was advanced in support of the postulated participation by the thienylmethyl ether [132]. The participation of sulfides, both from the two- and more remote positions, has been advanced on numerous occasions but recent NMR work calls such participation seriously into question [133,134].
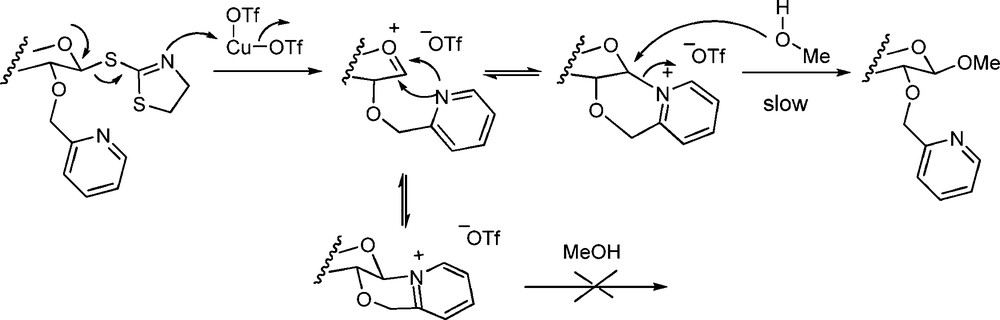
Participation by a 2-O-picolinyl ether.
7 Conclusion
In conclusion the mechanism of the glycosylation reaction is, with the exception of the case of neighboring group participation for the most part adequately described by Scheme 1 as a continuum with the position of any particular example on the continuum being a function of numerous factors. The very existence of a continuum of this kind mitigates against a clean demarcation between SN1 and SN2 mechanisms as has been possible recently for simpler systems with observable carbenium ions [135]. However, as knowledge of the field progresses and the possibility of observable glycosyl oxocarbenium ions looms larger such distinctions and even predictions become increasingly likely. The main deviation from this continuum is that of participation by esters and other intramolecular nucleophiles, in which kinetics factors as well as the more evident stereochemical ones must be taken into account. For a given donor-acceptor pair subtle modifications in the reaction conditions may favor either a bimolecular or a monomolecular rate-determining step. Glycosyl cations, for the moment hypothetical species, appear to have a role to play in most monomolecular reactions. Their actual observation by spectroscopic means would be a welcome advance in the field.