

1 Introduction
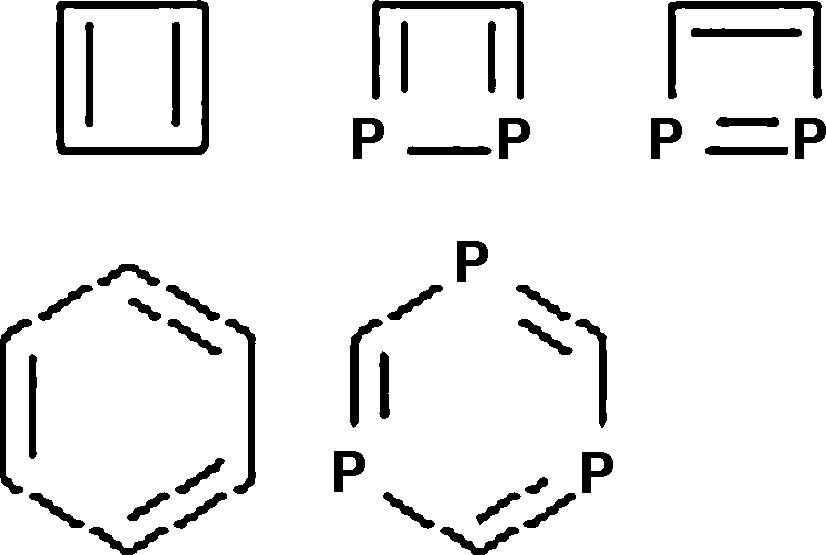
One of the interesting features of Mendeleev's periodic table is the diagonal rule: the chemical properties of a given element are similar to that in the bottom right diagonal. The chemistry of phosphorus is, for example, expected to be similar in many respects to that of carbon, as was pointed out in the book entitled “Phosphorus: the Carbon Copy” [1]. This similarity is also expressed in the geometric and electronic structures of the corresponding compounds. Both benzene and 1,3,5-triphosphabenzene (Scheme 1) are aromatic compounds, with equalized bond lengths and bond orders in the ring [2]. Also, cyclobutadiene and 1,2-diphosphacyclobutadiene are antiaromatic molecules, which have clearly distinguishable (nearly) single and double bonds [3,4]. However, the substitution of two CH groups in the cyclobutadiene by phosphorus introduces new structural features. First, diphosphacyclobutadiene has two constitution isomers, where the P-atoms are in 1,2 and 1,3 positions, respectively. Second, 1,2-diphosphacyclobutadiene has two distinguishable bond stretch isomers, that are indistinguishable in the cyclobutadiene parent compound.
.
One of the most recent achievements in the carbon chemistry is the successful isolation and characterization of graphene, a single graphite layer [5]. This compound has many interesting properties, including its planarity, exclusive band structure and magnetic properties. These make it a promising candidate for future developments in many fields including the semiconductor industry and other diverse applications, like ionic sieves [6]. The chemistry of graphene and graphene nano-ribbons is subjected to an extensive and actual research [7]. The existence of graphane, the hydrogenated derivative of graphene was proposed using theoretical methods [8], and was subsequently confirmed by experiments [9]. Graphene is a hydrocarbon with 1:1 carbon-hydrogen ratio, therefore formally it can be regarded as a polymer of acetylene. Fully fluorinated derivatives [10], and polysilanes, the silicon-analogue of graphane, were also prepared [11], and hydrogenated poly-aminoborane (BNH2) sheets were also investigated [12].
On the other hand, black phosphorus is also a layered material where the P6 rings in chair conformation form the two-dimensional lattices [13]. Thus, this compound is structurally similar to the graphane, even though their symmetries are different. Black phosphorus can formally be regarded as the polymer of P2, the all-phosphorus analogue of acetylene.
Phosphaethyne (HCP) stands between acetylene and P2. This compound is the simplest phosphaalkyne, which was synthesized in 1961 starting from phosphine (PH3) and graphite using a special reactor [14]. In its free form, this compound is stable only under its triple point (−124 °C), while polymerization occurs just above this temperature. The process becomes very rapid above −74 °C. It has to be noted, however, that the ether solution can be kept unchanged for months at −20 °C [15]. Similarly to the monomer, the freshly prepared polymer undergoes pyroforic reactions with oxygen or water. The aged polymer is fairly stable, however, at the above conditions. The polymer is a black compound, which according to element analysis has 1:1:1 C:P:H ratio, indicating that this is really the polymer of phosphaethyne and no side-products of the preparation reaction were incorporated. To the best of our knowledge, no further experiments were carried out using this polymer. Large oligomers of phenyl-phosphaethyne were investigated using NMR spectroscopy, and pointed out that they contained unsaturated carbon-phosphorus bonds [16]. Probably the presence of unsaturated bonds makes the freshly prepared compound pyroforic; however, during aging and equilibration, these bonds react and become saturated.
Nowadays several feasible synthetic methods of phosphaethyne exist [15,17]. In this article, we analyze the geometric and electronic structures of the one-dimensional (cis-polyacetylene-type) or two-dimensional (graphane-type) polymers of phosphaethyne, and we compare their properties with those of the well-known acetylene (graphane) and diphosphorus (black phosphorus). We hope that the calculated results reported in this paper could lead to further experimental study of poly-phosphaethynes.
2 Methods and computations
All computations were performed with the Gaussian 03 suite of programs [18] using PW91 generalized gradient approximation density functional [19], and the correlation consistent polarized triple-ζ cc-pVTZ basis set [20], without density fitting. The use of a full-electron Gaussian basis set provides a balanced and equal accuracy description of molecular and periodic systems. Also, one or two dimensional polymers can be computed by setting the appropriate exact boundary conditions for given dimensions. For periodic systems, one or two HCP units were considered depending on the unit cell. The accuracy of the method was compared to high level CBS-QB3 results for molecules. The molecules were visualized using the Jmol program [21].
3 Results and discussions
3.1 The oligomers
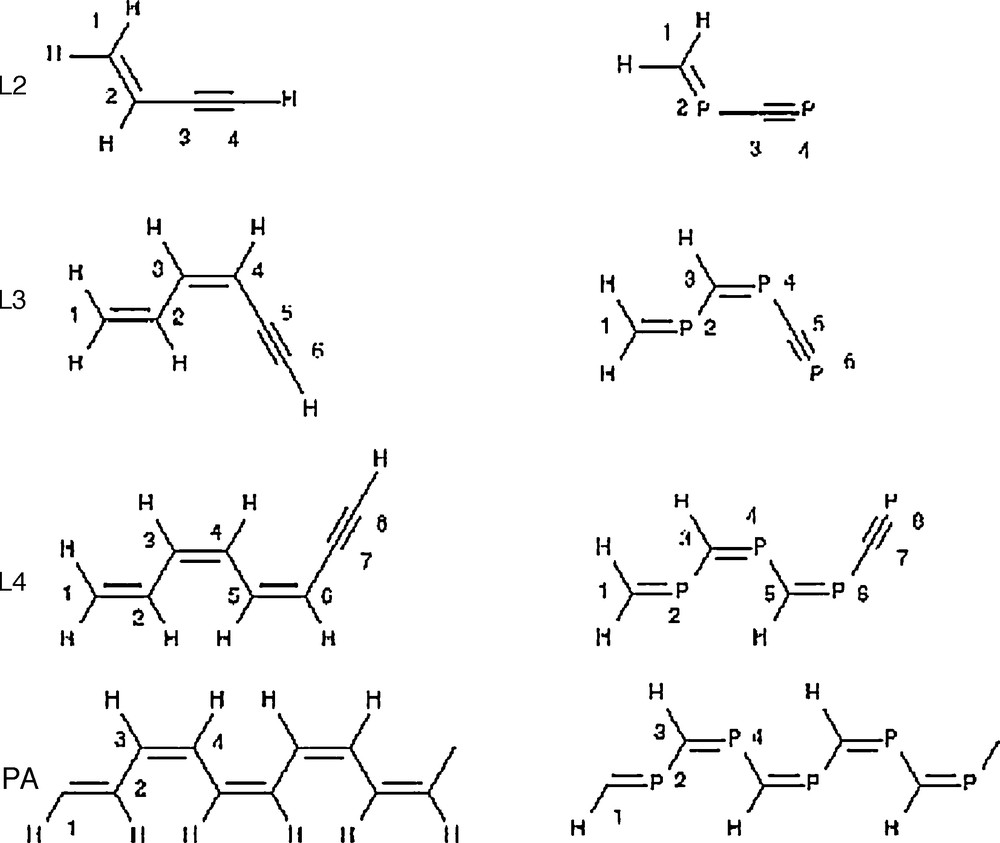
The computed geometries of acetylene, phosphaethyne and diphosphorus oligomers are summarized in Fig. 1. Their energetic stabilities compared to the monomers were measured using the dissociation energy of the given oligomer divided by the number of monomer units (Erel). These data are illustrated in Fig. 2. Isomers of acetylene and phosphaethyne dimers [4] and trimers [2b] were studied extensively in previous works and most of the small phosphorus cluster structures were also obtained [22]. Acetylene oligomers were also investigated, especially the isomers of benzene, where all possible isomers were aimed to be explored [23]. Here, we summarize the results briefly and compare the stabilities of these compounds.

Geometries of the computed oligomers of acetylene, phosphaethyne and diphosphorus.
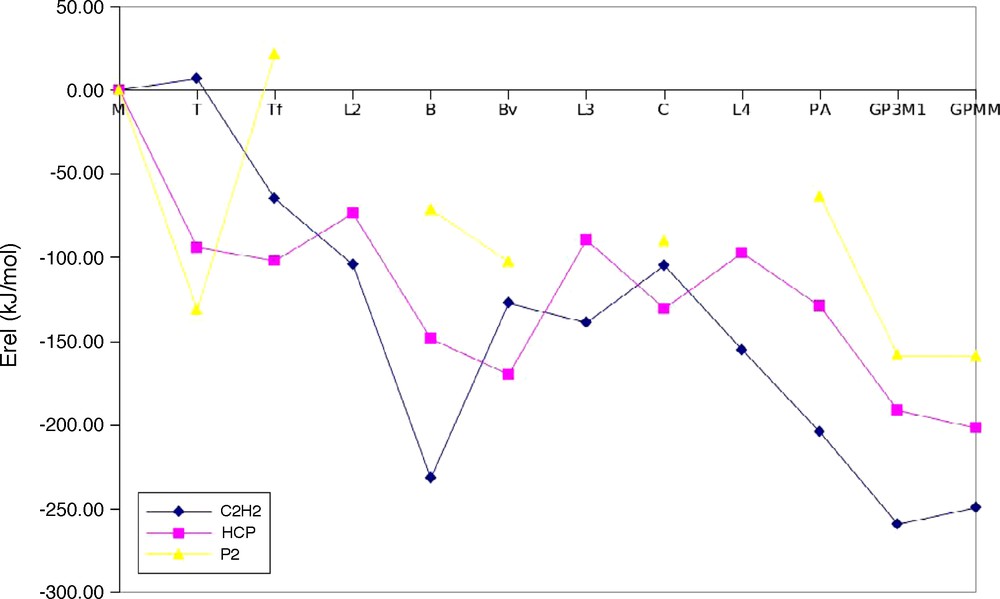
Dissociation energies of the acetylene, phosphaethyne and diphosphorus oligomers divided by the number of monomer units (Erel). For the notation of the compounds, see Figs. 1, 3 and 4. No low energy L2, L3 and L4 structures exist among pure phosphorus compounds.
The three most important factors which determine the relative stabilities of phosphorus compounds compared to carbon analogues are the following. First, due to presence of the lone pair and the inert s-orbital effect the chemical bonds around P are more pyramidal than along the carbon. It is well known that the HCH bond angle is 109.5° in methane, while it is only 92.4° (at PW91PW91/cc-pVTZ level of theory) in phosphine. Thus, P-compounds involving small bonding angles are more stable than the carbon analogues. This is well reflected in the tetrahedrane derivatives, as the Erel decreases as the CH groups are substituted by phosphorus successively. 1,2,-diphosphatriafulvene is more stable than triafulvene, 1,2,3-triphosphabenzvalene is more stable than benzvalene and tetraphosphacubane is more stable than cubane for the similar reason. Moreover, the lone pairs of phosphorus have stronger steric effects than the hydrogen in the CH group.
Second, the valence π orbitals of P are more diffuse than that of the carbon often resulting in smaller overlaps of atomic orbitals. This effect is, for example, responsible for the decreasing aromaticity as the CH groups of benzene are substituted by phosphorus. Therefore, in extended π systems the phosphorus atoms prefer close positions. Thus, the optimal position of the phosphorus atoms is determined by this fact, and also the steric effect of their lone pairs.
Third, phosphorus is more electropositive than carbon, which gives extra stabilization in, for e.g., different diphosphatriafulvenes [24]. Also, phosphorus readily forms pentavalent and ylide structures. For example, a formally pentavalent phosphorus can be found in 1,2,3,4-tetraphosphatriafulvene, but the compound is stabilized by an ylide structure, as showed in Scheme 2.
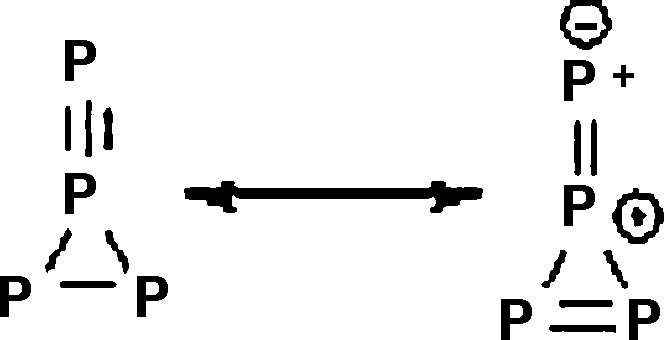
.
3.2 One-dimensional polyene derivatives
The two most important one-dimensional acetylene polymers are the cis- and the trans-polyacetylenes. However, due to geometrical reasons, trans-polyphosphaethyne is non-linear. Therefore, in this article we consider only the cis-isomer. Geometries of the optimized one-dimensional cis-polyacetylene-type (PA) polymers are displayed in Fig. 3.
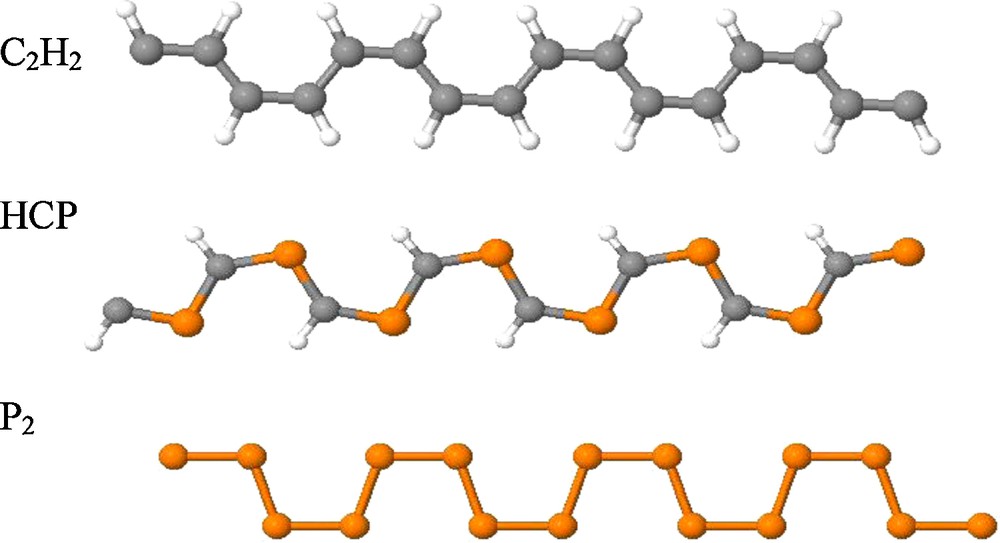
Geometries of the optimized structures of one dimensional cis-polymers (PA) of acetylene, phosphaethyne and diphosphorus.
The changes of the electronic structure during the transition from the monomers to the linear polymers are analyzed using the linear oligomers L2, L3 and L4 (Fig. 1). Unfortunately, no stable linear structures could be obtained in the case of pure phosphorus compounds. The geometric structures of these species are given in Fig. 3, while the relevant chemical bonding is depicted in Scheme 3. The computed interatomic distances, Wiberg-indices and HOMO-LUMO gaps are tabulated in Table 1.
.
Interatomic distances (in Å), Wiberg indices (in parentheses) and HOMO-LUMO gaps (H-L in eV) of L2, L3 and L4. For numbering, see Scheme 3.
| Bond | C2H2 | HCP | P2 | ||||||
| L2 | L3 | L4 | PA | L2 | L3 | L4 | PA | PA | |
| 1–2 | 1.343 | 1.345 | 1.346 | 1.377 | 1.683 | 1.686 | 1.684 | 1.718 | 2.094 |
| (1.87) | (1.85) | (1.83) | – | (1.79) | (1.76) | (1.76) | – | – | |
| 2–3 | 1.417 | 1.442 | 1.442 | 1.422 | 1.760 | 1.798 | 1.796 | 1.772 | 2.191 |
| (1.17) | (1.18) | (1.18) | – | (1.11) | (1.08) | (1.09) | – | – | |
| 3–4 | 1.213 | 1.359 | 1.363 | 1.377 | 1.569 | 1.704 | 1.710 | 1.718 | 2.094 |
| (2.79) | (1.70) | (1.67) | – | (2.64) | (1.60) | (1.58) | – | – | |
| 4–5 | – | 1.412 | 1.433 | 1.422 | – | 1.753 | 1.788 | 1.772 | 2.191 |
| – | (1.19) | (1.20) | – | – | (1.13) | (1.11) | – | – | |
| 5–6 | – | 1.215 | 1.363 | 1.377 | – | 1.571 | 1.707 | 1.718 | 2.094 |
| – | (2.77) | (1.67) | – | – | (2.60) | (1.57) | – | – | |
| 6–7 | – | – | 1.410 | 1.422 | – | – | 1.752 | 1.772 | 2.191 |
| – | – | (1.19) | – | – | – | (1.14) | – | – | |
| 7–8 | – | – | 1.215 | 1.377 | – | – | 1.571 | 1.718 | 2.094 |
| – | – | (2.76) | – | – | – | (2.59) | – | – | |
| H–L | 4.15 | 3.15 | 2.57 | 0.83* | 2.83 | 2.13 | 0.77 | 2.07* | 0.19 |
* Direct band-gap.
The computed bond lengths and Wiberg-indices fully support the electronic structures depicted in Scheme 3, namely, all oligomers have alternating single and double bonds and a triple bond in the end of the molecule. However, the Wiberg-indices indicate that the double bonds are conjugated to a small extent. The bond orders near the middle of the compound converge rapidly with the size of the oligomer, and the bonds 3–4 and 4–5 in L4 are close to those of the polymers. Therefore, it is expected that the corresponding Wiberg-indices of L4 approximate well the values of the polymers. As showed by the bond lengths, the geometry is disformed upon a Peierl's distortion. However, the Wiberg indices are somewhat smaller and larger than that of the double and single bonds, clearly indicating a significant electron delocalization. The band gap (HOMO-LUMO gap) converges slowly with the oligomer size.
The polymers of acetylene, phosphaethyne and diphosphorus are all direct-gap compounds, which means that the lowest-lying excitations are also possible by light irradiation. Cis-polyacetylene and cis-polyphosphaacetylene are considered as semiconductors, while the very small band-gap of the linear polymer of diphosphorus indicates that it is a highly unstable and light-sensitive compound. The band gap reduces monotonically as the CH groups of cis-polyacetylene are substituted by phosphorus atoms.
The relative energies of these compounds given in Fig. 2 show that poly-acetylene is the most stable thermodynamically. However, the thermodynamic stability of poly-phosphaethyne is comparable to the tetraphosphacubane. Tetraphosphacubane was synthesized with bulky substituents [3c,d] which prevented it from further reactions, making it also a kinetically stable compound. Therefore, it is expected that using protective groups instead of hydrogen, poly-phosphaethyne is also an interesting synthetic target. Its saturated analogue, poly-phosphaethene, was synthesized recently [25]. It has to be noted, however, that we have considered only the head-to-tail polymer of phosphaethyne, while other structures containing alternate head-to-head and head-to-tail parts are also possible which may give extra stabilization.
3.3 Two dimensional graphane sheets
Optimized geometries and bond lengths of the two dimensional graphane-like sheets are displayed in Fig. 4.
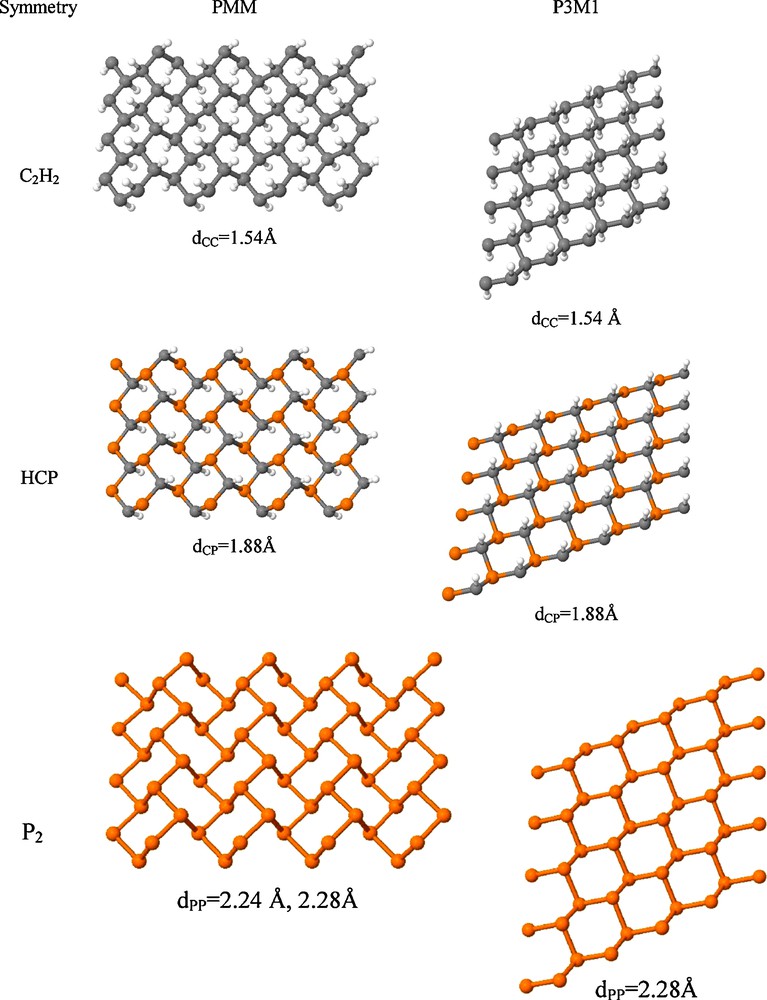
Geometries of the graphane-like (G) two dimensional polymers of acethylene, phosphaethyne and diphosphorus and the equilibrium bond lengths.
It was pointed out that graphane exists in two conformers, namely, the more stable chair (with P3M1 symmetry, denoted by GP3M1 in Fig. 1) and boat (PMM symmetry, denoted by GPMM in Fig. 1). The PMM conformation is analogous to the crystal structure of diamond, while the P3M1 conformation is analogous to the lonsdaelite allotrope of carbon [26]. Similarly, density functional computations demonstrated that the chair conformation of graphite monofluorite is the more stable [27]. In contrast, black phosphorus mainly exists in PMM symmetry layers. These findings are also reflected in the relative energies shown in Fig. 1, even though the energy difference between the pure phosphorus PMM and P3M1 is very small. It is interesting that the energy difference between both P3M1 and PMM structures decreases as the CH groups of graphane are consecutively substituted by P-atoms, thus giving a chemical possibility for tuning the crystal structure by changing the CH:P ratio (thus the acethylene:phosphaethyne ratio in copolymers).
In all cases considered, the graphane-like structures are thermodynamically more stable than the other oligomers or polymers examined. Graphane is even more stable than benzene, and graphane-like pure phosphorus structure is more stable than the P4 tetrahedron, in agreement with experimental findings. Therefore, it is expected that the graphane-like poly-phosphaethyne (GPP) can also be synthesized. GPP moieties may have been present in the originally synthesized phosphaethyne polymer. The direct band gap of GPP is 3.81 and 3.82 eV for the PMM and P3M1 isomers, respectively, somewhat larger than the reported values for graphane. The long bong lengths indicate single chemical bonds in all two-dimensional structures, as expected for fully saturated compounds.
4 Conclusion
Thermodynamic stability, electronic and structural properties of cis-polyacethylene and graphane-like polymers derived from phosphaethyne were studied using density functional theory methods. Their properties were compared to the corresponding oligomers and polymers of acetylene and diphosphorus. These compounds are thermodynamically stable, even though the one-dimensional cis-polyphosphaethyne needs kinetic stabilization by bulkier groups to be synthesized. Phosphaethyne-graphane may be present in the originally prepared polymers of phosphaethyne. Its promising properties, e.g., layered structure, large surface area, large band gap, chemically tunable properties by changing the acethylene:phosphaethyne ratio in copolymers, imply a wide range of potential applications.
Acknowledgements
The authors thank the Hungarian Scientific Research Foundation (OTKA) for financial support under the grant K76806. MTN thanks the K.U. Leuven Research Council (GOA and IDO programs) for continuing support.