

1 Introduction
The sugars that stem from natural chiral products present particular structures in which the chirality of each carbon atom bearing a hydroxyl group is well known. Thus, sugar derived structures and their derivatives are likely to present interesting properties in the field of materials. Reduced sugars hold OH functions regularly distributed on a carbon chain whose carbon atoms present well defined chiralities. The aim of this article is to describe some possibilities opened by three commercially available hexitols (mannitol, sorbitol, dulcitol) in the field of new materials. Moreover, these polyols are very cheap since they are by-products of industrial sugars used for food and the four regularly distributed OH functional groups can be used for chelation of ions and nanoparticles. The presence of these chiral moieties in nanostructured silica hybrids is of great interest for several potential applications: chiral stationary phase for enantiomeric separation, ion separation, catalytic materials for asymmetric synthesis and induction of chirality on nanoparticles presenting some physical properties…
The chemistry of organic–inorganic hybrid materials is expanding because it offers access to new kinds of nanomaterials. It consists in a “bottom-up” approach for the synthesis of hybrid materials. Moreover, this field of research bridges different types of chemistry (organic, inorganic, coordination…) to material science and might open also many possibilities by the encapsulation of chemical units presenting physical or biological properties. This methodology represents a very efficient and powerful tool since it makes the material science compatible with many different aspects of chemistry that were initially separated: solid state chemistry, organic and inorganic chemistry, organometallic and coordination chemistry, macromolecular chemistry and also biomolecules [1–7]. All the molecular organic precursors containing at least two Si(OR)3 groups can be transformed into silica-based hybrid materials by hydrolytic sol-gel polycondensation [1].
Obtaining chiral sol-gel materials has been an important challenge for several years [8]. The reason for this growing interest in chiral sol-gel materials mainly results from the various potential applications of these materials such as selective adsorption, catalysis, separation and sensors. The methods for inducing chirality in silicas and sol-gel materials can be divided into four approaches [8]: the silylation of silica surfaces by silanes bearing a chiral group [9,10] the use of chiral silanes as precursors in sol-gel polycondensation [11–18], the physical entrapment of chiral molecules and biomolecules during sol-gel polycondensation reaction [19,20] and the imprinting with chiral templates [21–23]. The design and synthesis of chiral hybrid materials with optical activity and enantioselectivity remains a challenge. The preparation of monodisperse metal nanoparticles can induce physical properties and catalytic properties highly depending on their size [24]. Supported gold nanoparticles have recently emerged as excellent catalysts for a broad range of organic transformations under mild conditions [25–27]. Materials containing gold (0) nanoparticles located within the pore channels have been prepared by anchoring an organogold precursor within the channels of functionalized ordered mesoporous silica followed by chemical reduction [28]. The interactions between metal ions and carbohydrates, namely the coordination of hydroxyl groups to metal ions, have been shown to be of fundamental importance in many biochemical processes. The study of sugar–metal ion interactions is one of the main objectives of carbohydrate coordination chemistry. Work has been reported on these interactions in the case of erythritol, galactitol (dulcitol) or glucitol (sorbitol) and a series of lanthanide chloride–galacticol complexes have been described [29–32].
The aim of the work presented here was to immobilize mannitol, sorbitol and dulcitol to form chiral hybrid silicas. In first instance, our wish was to use them for enantioselective purposes. As it was not possible to isolate the chiral molecular precursors (vide infra), thus to design correctly the final material (i.e. no control of the textural properties, specific surface area, porosity that are under kinetic control), we evaluated their potential for other applications such as the growth of monodisperse gold nanoparticles and their ability for lanthanides complexation.
2 Experimental
2.1 General
The commercially available mannitol, sorbitol and dulcitol were dried under vacuum before use. 3-iodopropyltriethoxysilane was prepared according to the literature procedure [33]. Reagent grade 3-(triethoxysilyl)propylisocyanate was commercially available and used without purification. Hydrogen tetrachloroaurate (III) hydrate, LaCl3 (8H2O), EuCl3 (6H2O) and YbCl3 were used as purchased. All reactions were carried out under argon using a vacuum line and Schlenk techniques. Solvents were dried and distilled just before use.
Infrared spectra were recorded on a FT-IR Perkin-Elmer 1600 neat or in KBr pellets. Raman spectra were recorded on a Horiba Jobin Yvon LabRAM ARAMIS neat (laser helium–neon 633 nm). The 29Si CP MAS NMR spectra were recorded on a Bruker FTAM 300 as were 13C CP MAS NMR spectra, in both cases, the repetition time was 10 and 5 s with contact times of 5 and 3 ms. The Tn notations are given for [R-Si(OSi)n(OR′)3 − n] environments. The specific surface areas of the xerogels were obtained in a Micromeritics Gemini III 2375 after one night vacuum (10−2 mbar) at 120 °C. Elemental analyses were performed by the Service central d’analyse (CNRS, Vernaison, France). Transmission electron microscopy (TEM) observations were carried out at 100 kV (JEOL 1200 EXII). Samples for TEM measurements were prepared by embedding the hydrid material in AGAR 100 resin, followed by ultramicrotomy techniques and deposition on copper grids.
2.2 Synthesis of material
2.2.1 General procedure for the synthesis of materials MX, SX, DX
To a solution of 1 g of polyol (M, S or D) (5.5 mmol) in dry DMF (5 ml) were added under argon 1.53 mL of freshly distilled triethylamine (11.1 mmol) and 2.71 mL of 3-(triethoxysilyl)propylisocyanate (11.1 mmol). The mixture was heated 90 °C for 3 days (mannitol, sorbitol) and 7 days (dulcitol). During this time a pale brown solid was appearing. The solvent was then removed under vacuum for 24 h. The resulting solid was ground with ethanol, then washed with ethanol under Soxhlet conditions, followed by acetone washing (Soxhlet) and then dried under vacuum to give a pale yellow powder.
2.2.1.1 MX: 1.98 g (79%)
Elem. Anal.: Found: C, 35.76; H, 6.09; N, 5.68; Si, 12.19 Calc. for C14H26N2O11Si2: C, 37.00; H, 5.73; N, 6.17; Si, 12.33%. FTIR νmax/cm−1 3318, 2932, 2876, 1688, 1530, 1247, 1073 and 1021. 13C CP-MAS NMR (δ, ppm): 157.8, 69.9, 58.4, 44.4, 23.8, 18.2, 10.3. 29Si CP MAS NMR: (δ, ppm): −44.5 (T0), −58.3 (T2), −66.3 (T3). Level of condensation at silicon 82%. BET surface area <10 m2g−1.
2.2.1.2 SX: 1.05 g (42%)
Elem. Anal. Found: C, 34.73; H, 6.13; N, 6.32; Si, 13.34 Calc. for C14H26N2O11Si2: C, 37.00; H, 5.73; N, 6.17; Si, 12.33%. FTIR νmax/cm−1 3325, 2929, 2890, 1693, 1537, 1249, 1096 and 1031. 13C CP MAS NMR: (δ, ppm) = 157.6, 70.2, 43.7, 23.8, 10.2. 29Si CP MAS NMR: (δ, ppm) = −67.4 (T3). Level of condensation at silicon >98%. BET surface area <10 m2g−1.
2.2.1.3 DX: 1.96 g (78%)
Elem. Anal.: Found: C, 37.87; H, 6.64; N, 5.58; Si, 11.18 Calc. for C14H26N2O11Si2: C, 37.00; H, 5.73; N, 6.17, Si, 12.33%. FTIR νmax/cm−1 3315, 2942, 1709, 1542, 1265 and 1078. 13C CP MAS NMR: (δ, ppm): 158.3, 60–80, 58.9, 44.2, 24.5, 18.5, 10.7. 29Si CP MAS NMR: (δ, ppm): −45.0 (T0), −59.2 (T2), −65.2 (T3). Level of condensation at silicon 76%. BET surface area <10 m2g−1.
2.2.2 Procedure for the synthesis of materials MY, SY, DY: optimized procedure
2.2.2.1 SY
To a solution of 5 g of sorbitol (27.44 mmol) and 9.68 g of K2CO3 (68.61 mmol) in DMF/H2O (3/1: v/v, 40 mL) was added, under argon, 18.25 g of 3-iodopropyltriethoxysilane (54.88 mmol). The mixture was heated at 100 °C for 4 days. Then the reaction mixture was decanted (the lower layer contained a white solid, the upper layer was homogeneous). The upper layer was collected and concentrated under vacuum at room temperature. The viscous oil became solid after several hours. The resulting solid was ground with ethanol, then washed with ethanol under Soxhlet conditions, followed by acetone washing (Soxhlet) and then dried under vacuum to give a white powder.
SY: 6.25 g (62%). Elem. Anal. Found: C, 33.94; H, 6.00; Si, 14.52 (N, 0.80; I, traces; K, traces). Calc. for C12H24O9Si2: C, 39.11; H, 6.56; Si, 15.24. FTIR νmax/cm−1 3340, 2941, 1024. 13C CP MAS NMR (δ, ppm): 72.1, 64.1, 52.6, 30.7, 25.8, 16.3, 9.2. 29Si CP MAS NMR (δ, ppm): −67.6. (T3). Level of condensation at silicon >95%. BET surface area <10 m2g−1.
2.2.2.2 MY, DY
To a solution of 1 g of mannitol or dulcitol (5.49 mmol) and 1.62 g of K2CO3 (11.72 mmol) in DMF/H2O (3/1: v/v, 8 mL) was added, under argon, 3.6 g of 3-iodopropyltriethoxysilane (10.83 mmol). The mixture was heated at 100 °C for 2 days. The same treatment used in the case of SY gave a white powder.
MY: 0.78 g (38%). Elem. Anal. Found: C, 28.46; H, 5.84; Si, 17.65 (N, 1.14; I, 1.44; K, 3.83). Calc. for C12H24O9Si2: C, 39.11; H, 6.56; Si, 15.24. FTIR νmax/cm−1 3360, 2931, 2876, 1008. 13C CP MAS NMR (δ, ppm): 71.0, 64.4, 52.9, 26.5, 16.9, 9.9. 29Si CP MAS NMR (δ, ppm): −67.2 (T3). Level of condensation at silicon = 92%. BET surface area <10 m2g−1.
DY: 1.29 g (64%). Elem. Anal.: Found: C, 30.33; H, 6.49; Si, 13.86 (N, 0.94; I, 1.07; K, 0.63). Calc. for C12H24O9Si2: C, 39.11; H, 6.56; Si, 15.24. FTIR νmax/cm−1 3260, 2934, 2880, 1026. 13C CP MAS NMR (δ, ppm): 72.1, 70.6, 64.1, 52.1, 26.4, 16.8, 10.2. 29Si CP MAS NMR (δ, ppm): −59.9(T2), −67.6. (T3). Level of condensation at silicon = 91%. BET surface area <10 m2g−1.
2.2.3 Synthesis of the gold nanoparticles: optimized procedure
To a suspension of 50 mg of X or Y in deionized water (18 mL) was added a solution of HAuCl4·3H2O in water (2 mL) with various Au/polyol ratios (Table 1). The resulting mixture was stirred at different temperatures and times (Table 1). The yellow solution became colorless and the white solid turned yellow then beige or purple. After filtration, the solid was washed several times with water, then dried under vacuum at room temperature and analyzed by TEM.
Experimental conditions for gold complexation and nanoparticles formation within SX using hydrogenotetrachloroaurate (III) hydrate (HAuCl4·3H2O) as gold precursor.
| Entry | Au/polyol ratio | Temperature, °C (time, h) | Nanoparticles size (ϕ nm) |
| 1 | 0.2 | 60 (18) | 20–30 |
| 2 | 0.2 | 60 (3)a | <10 |
| 3 | 0.1 | 40 (3.5) + 90 (1.5) | 5–25 |
| 4 | 0.1 | 40 (3) + 50 (3.5) | 1–5 and 20 |
| 5 | 0.04 | 60 (18) | <10 |
| 6 | 0.04 | 60 (3)a | <10 |
| 7 | 0.02 | 40 (6) | <10 |
| 8 | 0.02 | 40 (4)b | 5 and 20–30 |
a Followed by NaBH4 reduction at 60 °C during 15 h.
b Followed by treatment with citric acid trisodium salt during 2 h at 40 °C.
2.2.4 Ions complexation
To a suspension of one equivalent of X or Y in absolute ethanol (C = 0.136 M) was added 1.5 equivalents of LaCl3 (8H2O), or EuCl3 (6H2O) or YbCl3 (Table 3). The resulting mixture was stirred at 90 °C overnight. The suspension was then filtered and washed by ethanol. The filtrate was concentrated under vacuum at room temperature to obtain a residue, which was dissolved in 50 mL of a buffer solution (pH = 5.5) and titrated by a solution of EDTA (C = 0.002 M) with xylenol orange sodium salt as colored indicator. This titration gave the amount of lanthanide, which was not complexed to the osidic core (nosidic core). The amount (nLn) of complexed lanthanide was then deduced by difference (Table 3).
3 Results and discussion
The three polyols, mannitol M, sorbitol S and dulcitol D, that are by-products of the sugar industry, are very attractive molecular precursors for obtaining new silica hybrid materials. Indeed, these three polyols are very cheap, they present natural chirality and differ only by the stereochemistry around the four central carbon atoms (Fig. 1). Mannitol M exhibits an axis of symmetry, dulcitol D, a centre of symmetry and sorbitol S no symmetry.
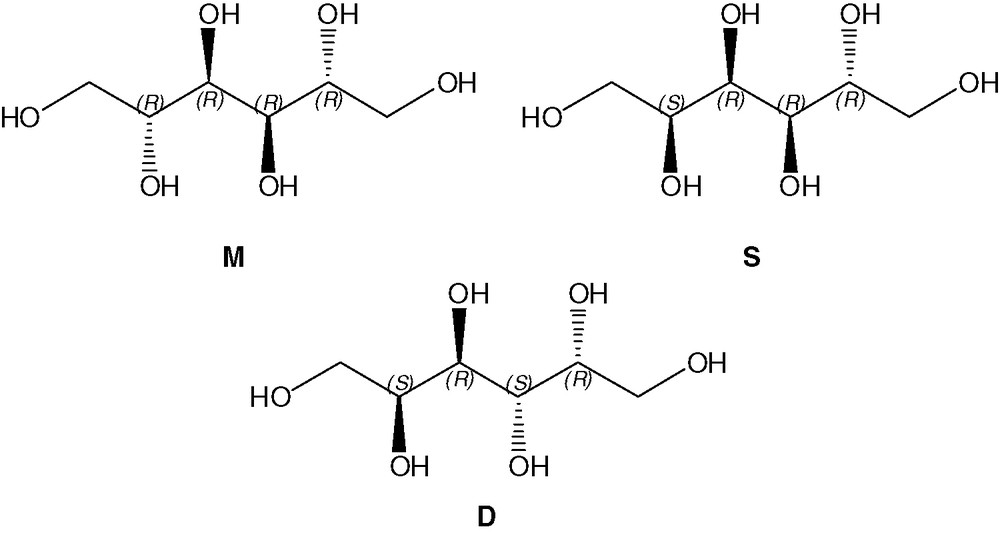
Molecular precursors M, S, D.
Taking into account that, generally, primary alcohols react faster than secondary ones, two different routes were explored to introduce trialkoxysilyl hydrolysable groups on the polyols core (route A, Fig. 2 and route B, Fig. 4).

Route A.
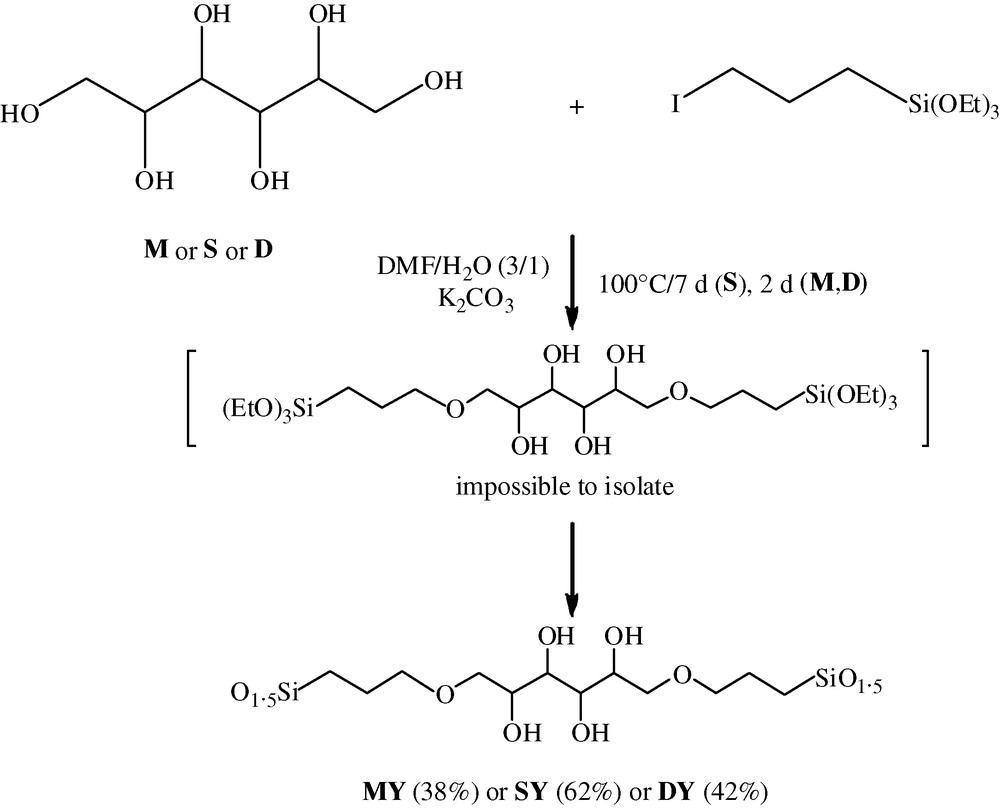
Route B.
3.1 Route A
The polyols (M, S or D) were treated by two equivalents of 3-(triethoxysilylpropyl)isocyanate and two equivalents of triethylamine in dry DMF under reflux during 3 days (M, S) or 7 days (D), leading to the formation of carbamate bridges between the polyol core and the hydrolysable alkoxy groups. Under these conditions, hard solids appeared spontaneously in the reaction mixture. These solids were washed with ethanol in order to eliminate by-products. As observed on 29Si NMR spectra (see below and the Supplementary material), the polycondensed materials MX, SX, DX were directly isolated (Fig. 2).
The solids MX, SX, DX were characterized by spectroscopic techniques, which clearly showed that polycondensation occurred. The 13C CP MAS NMR spectra revealed the presence of the organic units in the inorganic network. The broad signal centred at ∼70 ppm was attributed to the organic osidic core in all cases. The carbon atoms of the carbamate arms appeared at 10 ppm (CONHCH2CH2CH2Si), 23.8 ppm (CONHCH2CH2CH2Si), 43–44 ppm (CONHCH2CH2CH2Si) and ∼158 ppm (CONHCH2CH2CH2Si). In the case of MX and DX weak signals appeared at ∼18.2 ppm (SiOCH2CH3), and ∼58.4 ppm (SiOCH2CH3), indicative of some residual triethoxysilyl groups. The level of condensation at silicon was deduced from the 29Si CP MAS NMR spectra. This level was estimated in first approximation by deconvolution of the spectra [34,35]. Moreover, since materials of the same precursor are compared, it can be assumed that relative peak intensity can be used as indicative of the order of magnitude for the level of condensation (LC), which was calculated according to the general equation: LC = [0.5(T1 × area) + 1.0(T2 × area) + 1.5(T3 × area)]/1.5. These spectra exhibited two weak signals at ∼−45 ppm and ∼−58 ppm, (T0 substructure [C–Si(OR)3]) and T2 substructure [C–Si(OR)(OSi)2]) and a major one at −66 ppm, (T3 substructure [C–Si(OSi)3]) in the case of MX and DX, (levels of condensation respectively 82 and 76%). In the case of SX, only one signal at ∼−67 ppm (T3 substructure) was present (level of condensation 98%).
Certainly because of the poor solubility of dulcitol in the reaction mixture, the reaction time was 9 days and DX appeared less polycondensed (76%). Hydrogen bond association was evidenced by FTIR spectroscopy. In all cases, a broad absorption band at ∼3320 cm−1 was attributed to hydrogen bonded OH and NH of carbamate groups. This was in agreement with the shift of νC O and δNH stretching absorptions towards respectively ∼1690 and ∼1530 cm−1. [36] This shift was slightly different, considering MX, SX, or DX, since the osidic cores differ by their stereochemistry around the four central carbon atoms and by the symmetry of the core.
During the solids formation, first the polyol reacted with 3-(triethoxysilylpropyl)isocyanate, leading to carbamate units M′ (Fig. 3), that then polycondensed to give MX. In order to isolate the precursor M′, an attempt at purification by washing the crude solid with dry toluene was investigated in the case of MX. It allowed us to isolate a slightly polycondensed material M′ that exhibited already H-bonds association between carbamate units (νC O at 1698 and 1660 cm−1 and δNH at 1529 cm−1). Moreover, the νCH stretching vibration bands were more intense in the case of M′ compared to MX, indicating the presence of a greater amount of non polycondensed ethoxy groups in M′ (cf. ESI). The direct formation of polycondensed solids can be connected with the existence of a tight network of these observed hydrogen bonds, on one hand between the four OH groups of the osidic core, and on the other hand, between the carbamate groups (Fig. 3). This favored the aggregation of the molecules into chains by minimizing the entropy. Thus, the Si(OEt)3 groups could react more easily one with each other due to their closeness and because the formation of the SiOSi bonds represents an energetic gain. This higher level of condensation at silicon observed for SX can be attributed to a difference in the self-assembling of the organic cores due to the different configurations of the polyols.
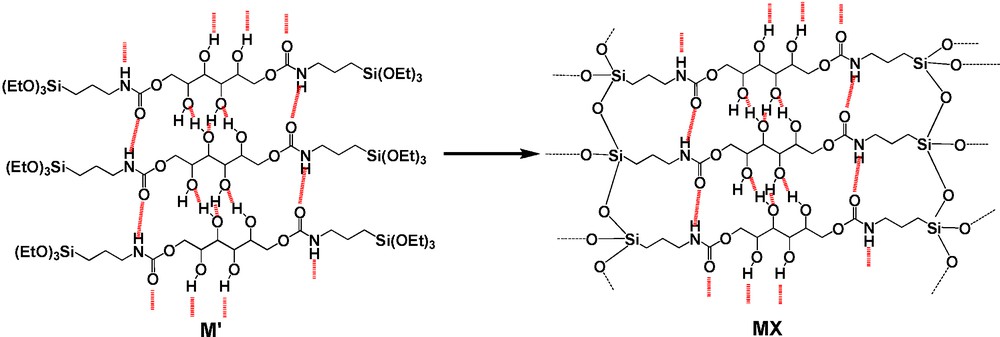
Hydrogen bonds network between hydroxy and carbamate groups.
However, the XRD pattern of the solids exhibited broad signals (no Bragg peaks). All these solids exhibited no significant specific surface areas (<10 m2/g).
3.2 Route B
This route (Fig. 4) involved the reaction of the polyols (M, S or D) with 2 equivalents of 3-iodopropyltriethoxysilane in the presence of K2CO3 in DMF at 100 °C during 4 days (S) or 2 days (M, D).
In this case, the polycondensed solids MY, SY, DY were also directly obtained. However an optimization of the experimental procedure was necessary since the reaction occurred in heterogeneous conditions (K2CO3 is insoluble in DMF). In the case of mannitol, when dry DMF was employed, the whole reaction mixture solidified during the heating. Therefore the obtained solid MY (overall yield 49%) was a co-gel containing 24% of I(CH2)3SiO1.5 as determined by elemental analysis (%I = 10.60, %Si = 9.76). When water was added (DMF/water: 3/1), the resulting biphasic mixture permitted the coupling between the iodide compound and the polyol. Then the liquid phase was collected to carry on the formation of solids Y, which contained only traces of I(CH2)3SiO1.5 (See experimental data).
The 13C CP MAS NMR spectra revealed the presence of the organic units in the inorganic network. The broad signal centred at ∼70 ppm was attributed to the organic osidic core. The carbon atoms of the propyl arms were present at 9–10 ppm (OCH2CH2CH2Si), ∼26 ppm (OCH2CH2CH2Si), ∼64 ppm (OCH2CH2CH2Si). In the case of solids Y, weak signals appeared at ∼16.3 ppm (SiOCH2CH3), and ∼52.6 ppm (SiOCH2CH3), indicative of some residual triethoxysilyl groups. 29Si CP MAS NMR spectra exhibited one signal at ∼−67 ppm, indicative of a high level of condensation (>95%), certainly due to the synthetic conditions (100 °C in an aqueous basic medium).
As we observed for materials X, these materials also exhibited no significant specific surface areas (<10 m2/g).
3.3 Synthesis of the gold nanoparticles
The methodology generally employed was based on metallic precursor impregnation into the support with subsequent reduction/decomposition of the metal precursor to generate the nanoparticles. Recently, in homogeneous conditions, different sugars have been successfully employed as reducing as well as stabilizing agents for the nanoparticle evolution [37].
We studied the gold complexation and nanoparticle formation within the osidic core of hybrid solids X and Y according to a previously described procedure [28]. The gold precursor used was the hydrogenotetrachloroaurate (III) hydrate (HAuCl4·3H2O). We prospected on the solid SX the influence of the use of reducing agents and that of the Au/polyol ratio on the size and the density of nanoparticles (Table 1).
The main observations arising from these preliminary experiments are the following:
- i) for the same Au/polyol ratio, the use of a reducing agent was not necessary to form nanoparticles as previously reported [37] (Table 1, entries 1 and 2; 5 and 6), the Au/polyol ratio influenced only the size of particles;
- ii) the use of citric acid trisodium salt increased the nanoparticle size (Table 1, entries 7 and 8);
- iii) a higher temperature favored nanoparticle aggregation (Table 1, entries 3 and 4);
- iv) decreasing the ratio Au/polyol induced a decrease in density and size of nanoparticles (Table 1, entries 1 and 5; 4 and 7).
TEM images of nanoparticles (Table 1, entries 4 and 8) are given in Fig. 5, the other ones are given in the supplementary material. These particles appeared to be stable since TEM imaging performed after 3 months were unchanged.

TEM images of gold nanoparticles within SX, a) Table 1, entry 4; b) Table 1, entry 8.
The experimental conditions reported in entry 4, (Table 1), which allowed the formation of two distinct populations of nanoparticles, were applied to all the solids X and Y (Table 2) in order to evaluate the influence of the stereochemistry and the spacers of the osidic core on the size distribution of gold nanoparticles.
Influence du cœur osidique sur la complexation et la formation de nanoparticules d’or (Au/polyol ratio : 0.1 ; température [durée] 40 °C [3 heures] + 50 °C [3.5 heures]).
| Entry | Materials | Nanoparticles size (ϕ nm) |
| 1 | SX | 1–5 and 20 (2 populations) |
| 2 | MX | Broad range 1–25 (geometric shapes for the big ones) |
| 3 | DX | Broad range 1–15 |
| 4 | SY | Broad range 10–50 |
| 5 | MY | Broad range 10–75 (aggregates) |
| 6 | DY | Broad range 25–60 |
The main observation arising from these results is the great influence of the spacer on the nanoparticle size. Smaller nanoparticles were formed in the case of carbamate spacer (solids X, Table 2, entries 1–3). That is probably due to the presence of the tight H-bonds network between carbamate groups, which hindered the access to complexation sites thus controlling the growth of particles. Moreover, it was easier to incorporate nanoparticles in Y solids where the complexation sites were more accessible because of the absence of H-bond network between the more flexible spacers. Bigger nanoparticles formed (Table 2, entries 4–6).
In the case of SX, it is possible to control the size distribution (between 1 and 5 nm), despite the presence of larger particles certainly due to the agglomeration of the smallest ones. Nevertheless, we demonstrated the ability of these materials to immobilize and stabilize nanoparticles avoiding the use of reducing agent. TEM images are given in Fig. 6 and in the Supplementary material.

TEM images of gold nanoparticles within materials, a) Table 2, entry 3; b) Table 2, entry 5.
3.4 Ions complexation
The study of sugar–metal ion interactions is one of the main objectives of carbohydrate coordination chemistry [29–32]. Considering the results reported on these interactions between various polyols and a series of lanthanide chlorides, it appeared interesting to test the complexation of different lanthanides on the hybrid solids X and Y in heterogeneous conditions. We investigated three lanthanide chlorides, LaCl3 (8H2O), or EuCl3 (6H2O) or YbCl3. The percentages of complexation observed after 15 h were determined by titration (see experimental) and are reported in Table 3.
The main observation arising from these preliminary experiments is that the materials MX, SX and DX are very weak complexing reagents compared with MY, SY and DY. As in the case of the growth of gold nanoparticles, this difference can be explained by the hindrance around the coordination sites due to the presence of a very tight and strong H-bonding network between the carbamate units. The degree of complexation of lanthanide ions seemed to be influenced by the size of their ionic radius (La3+, 1.032 Å; Eu3+, 0.947 Å; Yb3+, 0.868 Å) [38] on one hand, and the stereochemistry and the symmetry of the osidic core on the other hand. We observed that mannitol derivatives showed a better affinity for middle size ions. Dulcitol derivatives seemed to be better ligands for biggest lanthanides while smallest ones were better complexed by sorbitol derivatives (Table 3). Furthermore, the presence of an axis of symmetry seemed to be more efficient than that of a centre of symmetry since mannitol exhibited the best complexing ability. In the case of the materials exhibiting the highest level of complexation, Raman spectroscopy was studied. The spectra of MY@La and MY@Eu, compared to that of MY clearly evidenced that ions complexation had occurred as shown in Fig. 7. After complex formation, the spectra differed since new bands appeared at ∼3600 and ∼860 cm−1 for both MY@La and MY@Eu, and respectively at 1662 and 1582 cm−1 as previously reported in the case of similar polyol-lanthanide complexes [29]. In the case of SY@Yb, new bands appeared 1603, 1585 and 1159 cm−1. These new bands are attributed to the coordination of the lanthanides with the polyols [29]. They appeared in our case despite the low levels of complexation (Table 3).
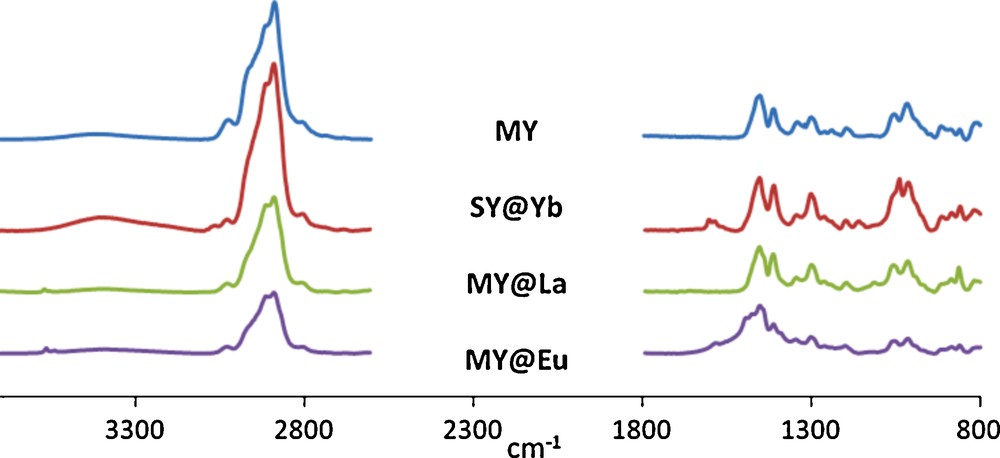
Raman spectra of materials@lanthanide complexes.
4 Conclusion
We report here a valorization of cheap polyols that are by-products of sugar manufacturing by using them for the preparation of new silica hybrid materials. The strong hydrogen bonds from the polyols moieties on the one hand, and the carbamate units on the other hand play a fundamental role allowing a one step synthesis of these materials, particularly by lowering the entropy of the system. These hybrid solids appeared to be efficient stabilizing matrices for gold nanoparticles, the polyol units avoiding the use of reducing agents. The solids bearing carbamate bridges X allowed a better control of the nanoparticles size and distribution. Lanthanide ions were complexed with different levels of complexation (up to 45%). In this case, solids X were less efficient than solids Y. The level of complexation was different depending on the nature of the polyol. These results might be useful for lanthanides separation. Work is in progress to control the molecular precursors formation, thus to design the textural properties of the solids in order to use them for enantioselective purposes. The optimization and the extension of complexation reactions to other metal ions and the study of the selectivity are also in progress.
Electronic supporting information available: 13C and 29Si solid state NMR and FTIR spectra of MX, SX, DX, M′, MY, SY, DY and TEM images of gold nanoparticles described in Tables 1 and 2.